

Abstract
Varenicline, a widely used and successful smoking cessation agent, acts as a partial agonist at nicotinic acetylcholine receptors. Here, we explore the effects of varenicline at human and mouse 5-Hydroxytryptamine3 (5-HT3) receptors. Application of varenicline to human 5-HT3 receptors expressed in Xenopus laevis oocytes reveal it is almost a full agonist (Rmax = 80%) with an EC50 (5.9 μM) 3-fold higher than 5-HT. At mouse 5-HT3 receptors varenicline is a partial agonist (Rmax = 35%) with an EC50 (18 μM) 20-fold higher than 5-HT. Displacement of the competitive 5-HT3 receptor antagonist [3H]granisetron reveals similar IC50 values for varenicline at mouse and human receptors expressed in human embryonic kidney 293 cells, although studies in these cells using a membrane potential-sensitive dye show that again varenicline is a 4- or 35-fold less potent agonist than 5-HT in human and mouse receptors, respectively. Thus the data suggest that the efficacy, but not the affinity, of varenicline is greater at human 5-HT3 receptors compared with mouse. Docking studies provide a possible explanation for this difference, because they suggest distinct orientations of the ligand in the mouse versus human 5-HT3 agonist binding sites. Additional binding selectivity studies in a broad panel of recombinant receptors and enzymes confirmed an interaction with 5-HT3 receptors but revealed no additional interactions of varenicline. Therefore, activation of human 5-HT3 receptors may be responsible for some of the side effects that preclude use of higher doses during varenicline treatment.
Introduction
The 5-HT3 receptor is a member of the Cys-loop ligand-gated ion channel family, of which the nACh receptor is the prototypic member. These receptors, which play roles in synaptic transmission in both the central and peripheral nervous systems, are pentameric assemblies of one, or, more usually, several subunits, which surround a central ion-conducting pore. Five 5-HT3 receptor subunits (A–E) have been identified to date, and whereas the A subunit can form functional homomeric receptors, subunits B to E function only as heteromeric receptors in combination with the A subunit (Davies et al., 1999; Niesler et al., 2003). In the human nervous system, 5-HT3A subunits have been found throughout the adult brain, but are also widely distributed in extraneuronal cells (Miyake et al., 1995; Fiebich et al., 2004). 5-HT3B receptor subunits are not as widespread, but are still detectable in a range of tissues (Davies et al., 1999; Dubin et al., 1999; Niesler et al., 2003; Tzvetkov et al., 2007). The other subunits have been less well studied, and their functional significance is not yet clear (Thompson et al., 2010a).
5-HT3 receptor antagonists are in use clinically, primarily for controlling chemotherapy- and radiotherapy-induced nausea and vomiting, and in postoperative nausea and vomiting. Alosetron is an effective symptomatic treatment for irritable bowel syndrome. In addition, studies have revealed a diversity of potential disease targets that might be amenable to alleviation by 5-HT3 receptor-selective compounds; these include addiction, pruritis, emesis, fibromyalgia, migraine, rheumatic diseases, and neurological phenomena such as anxiety, psychosis, nociception, and cognitive function (Thompson and Lummis, 2007).
Structural details of the 5-HT3 receptor at the molecular level are unresolved, but a wealth of evidence shows that the structure of these receptors is closely related to the structure of the nACh receptor (see Thompson et al., 2010a and Reeves and Lummis, 2002 for reviews). Consequently, the 5-HT3 receptor is thought to be well represented by cryo-electron microscope images of the nACh receptor and crystal structures of the acetylcholine binding protein (AChBP), a protein homologous with the extracellular domain of the nACh receptor (Brejc et al., 2001). Perhaps unsurprisingly, because the 5-HT3 and nACh receptors have similar functions (agonist binding results in the activation of a cation-selective ion channel), there are some overlaps in their pharmacological profiles; d-tubocurarine for example, which classically is considered to be a nACh receptor antagonist, is also a highly potent competitive, 5-HT3 receptor antagonist.
Varenicline, a successful smoking cessation agent, is a derivative of cytisine, a nACh receptor agonist (Coe et al., 2005; Lowe et al., 2010). Varenicline is also a nACh receptor agonist, and, like cytisine, it has different potencies and efficacies at different subtypes of nACh receptors. At the α4β2 receptor, varenicline is a potent partial agonist, whereas at the α3β4 receptor it is a weak partial agonist. However, at the α7 nACh receptor it is a full agonist, albeit with a comparatively high EC50 (Mihalak et al., 2006). Here, we explore the effect of varenicline at the 5-HT3 receptor, a protein with striking homology to the nAChR. The data show that varenicline is a potent agonist at the human but not the mouse 5-HT3 receptor.
Materials and Methods
Materials.
All cell culture reagents were obtained from Invitrogen (Paisley, UK), except fetal calf serum, which was from Labtech International (Ringmer, U.K.). [3H]granisetron (63.5 Ci/mmol) was from PerkinElmer Life and Analytical Sciences (Waltham, MA). All other reagents were of the highest obtainable grade.
Cell Culture.
Human embryonic kidney (HEK) 293 cells were maintained on 90-mm tissue culture plates at 37°C and 7% CO2 in a humidified atmosphere. They were cultured in Dulbecco's modified Eagle's medium/nutrient mix F12 (1:1) with GlutaMAX I media containing 10% fetal calf serum. For radioligand binding studies cells were transfected by using electroporation and plated in 90-mm dishes. For FlexStation studies, cells were transfected and then plated in 96-well plates. Cells were incubated 1 to 2 days before assay.
Oocytes.
Harvested stage V to VI Xenopus laevis oocytes were washed in four changes of ND96 (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 5 mM HEPES, pH 7.5), defolliculated in 1.5 mg · ml−1 collagenase type 1A for approximately 2 h, washed again in four changes of ND96, and stored in ND96 containing 2.5 mM sodium pyruvate, 50 mM gentamycin, and 0.7 mM theophylline. Human 5-HT3A (GenBank accession number P46098) and mouse 5-HT3A (GenBank accession number Q6JIJ7) receptor subunit cDNAs were cloned into pGEMHE for oocyte expression (Liman et al., 1992). cRNA was transcribed in vitro from linearized plasmid cDNA template using the mMessage mMachine T7 transcription kit (Ambion, Austin, TX). Stage V and VI oocytes were injected with 20 ng of cRNA, and currents were recorded 2 to 3 days postinjection.
Electrophysiology.
Using a two-electrode voltage clamp, X. laevis oocytes were clamped at −60 mV with an OpusXpress (Molecular Devices, Wokingham, UK) as described previously (Lummis et al., 2005). Analysis and curve fitting was performed using Prism version 3.0 (GraphPad Software Inc., San Diego, CA; www.graphpad.com). Concentration-response data for each oocyte were normalized to the maximum current for that oocyte. The mean and S.E.M. for a series of oocytes were plotted against agonist concentration and iteratively fitted to the four-parameter logistic equation using Prism. Values are presented as mean ± S.E.M.
Radioligand Binding.
This was undertaken as described previously (Price and Lummis, 2004) with minor modifications. In brief, transfected HEK293 cell membranes were incubated in 0.5 ml of HEPES buffer containing the 5-HT3 receptor antagonist [3H]granisetron (0.1 nM). Nonspecific binding was determined using 1 μM quipazine. Data were analyzed by iterative curve fitting using Prism. Values are presented as mean ± S.E.M.
FlexStation Analysis.
This technique uses fluorescent voltage-sensitive dyes to detect changes in the membrane potential and has been used to examine various ion channels including 5-HT3 receptors (Price and Lummis, 2005; Thompson et al., 2010b). The methods were as described previously (Price and Lummis, 2005). In brief, fluorescent membrane potential dye (Molecular Devices) was diluted in Flex buffer (10 mM HEPES, 115 mM NaCl, 1 mM KCl, 1 mM CaCl2, 1 mM MgCl2, and 10 mM glucose, pH 7.4) and added to transfected cells grown on a 96-well plate. The cells were incubated at room temperature for 45 min and then fluorescence was measured in a FlexStation (Molecular Devices) at 2-s intervals for 200 s. Control (Flex buffer), 5-HT, or varenicline (0.001 μM–1.0 mM) was added to each well after 20 s. The percentage change in fluorescence was calculated as F (peak fluorescence) minus Fmin (baseline fluorescence at 20 s) divided by Fmax (peak fluorescence at 30 μM 5-HT). Concentration-response data were fitted to the four-parameter logistic equation using Prism software.
Modeling and Docking.
Amino acid sequences of the human and mouse 5-HT3A receptor subunits were aligned with that of AChBP using Fugue (http://www-cryst.bioc.cam.ac.uk/fugue). Three-dimensional homology models of human and mouse 5-HT3A receptors were generated using MODELLER (University of California, San Francisco) based on the crystal structure of AChBP with carbamoylcholine docked (Protein Data Bank ID1UV6) as described previously (Thompson et al., 2008). The protonated form of varenicline was constructed in Chem3DUltra (CambridgeSoft Corporation, Cambridge, MA), energy-minimized using the MM2 force field, and docked into the models using GOLD (The Cambridge Crystallographic Data Centre, Cambridge, UK). The binding site was defined as being within 10 Å of W183, a critical ligand binding residue (e.g., Reeves et al., 2002). Ten genetic algorithm runs were performed, and the structures were categorized using their final ligand orientations.
Binding to Other Targets.
A survey was conducted at NovaScreen/Caliper Life Sciences, Inc. (Hanover, MD). Assays included the nicotinic drug [3H]epibatidine binding to α-bungarotoxin-insensitive nACh receptors, the 5-HT3 inhibitor [N-methyl-3H]GR65630 to mouse receptors in N1E-115 cells, and human receptors expressed in a clonal cell line.
Results
This study sought to determine whether varenicline is an agonist at human 5-HT3 receptors. Because the homomultimeric human 5-HT3A receptor is ∼20-fold more sensitive to agonists than the heteromultimeric 5-HT3AB receptor (Thompson and Lummis, 2008), we experimented on the former.
Oocyte Expression Studies.
Application of 5-HT to X. laevis oocytes expressing human or mouse 5-HT3A receptors produced concentration-dependent, rapidly activating inward currents. Plotting current amplitude against a series of 5-HT concentrations allowed curves to be fitted (Fig. 1). The pEC50 values and Hill slopes determined from these curves are in Table 1.
Fig. 1.

A and B, typical responses of a single oocyte expressing human 5-HT3A receptors to application of various 5-HT (A; 0.3–100 μM) or varenicline (VAR) (B; 0.3–100 μM) concentrations. C, concentration response curves are shown (mean ± S.E.M.; n = 7–10).
TABLE 1.
Parameters for 5-HT3 receptors expressed in X. laevis oocytes assayed using two-electrode voltage clamp
Data are mean ± S.E.M.
| Agonist | pEC50 | EC50 | nH | n |
|---|---|---|---|---|
| μM | ||||
| Human 5-HT3A | ||||
| 5-HT | 5.70 ± 0.03 | 2.0 | 2.2 ± 0.2 | 10 |
| Varenicline | 5.23 ± 0.06 | 5.9 | 1.8 ± 0.3 | 10 |
| Mouse 5-HT3A | ||||
| 5-HT | 5.99 ± 0.03 | 1.0 | 1.9 ± 0.2 | 7 |
| Varenicline | 4.74 ± 0.08 | 18.3 | 1.2 ± 0.2 | 7 |
The effect of varenicline on human 5-HT3A receptors expressed in oocytes revealed that it was a relatively potent agonist with application of 10 μM routinely eliciting peak current responses of 2 to 10 μA. Challenging the receptor with a range of varenicline concentrations revealed an EC50 of 5.9 μM, compared with an EC50 of 2.0 μM for 5-HT (Table 1). Examination of the maximum responses to the two compounds revealed that varenicline had an Imax value 79 ± 3% of that of 5-HT, indicating it is a near full agonist (Fig. 1). There were no significant differences in the Hill slopes.
Application of varenicline to mouse 5-HT3A receptors expressed in oocytes revealed this compound was also an agonist, but it had a considerably lower potency than 5-HT, with an EC50 close to 20 μM. The Hill slope was also significantly reduced. Examination of the maximum responses to the two compounds revealed that the maximum response to varenicline was 22 ± 2% of that of 5-HT, indicating it is a partial agonist (Fig. 2).
Fig. 2.

A and B, typical responses of a single oocyte expressing mouse 5-HT3A receptors to application of various 5-HT concentrations (A; 0.1–30 μM) or varenicline (B; 0.3–300 μM). C, concentration response curves are shown (mean ± S.E.M.; n = 7–10).
FlexStation Expression Studies.
Application of varenicline to HEK293 cells transfected with human 5-HT3A subunit DNA and analyzed using membrane potential fluorescent dye in the FlexStation revealed an EC50 ∼4-fold higher than that of 5-HT, i.e., comparable with the data obtained in oocytes (Table 2; Fig. 3). Cells expressing mouse 5-HT3A receptors had an EC50 for varenicline ∼ 35-fold higher than 5-HT and again there was a decrease in the Hill slope. Relative EC50 values determined using the FlexStation are therefore similar, but absolute EC50 values are lower than those determined using voltage clamp, as we (and others) have observed previously (e.g. Price and Lummis 2005). This is expected because different phenomena are being measured. For electrophysiological studies, EC50 values represent the agonist concentration required to open 50% of channels, whereas in fluorescent studies they represent the agonist concentration required to depolarize the membrane potential to 50% of its original value.
TABLE 2.
Parameters derived from 5-HT3 receptors expressed in HEK293 cells assayed using membrane potential-sensitive dye
Data are mean ± S.E.M.
| Agonist | pEC50 | EC50 | nH | n |
|---|---|---|---|---|
| μM | ||||
| Human 5-HT3A | ||||
| 5-HT | 7.13 ± 0.08 | 0.07 | 2.7 ± 0.6 | 6 |
| Varenicline | 6.57 ± 0.06 | 0.27 | 2.9 ± 0.9 | 6 |
| Mouse 5-HT3A | ||||
| 5-HT | 6.85 ± 0.03 | 0.14 | 3.0 ± 0.4 | 4 |
| Varenicline | 5.31 ± 0.08 | 4.93 | 1.8 ± 0.2 | 4 |
Fig. 3.

Typical FlexStation responses of HEK293 cells transfected with human 5-HT3A receptors. Dose-dependent increases in fluorescence in cells loaded with membrane potential fluorescent dye were observed with both 5-HT (0.03, 0.1, or 0.3 μM) or varenicline (0.1, 0.3, or 1 μM), which were added at *. AU, arbitrary fluorescent units.
Radioligand Binding Studies.
Human and mouse 5-HT3A receptors were expressed in HEK293 cells, and competition of varenicline with the competitive antagonist [3H]granisetron was explored. IC50 values for varenicline and 5-HT were similar at both receptor types (Table 3; Fig. 4). Thus, given the large differences in EC50 values, these data suggest that varenicline has different efficiencies of transducing the binding information to channel opening in the two species.
TABLE 3.
IC50 values derived from radiolabeled antagonist-inhibition curves
Data are mean ± S.E.M.
| 5-HT IC50 | n | Varenicline IC50 | n | |
|---|---|---|---|---|
| μM | μM | |||
| Human 5-HT3A | 0.09 ± 0.01 | 5 | 0.58 ± 0.10 | 4 |
| Mouse 5-HT3A | 0.11 ± 0.03 | 4 | 0.53 ± 0.14 | 4 |
Fig. 4.

Typical experiments showing varenicline displacement of [3H]granisetron binding to human and mouse 5-HT3A receptors expressed in HEK293 cells. Parameters obtained from these data are shown in Table 3.
Modeling and Docking Studies.
Many critical residues in the ligand binding site of human and mouse 5-HT3A receptors are identical, but homology models of their binding sites reveal several subtle differences (Fig. 5). These differences arise from differing side chains, especially in loop C, which in turn lead to differing orientations of identical residues. Several types of data suggest that loop C closes over the bound agonist; this may be a critical step in channel gating. Docking of varenicline reveals different orientations in human and mouse 5-HT3A receptor binding sites (Fig. 6). Relative to the loops that form the binding site, the angle of the relatively planar varenicline molecule differs by nearly 90°. Differing orientations of an agonist in the binding site could markedly affect C-loop closure and hence subsequent activation. Thus, these docking studies data provide a possible explanation for the different efficacies of varencline at human and mouse receptors.
Fig. 5.

A and B, an overlay of the human (orange) and mouse (white) 5-HT3 A+A- binding interface, with differing residues and some critical binding residues highlighted. For clarity, the C-loop is rendered as a wire. The interface is shown from the membrane (A) and the extracellular compartment (B). C, an alignment of the loop C and loop E residues, some of which differ.
Fig. 6.
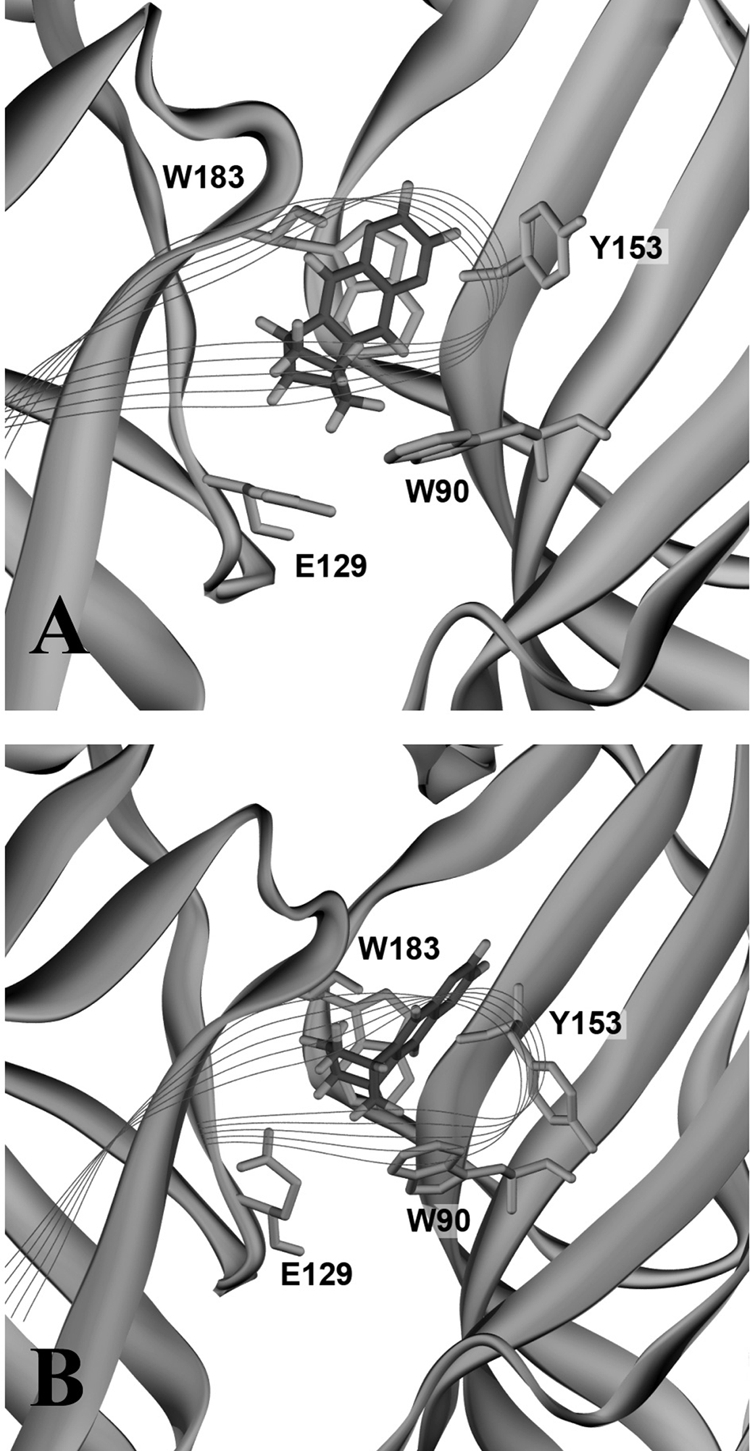
Varenicline docked into mouse (A) and human (B) 5-HT3A receptors. The favored docked poses for varenicline lie in the same general regions but have quite distinct orientations.
Lack of Binding to Other Sites.
The binding assays for other targets were performed using 10 μM varenicline and included 31 neurotransmitter-related targets, two steroid-related targets, six ion channels, nitric-oxide synthase, three prostaglandin receptors, four growth factor receptors, 13 brain/gut peptide receptors, and five enzymes (Supplemental Table 1). Positive results were obtained by competition for nACh receptors. Displacement of specific binding was also obtained in assays for mouse and human 5-HT3 receptors. No other targets were detected.
Discussion
This study shows that varenicline is a relatively potent agonist of human 5-HT3A receptors and binds to these receptors at clinically relevant concentrations (32–131 nM; Rollema et al., 2010). It is, however, only a weak agonist at murine 5-HT3A receptors. In addition to differences in EC50, we observed large differences in efficacy. Varenicline was a near full agonist at human receptors (Rmax/Rmax 5-HT = ∼80%) but a partial agonist at mouse receptors (Rmax/Rmax 5-HT ∼35%), possibly because of different orientations of varencline in the two different binding sites. Varenicline is widely used as a smoking cessation drug, an effect exerted through its action at nACh receptors (Faessel et al., 2006). However, its high potency and efficacy at human 5-HT3A receptors suggests there may also be 5-HT3-mediated effects in patients taking varenicline. One such effect may be nausea, which is the most common adverse event reported for varenicline in smoking cessation trials, and occurs at rates approaching 40% of patients (Aubin et al., 2008; Faessel et al., 2009). Nausea rarely causes patients to discontinue treatment but it does cause distress. It is noteworthy that early investigations established that nausea also limits the doses of varenicline tolerated by patients (Faessel et al., 2006). If nausea did not occur at higher doses, varenicline might be a more effective smoking cessation drug. The present data also show that 5-HT3 agonism begins to increase at precisely the high end of clinically useful doses. Many studies show that 5-HT3 receptor antagonists are antiemetic; some studies also show that 5-HT3 receptor agonists directly cause emesis (Miller and Nonaka, 1992).
Previous commentaries suggest that varenicline side effects arise primarily from actions on α3β4 and/or α7 nACh receptors, where oocyte expression reveals EC50 values of 55 and 18 μM, respectively (Coe et al., 2005; Mihalak et al., 2006). We now suggest that the 5-HT3 receptor is a possible target for some side effects. Our oocyte expression experiments show that varenicline functions as a nearly full (∼80%) 5-HT3A receptor agonist at human 5-HT3 receptors. The EC50 is ∼ 6 μM for the human 5-HT3A receptor, which is in the same range as, or lower than, the values for nACh receptors. Some nerve terminals coexpress both 5-HT3 and α4β2 nACh receptors (Nayak et al., 2000), leading to the possibility of synergistic activation. In our comprehensive survey of potential targets, no binding was detected to sites other than nACh and 5-HT3 receptors.
The data suggest that, at the submicromolar varenicline concentrations found in the plasma of patients, α4β2 and 5-HT3 receptors are activated to approximately the same extent by varenicline (Table 4). We assume that varenicline has a Hill slope (nH) of 1.8 for the human 5-HT3 receptor (Table 1), and 1.4 for the α4β2 receptor [based on H. A. Lester's unpublished results, which show a higher EC50 than measured by Mihalak et al. (2006)]. The clinically used 1- and 2-mg daily doses of varenicline result in steady-state plasma varenicline concentrations of 8.5 and 15 ng/ml (40 and 71 nM, respectively) (Faessel et al., 2006). The calculations of Table 4 suggest that varenicline at these doses produces very low fractional activation of several nACh receptors, including the α4β2 thought to participate in the therapeutic effects of varenicline and the α3β4 or α7 nACh receptors previously considered as targets for the unpleasant side effects of varenicline (Coe et al., 2005; Mihalak et al., 2006). The fractionally small 5-HT3 receptor activation is of the same order as for nACh receptors if calculated from the 5-HT3 receptor oocyte data. The α7 nACh receptor seems most strongly activated; however, the Hill coefficients measured by Mihalak et al. (2006) are surprisingly low for agonist activation of α7; and if the usual estimates of >2 are used, then α7 activation resembles that for other receptors in Table 4.
TABLE 4.
Calculated maximal activation at several receptors for two clinically effective daily doses of varenicline
Values were obtained using a simplified Hill equation appropriate for fractional activation of receptors by agonist at concentrations much lower than EC50: fractional activation = peak response/(1 + (EC50/[agonist])nH.
| Receptor | EC50, | nH | Efficacy | Fractional Activation |
||
|---|---|---|---|---|---|---|
| Plasma Concentration | ||||||
| 8.6 ng/ml*(1 mg daily) | 15 ng/ml*(2 mg daily) | |||||
| μM | ||||||
| 5-HT3@ | Oocyte | 2.0 | 1.8 | 0.80 | 1.0E-04 | 2.8E-04 |
| 5-HT3@ | FlexStation | 0.27 | 2.9 | 0.80 | 3.3E-03 | 1.7E-02 |
| a4β2#& | 2.5 | 1.4 | 0.13 | 4.1E-04 | 8.9E-04 | |
| α3β4# | 55 | 2 | 0.75 | 4.1E-07 | 1.3E-06 | |
| α7# | 1.8 | 1 | 0.93 | 2.1E-02 | 3.7E-02 | |
Data from Faessel et al., 2006.
Data from this study.
Data from Mihalak et al., 2006.
H. A. Lester, unpublished data.
It has been appreciated for some time that smoked concentrations of nicotine produce fractionally small activation of nAChRs; other agonist-receptor interactions, such as desensitization and pharmacological chaperoning, may therefore underlie some of the mechanisms of nicotine and varenicline (Lester et al., 2009; Miwa et al., 2011). In addition, when both varenicline and a full agonist are present, varenicline can act to decrease a full agonist-mediated response to the level of the lower, maximal varenicline response (Rollema et al., 2007). As a partial agonist, varenicline is especially advantageous in smoking cessation, because it decreases activation when nicotine is present (i.e., decreasing the pleasurable aspects of smoking), but activates weakly when nicotine is absent (i.e., during abstinence), presumably countering withdrawal symptoms.
Because varenicline is nearly a full agonist at 5-HT3 receptors, it is unlikely to produce an analogous decreased response when both varenicline and 5-HT are present. Antagonists of 5-HT3 receptors are used clinically as antiemetics. Some literature suggests that 5-HT3 receptor agonists directly cause emesis (Miller and Nonaka, 1992). Our present suggestion, that the clinically relevant emetic side effects of varenicline involve 5-HT3 receptor activation, favors the explanation that varenicline's emetic side effects arise via activation rather than antagonism of 5-HT3 receptors. Using the EC50 values obtained from oocyte data indicates that receptor activation is low, but if we use the values obtained from FlexStation data it is considerably higher (assuming an efficacy of 0.8 as in the oocyte data; see Table 4). Because the latter is derived from changes in membrane potential rather than ion flux, it may well be more representative of the situation in vivo. Because the concentration of varenicline in the human brain is sufficient to activate nAChR (albeit less effectively than desensitizing them; Rollema et al., 2010), it is at a sufficiently high concentration to activate 5-HT3 receptors. In addition, as mentioned above, there are potentially alternative mechanisms such as desensitization and/or pharmacological chaperoning. Pharmacological chaperoning by agonists is much stronger than by antagonists at nAChRs (Lester et al., 2009; Miwa et al., 2011). If the same is true of 5-HT3 agonists at 5-HT3 receptors, the varenicline-5-HT3 receptor interaction and/or the downstream sequelae must therefore be considered as a cause of the nausea that limits the effectiveness of varenicline therapy.
Supplementary Material
This work was supported by the Wellcome Trust [Grant 81925] (to A.J.T. and S.C.R.L.); the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant NS11756] (to H.A.L.); and the National Institutes of Health National Institute of General Medical Sciences [Grant GM19375] (to H.A.L.). S.C.R.L. is a Wellcome Trust Senior Research Fellow in Basic Biomedical Science.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.111.185306.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
- 5-HT
- 5-hydroxytryptamine
- nACh
- nicotinic acetylcholine
- AChBP
- acetylcholine binding protein
- HEK
- human embryonic kidney
- ND96
- 96 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 5 mM HEPES, pH 7.5
- VAR
- varenicline.
Authorship Contributions
Participated in research design: Lummis, Thompson, Bencherif, and Lester.
Conducted experiments: Lummis and Thompson.
Performed data analysis: Lummis, Thompson, and Lester.
Wrote or contributed to the writing of the manuscript: Lummis, Thompson, Bencherif, and Lester.
References
- Aubin HJ, Bobak A, Britton JR, Oncken C, Billing CB, Jr, Gong J, Williams KE, Reeves KR. (2008) Varenicline versus transdermal nicotine patch for smoking cessation: results from a randomised open-label trial. Thorax 63:717–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. (2001) Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411:269–276 [DOI] [PubMed] [Google Scholar]
- Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, Sands SB, Davis TI, Lebel LA, Fox CB, et al. (2005) Varenicline: an α4β2 nicotinic receptor partial agonist for smoking cessation. J Med Chem 48:3474–3477 [DOI] [PubMed] [Google Scholar]
- Davies PA, Pistis M, Hanna MC, Peters JA, Lambert JJ, Hales TG, Kirkness EF. (1999) The 5-HT3B subunit is a major determinant of serotonin-receptor function. Nature 397:359–363 [DOI] [PubMed] [Google Scholar]
- Dubin AE, Huvar R, D'Andrea MR, Pyati J, Zhu JY, Joy KC, Wilson SJ, Galindo JE, Glass CA, Luo L, et al. (1999) The pharmacological and functional characteristics of the serotonin 5-HT3A receptor are specifically modified by a 5-HT3B receptor subunit. J Biol Chem 274:30799–30810 [DOI] [PubMed] [Google Scholar]
- Faessel H, Ravva P, Williams K. (2009) Pharmacokinetics, safety, and tolerability of varenicline in healthy adolescent smokers: a multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 31:177–189 [DOI] [PubMed] [Google Scholar]
- Faessel HM, Gibbs MA, Clark DJ, Rohrbacher K, Stolar M, Burstein AH. (2006) Multiple-dose pharmacokinetics of the selective nicotinic receptor partial agonist, varenicline, in healthy smokers. J Clin Pharmacol 46:1439–1448 [DOI] [PubMed] [Google Scholar]
- Fiebich BL, Akundi RS, Seidel M, Geyer V, Haus U, Müller W, Stratz T, Candelario-Jalil E. (2004) Expression of 5-HT3A receptors in cells of the immune system. Scand J Rheumatol Suppl 119:9–11 [PubMed] [Google Scholar]
- Lester HA, Xiao C, Srinivasan R, Son CD, Miwa J, Pantoja R, Banghart MR, Dougherty DA, Goate AM, Wang JC. (2009) Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry. Implications for drug discovery. AAPS J 11:167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liman ER, Tytgat J, Hess P. (1992) Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 9:861–871 [DOI] [PubMed] [Google Scholar]
- Lowe JA, 3rd, DeNinno SL, Coe JW, Zhang L, Mente S, Hurst RS, Mather RJ, Ward KM, Shrikhande A, Rollema H, et al. (2010) A novel series of [3.2.1] azabicyclic biaryl ethers as α3β4 and α6/4β4 nicotinic receptor agonists. Bioorg Med Chem Lett 20:4749–4752 [DOI] [PubMed] [Google Scholar]
- Lummis SC, Beene D, Harrison NJ, Lester HA, Dougherty DA. (2005) A cation-π binding interaction with a tyrosine in the binding site of the GABAC receptor. Chem Biol 12:993–997 [DOI] [PubMed] [Google Scholar]
- Mihalak KB, Carroll FI, Luetje CW. (2006) Varenicline is a partial agonist at α4β2 and a full agonist at α7 neuronal nicotinic receptors. Mol Pharmacol 70:801–805 [DOI] [PubMed] [Google Scholar]
- Miller AD, Nonaka S. (1992) Mechanisms of vomiting induced by serotonin-3 receptor agonists in the cat: effect of vagotomy, splanchnicectomy or area postrema lesion. J Pharmacol Exp Ther 260:509–517 [PubMed] [Google Scholar]
- Miwa JM, Freedman R, Lester HA. (2011) Neural systems governed by nicotinic acetylcholine receptors: emerging hypotheses. Neuron 70:20–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake A, Mochizuki S, Takemoto Y, Akuzawa S. (1995) Molecular cloning of human 5-hydroxytryptamine3 receptor: heterogeneity in distribution and function among species. Mol Pharmacol 48:407–416 [PubMed] [Google Scholar]
- Nayak SV, Rondé P, Spier AD, Lummis SC, Nichols RA. (2000) Nicotinic receptors co-localize with 5-HT3 serotonin receptors on striatal nerve terminals. Neuropharmacology 39:2681–2690 [DOI] [PubMed] [Google Scholar]
- Niesler B, Frank B, Kapeller J, Rappold GA. (2003) Cloning, physical mapping, and expression analysis of the human 5-HT3 serotonin receptor-like genes HTR3C, HTR3D and HTR3E. Gene 310:101–111 [DOI] [PubMed] [Google Scholar]
- Price KL, Lummis SC. (2004) The role of tyrosine residues in the extracellular domain of the 5-hydroxytryptamine3 receptor. J Biol Chem 279:23294–23301 [DOI] [PubMed] [Google Scholar]
- Price KL, Lummis SC. (2005) FlexStation examination of 5-HT3 receptor function using Ca2+- and membrane potential-sensitive dyes: advantages and potential problems. J Neurosci Methods 149:172–177 [DOI] [PubMed] [Google Scholar]
- Reeves DC, Lummis SC. (2002) The molecular basis of the structure and function of the 5-HT3 receptor: a model ligand-gated ion channel (review). Mol Membr Biol 19:11–26 [DOI] [PubMed] [Google Scholar]
- Rollema H, Chambers LK, Coe JW, Glowa J, Hurst RS, Lebel LA, Lu Y, Mansbach RS, Mather RJ, Rovetti CC, et al. (2007) Pharmacological profile of the α4β2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology 52:985–994 [DOI] [PubMed] [Google Scholar]
- Rollema H, Shrikhande A, Ward KM, Tingley FD, 3rd, Coe JW, O'Neill BT, Tseng E, Wang EQ, Mather RJ, Hurst RS, et al. (2010) Pre-clinical properties of the α4β2 nicotinic acetylcholine receptor partial agonists varenicline, cytisine and dianicline translate to clinical efficacy for nicotine dependence. Br J Pharmacol 160:334–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Lummis SC. (2007) The 5-HT3 receptor as a therapeutic target. Expert Opin Ther Targets 11:527–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Lummis SC. (2008) Antimalarial drugs inhibit human 5-HT3 and GABAA but not GABAC receptors. Br J Pharmacol 153:1686–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Lester HA, Lummis SC. (2010a) The structural basis of function in Cys-loop receptors. Q Rev Biophys 43:449–499 [DOI] [PubMed] [Google Scholar]
- Thompson AJ, Verheij MH, Leurs R, De Esch IJ, Lummis SC. (2010b) An efficient and information-rich biochemical method design for fragment library screening on ion channels. Biotechniques 49:822–829 [DOI] [PubMed] [Google Scholar]
- Tzvetkov MV, Meineke C, Oetjen E, Hirsch-Ernst K, Brockmöller J. (2007) Tissue-specific alternative promoters of the serotonin receptor gene HTR3B in human brain and intestine. Gene 386:52–62 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.