

Abstract
Failures to arrest growth in response to senescence or transforming growth factor β (TGF-β) are key derangements associated with carcinoma progression. We report that activation of telomerase activity may overcome both inhibitory pathways. Ectopic expression of the human telomerase catalytic subunit, hTERT, in cultured human mammary epithelial cells (HMEC) lacking both telomerase activity and p16INK4A resulted in gaining the ability to maintain indefinite growth in the absence and presence of TGF-β. The ability to maintain growth in TGF-β was independent of telomere length and required catalytically active telomerase capable of telomere maintenance in vivo. The capacity of ectopic hTERT to induce TGF-β resistance may explain our previously described gain of TGF-β resistance after reactivation of endogenous telomerase activity in rare carcinogen-treated HMEC. In those HMEC that overcame senescence, both telomerase activity and TGF-β resistance were acquired gradually during a process we have termed conversion. This effect of hTERT may model a key change occurring during in vivo human breast carcinogenesis.
An indefinite lifespan is one of the most common tumor-associated properties and is likely crucial for malignant progression, because tumor cells may need to overcome the proliferative constraints imposed by senescence to accumulate the multiple errors necessary for invasive and metastatic behavior. The finite lifespan of normal human somatic cells is thought to be due to their lack of telomerase activity, resulting in continued telomere shortening with cell division. In contrast, human tumor tissues frequently contain cells that display an indefinite lifespan in culture, and most human carcinomas express high telomerase activity, which allows the telomeric ends of linear chromosomes to be maintained (1). Tumor-derived cells contain many other cellular derangements, including changes in expression of tumor suppressor genes, protooncogenes, genomic instability, acquisition of growth factor and anchorage independent growth (AIG), and alterations in cell-cycle regulatory proteins. In cancers derived from epithelial cells, malignancy is commonly correlated with a loss or reduction in the ability of the multifunctional cytokine transforming growth factor β (TGF-β) to inhibit growth (2).
Whether acquisition of telomerase activity and immortality simply confer a greater time frame for accumulation of tumor-promoting errors, or intrinsically contribute to cancer formation, has not been determined. Previously, it was not possible to separate immortal from malignancy-associated properties due to the obligate derivation of immortal cells from tumors or abnormal tissues, or by cell exposure to viral oncogenes or physical carcinogens. More recently, finite lifespan cells exposed to the human catalytic subunit of the telomerase complex, hTERT, have been rendered immortal (3), making it possible to determine whether hTERT-induced immortality confers other alterations in cellular growth control. Initial published reports on hTERT-immortalized human cells showed no alterations in growth control in response to serum deprivation, high cell density, specific pharmacological inhibitors, or oncogenic ras, nor were gross chromosome instability, AIG, or tumorigenicity reported (4–6). However, ectopic hTERT expression in conjunction with ectopic expression of the oncogenes SV40-T and H-ras, was able to malignantly transform normal human cells (7), and long-term culture of hTERT-transduced human mammary epithelial cells (HMEC) was reported to be associated with increased expression of c-myc (8).
Our laboratory has been studying chemical carcinogen-induced immortal transformation of cultured HMEC. Benzo[a]pyrene exposure of reduction mammoplasty-derived HMEC from specimen 184 led to several extended life cultures that ceased growth when their mean terminal restriction fragment (TRF) length was ≈5 kb. Two cells overcame senescence and gave rise to two distinct immortal lines, 184A1 and 184B5 (9). Surprisingly, even after overcoming senescence, early passages of these clonally derived immortal lines did not express telomerase activity and displayed continued telomere erosion. When telomeres in these cells became critically short (mean TRF ≤ 3 kb), a conversion process began. Mass cultures underwent an extended period of slow heterogeneous growth, associated with high expression of the cyclin-dependent kinase inhibitor p57. Although mass cultures always maintained indefinite growth, most individual cells ceased proliferation. We thus termed early passage cells of these immortal lines conditionally immortal. During the conversion process, conditional immortal cells very gradually converted to a fully immortal phenotype with telomerase activity, stable telomeres (≈3–7 kb mean TRF), no p57 expression, and good uniform growth in the absence or presence of TGF-β (10). The prolonged and incremental nature of the conversion process, as well as its reproducible manifestation in both mass cultures and repeatedly subcloned populations, suggests an epigenetic component to this process. Based on these studies, we have postulated that the continued growth of conditionally immortal HMEC, which have already overcome senescence, uncovers an inherent epigenetic mechanism that reactivates telomerase as the telomere length becomes critically short. In normal HMEC, stringent senescence prevents continued growth with short telomeres, so this mechanism is not encountered (10, 11).
Conversion of conditionally immortal HMEC to full immortality was consistently associated with acquisition of the ability to maintain growth in the presence of TGF-β. This ability was acquired gradually over 10–20 passages after the first detectable telomerase activity (ref. 10 and unpublished work). These data prompted us to examine the TGF-β responses of finite lifespan and conditionally immortal HMEC after ectopic introduction of hTERT. Our results indicate that the expression of hTERT alone can be responsible for inducing resistance to TGF-β inhibition in HMEC lacking expression of the cyclin-dependent kinase inhibitor p16. After hTERT transduction, finite lifespan p16(−) HMEC became immortal and acquired TGF-β growth resistance within 1–3 passages; however, finite lifespan p16(+) HMEC did not become immortal nor acquire TGF-β resistance. hTERT-transduced early-passage conditionally immortal HMEC acquired TGF-β resistance gradually over 10–20 passages, similar to the kinetics seen after endogenous reactivation of hTERT during conversion.
Methods
Cell Culture.
HMEC strains 184 and 161 were obtained from reduction mammoplasty tissue and cultured in serum-free MCDB 170 medium (BioWhittaker) or serum-containing MM medium as described (13, 14). In MCDB 170, HMEC undergo a self-selection process after ≈15–25 population doublings (PD), in which most cells senesce but a small number maintain good growth. Postselection HMEC do not express p16 mRNA and protein, associated with hypermethylation of the p16 promoter (15), and cease growth after an additional ≈40–60 PD. In MM, HMEC senesce after ≈15–25 PD, with high levels of p16 expression. Indefinite lifespan 184A1 and 184B5 were derived from benzo[a]pyrene-exposed 184 HMEC as described (9). These lines retain wild-type p53 and TGF-β receptors and respond to TGF-β with induction of extracellular matrix-related proteins (16). Neither line shows sustained AIG, tumorigenicity, or gross genomic instability. Complete details on the derivation and culture of these HMEC can be found at www.lbl.gov/∼mrgs.
Retroviral Transduction.
HMEC were infected with LXSN or pBABE retroviruses containing hTERT (17, 18), hTERT with the mutations R631A, D712A, and D868A, or hTERT with a carboxyl-terminal hemagglutinin epitope tag (from Robert Weinberg, Whitehead Institute, Cambridge, MA), and selected in G418 (GIBCO) or puromycin (Sigma) as described (11).
Growth Assays.
Growth in MCDB 170 ± TGF-β was assayed as described (10). Briefly, colony-forming efficiency (CFE) and growth capacity were assayed in single cell-derived colonies grown for 14–20 days, labeled with [3H]thymidine for 24 h, and then visualized by autoradiography (19). CFE was determined by counting the number of colonies of >50 cells, and growth capacity was determined by counting the percentage of labeled nuclei in these colonies. Some cultures received 5 ng/ml TGF-β (R&D Systems, Minneapolis) in the presence of 0.1% BSA (Sigma) for 10–14 days once the largest colonies contained ≈100 cells, and growth capacity per colony was determined as above. Rare TGF-β-resistant cells were detected as described (10). Growth in MM was assayed by seeding 5 × 104 cells in 35-mm dishes for 1–8 days, then labeling and visualizing by autoradiography as above. Where indicated, TGF-β was added 24 h after seeding. Growth curves were obtained by seeding 5 × 104 cells into 35-mm dishes. The number of attached cells was determined 16 h later, at which time cultures were refed with medium containing either 5 ng/ml TGF-β in BSA or BSA alone. Cell number was determined from triplicate dishes. We define TGF-β resistance as ability to maintain growth in TGF-β, to distinguish between finite lifespan HMEC, which are incapable of maintaining growth in TGF-β, and immortal HMEC, which can sustain growth but may grow slower in response to TGF-β.
AIG was assayed by suspending 105 single cells in 5 ml of a 1.5% methylcellulose solution in MCDB 170, seeded onto 60-mm Petri dishes coated with PolyHEMA (Sigma). Plates were fed once a week, and colonies >75 μm in diameter were counted after 3 weeks.
Telomerase Assays and TRF Analysis.
Telomerase assays were performed as described (11) with the TRAP-EZE telomerase detection kit (Intergen, Purchase, NY) and 2 μg of protein per assay. Telomerase products were stained with Sybr-green (Molecular Probes) and detected by using a storm fluorescent imaging system (Molecular Dynamics).
DNA isolation and mean TRF analysis were performed as described (11) with 3 μg of digested cellular genomic DNA resolved on 0.5% agarose gels and hybridized to a 32P-labeled telomere specific probe (CCCTAA)4. Signal was detected by using a PhosphorImager system and quantitated by using the imagequant software program (Molecular Dynamics). Mean TRF length was calculated as described (20).
Western Blot Analysis.
Protein lysates were collected from cells fed 24 h before harvest and processed as described (11). Fifty-microgram samples were resolved on a 4–12% polyacrylamide gradient gel and transferred to nitrocellulose membrane. Binding of mAb JC8 against p16 (from James Koh, University of Vermont, Burlington) was detected as described (11).
Immunohistochemical Analysis.
Cultures were washed twice with PBS and fixed for 30 min with 4% formaldehyde. Cells were permeablized with 0.1% Triton X-100 for 5 min, blocked for 30 min in PBS containing 5% goat serum, and incubated with p16 antibody JC8 for 60 min. Antibody binding was visualized by using the MultiLink horseradish peroxidase detection system (BioGenex Laboratories, San Ramon, CA).
Plasminogen Activator Inhibitor-1 (PAI-1) Assay.
PAI-1 levels in protein lysates prepared from cells grown for 5 days ± TGF-β were measured with the PAI-1 ELISA kit (American Diagnostics, Greenwich, CT).
Results
hTERT Transduction into Postselection Finite Lifespan, and Conditionally Immortal HMEC Confers Resistance to TGF-β-Induced Growth Inhibition.
Postselection finite lifespan HMEC strains 184, at passages 11 and 18, and 161, at passage 5, and conditional immortal 184A1, at passages 12 and 22, were exposed to hTERT via retroviral vectors or to control LXSN vector (see Fig. 1 A and B). These cell types do not express p16 (15, 21). All hTERT-transduced cultures showed telomerase activity when examined one passage after infection, whereas cultures exposed to vector alone showed no activity (data not shown). The control finite lifespan cultures senesced as expected while hTERT-exposed postselection HMEC have maintained rapid continuous growth through passage 36. The hTERT-transduced HMEC maintained growth past confluence, leading to ridged and dome-like structures; however, neither 184 HMEC transduced at passage 11 with hTERT [hTERT-184(11p)] nor 184A1 transduced at passage 12 [hTERT-184A1(12p)] displayed any AIG.
Figure 1.

Schematic chart of hTERT transduction of (A) postselection p16(−) 184 HMEC at passages 11 and 18; (B) conditional immortal 184A1 (preconversion at passage 12, early conversion at passage 22); (C) preselection p16(+) 184 HMEC at passage 3.
hTERT transduction of good growing preconversion conditional immortal 184A1 at passage 12 (mean TRF ≈4–5 kb) completely eliminated the slow heterogeneous growth phase characteristic of the 184A1 conversion process during passages 16–30 (10), as well as the associated elevated p57 protein expression (22). As seen in Table 1, hTERT-184A1(12p) sustained uniform good growth whereas control LXSN-184A1(12p) underwent the slow-growth phase. However, hTERT transduction of conditional immortal 184A1 at passage 22, which had already begun the conversion process (mean TRF <2.5 kb, poor heterogeneous growth, elevated p57 levels) resulted neither in an increased CFE or growth rate in mass culture (Table 1), nor reduced p57 expression (data not shown), relative to control cultures. Those cells capable of colony formation did show a slight initial increase in labeling index (LI) per colony. Both hTERT-184A1(22p) and LXSN-184A1(22p) cultures showed a slowly increasing growth rate and CFE through passage 54. Both LXSN-184A1 controls showed the gradual acquisition of telomerase activity normally seen during conversion of conditional immortal 184A1 to full immortality (data not shown).
Table 1.
Colony growth of postselection HMEC ± hTERT and ± TGF-β
| Passage cell | LI,
%
|
CFE, % | |||||||
|---|---|---|---|---|---|---|---|---|---|
| TGFβ (−)
|
TGFβ (+)
|
||||||||
| <10 | 10–25 | 26–50 | >50 | <10 | 10–25 | 26–50 | >50 | ||
| 184(11p) | |||||||||
| 12 hTERT | 0 | 0 | 0 | 100 | 4 | 13 | 18 | 63 | 12.1 |
| 13 LXSN | 54 | 23 | 12 | 10 | 100 | 0 | 0 | 0 | 2.1 |
| hTERT | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 100 | 7 |
| 20 hTERT | 0 | 0 | 0 | 100 | 0 | 0 | 4 | 96 | 40 |
| 184(18p) | |||||||||
| 19 LXSN | 0 | 1 | 20 | 79 | 100 | 0 | 0 | 0 | 11.4 |
| hTERT | 0 | 0 | 0 | 100 | 0 | 0 | 5 | 95 | 15.57 |
| 161(5p) | |||||||||
| 6 LXSN | 10 | 17 | 46 | 27 | 100 | 0 | 0 | 0 | 11.4 |
| hTERT | 0 | 0 | 0 | 100 | 26 | 11 | 22 | 41 | 12.5 |
| 10 LXSN | 27 | 24 | 38 | 11 | 100 | 0 | 0 | 0 | 2.6 |
| hTERT | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 100 | 29.2 |
| 184(12p) | |||||||||
| 13 Babe | 41 | 39 | 19 | 1 | 100 | 0 | 0 | 0 | 8.8 |
| hTERTmut | 25 | 10 | 40 | 25 | 100 | 0 | 0 | 0 | 4.4 |
| hTERT-HA | 27 | 27 | 37 | 9 | 88 | 5 | 3 | 4 | 6.2 |
| hTERT | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 100 | 18.0 |
Single cells were seeded, and the LI ± TGF-β in colonies of >50 cells was determined. Number in parentheses indicates passage level of cell transduction with hTERT or empty vector (LXSN).
Because conversion of our carcinogen-exposed HMEC lines is associated with gradual acquisition of TGF-β resistance (10), we tested the hTERT-transduced postselection and conditional immortal HMEC for growth responses to TGF-β. All of the postselection hTERT-HMEC showed sustained, uniform growth in TGF-β within a few passages of hTERT transduction (Table 1, Figs. 2 and 3A, and Fig. 6, which is published as supplemental data on the PNAS web site, www.pnas.org). This rapid acquisition of good growth in TGF-β by the postselection hTERT-HMEC differed from the slow incremental acquisition seen during conversion of conditional immortal HMEC. At early passages after infection, the hTERT-HMEC displayed some morphological changes characteristic of TGF-β exposure (large flattened morphology, ruffled edges) (23), whereas later passages retained small refractile appearance of untreated cells even after TGF-β exposure. Although TGF-β reduced the growth rate in the hTERT-HMEC, this reduced rate was still equivalent to good growing postselection HMEC without TGF-β exposure (Fig. 2). Control LXSN-HMEC did not maintain growth in the presence of TGF-β (Table 1 and Fig. 2).
Figure 2.

Growth of postselection HMEC ± hTERT and ± TGF-β. 184 HMEC infected at passage 11 with either hTERT-containing [hTERT-184(11p)] or control retroviruses [LXSN-184(11p)] and uninfected 184 HMEC were assayed for growth ± TGF-β at the indicated passages.
Figure 3.

hTERT expression induces TGF-β resistance in (A) postselection and (B) conditionally immortal HMEC. Data from Tables 1 and 2 and from ref. 10 and unpublished work for the p53(+) line 184A1 and the p53(−/−) line 184AA3 are plotted to illustrate the rapid gain of TGF-β resistance in postselection HMEC vs. the gradual gain of resistance, with similar kinetics after telomerase activation, in p53(+) and (−/−) immortal HMEC lines and in hTERT-transduced 184A1. Closed arrows indicate passage of infection; open arrows indicate passage when telomerase activity was first detected in the uninfected lines.
Exogenous introduction of hTERT into conditional immortal 184A1 also conferred TGF-β resistance, well before it would have been acquired as part of the conversion process. However, TGF-β resistance was acquired gradually over 10–20 passages, similar to the kinetics observed in our five immortally transformed HMEC lines after endogenous reactivation of hTERT during conversion (Table 2 and Fig. 3B). No significant difference in kinetics was seen between hTERT-184A1(12p) and hTERT-184A1(22p), indicating that the acquisition of good growth in TGF-β after hTERT expression was distinct from attainment of good growth and increased CFE in the absence of TGF-β.
Table 2.
Colony growth of conditionally immortal 184A1 HMEC ± hTERT and ± TGF-β
| Passage cell | LI, %
|
CFE, % | |||||||
|---|---|---|---|---|---|---|---|---|---|
| TGFβ
(−)
|
TGFβ (+)
|
||||||||
| <10 | 10–25 | 26–50 | >50 | <10 | 10–25 | 26–50 | >50 | ||
| 184A1(12p) | |||||||||
| 17 LXSN | 10 | 8 | 33 | 49 | 100 | 0 | 0 | 0 | 1.8 |
| hTERT | 0 | 0 | 1 | 99 | 86 | 9 | 4 | 10 | 29.2 |
| 21 LXSN | 7 | 3 | 15 | 75 | 100 | 0 | 0 | 0 | 5.0 |
| hTERT | 0 | 0 | 0 | 100 | 23 | 8 | 9 | 60 | 21.9 |
| 25 LXSN | 8 | 19 | 35 | 38 | 97 | 3 | 0 | 0 | 10.3 |
| hTERT | 0 | 0 | 0 | 100 | 1 | 6 | 36 | 57 | 17.3 |
| 29 LXSN | 0 | 0 | 0 | 100 | 86 | 14 | 0 | 0 | 24.4 |
| hTERT | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 100 | 15.2 |
| 35 LXSN | ND | 60 | 25 | 11 | 4 | 24.4 | |||
| 41 LXSN | ND | 57 | 12 | 15 | 16 | 25.5 | |||
| 47 LXSN | ND | 34 | 18 | 30 | 21 | ND | |||
| 184A1(22p) | |||||||||
| 24 hTERT | 0 | 0 | 0 | 100 | 100 | 0 | 0 | 0 | 4.1 |
| 25 LXSN | 24 | 2 | 28 | 46 | ND | 2.9 | |||
| 27 LXSN | 24 | 8 | 25 | 43 | 100 | 0 | 0 | 0 | 3.3 |
| 33 LXSN | 0 | 0 | 5 | 95 | 100 | 0 | 0 | 0 | 8.5 |
| hTERT | 0 | 0 | 0 | 100 | 63 | 8 | 8 | 21 | 10.1 |
| 43 LXSN | 0 | 0 | 5 | 100 | 70 | 17 | 7 | 6 | 12.8 |
| hTERT | 0 | 0 | 0 | 100 | 17 | 18 | 11 | 54 | 11.2 |
| 54 LXSN | 1 | 3 | 17 | 79 | 25 | 23 | 43 | 9 | 15.0 |
| hTERT | 0 | 0 | 0 | 100 | 0 | 2 | 22 | 76 | 17.6 |
Single cells were seeded and the labeling index ± TGF-β in colonies of >50 cells determined; ND, not determined. Number in parentheses indicates passage level of cell transduction with hTERT or empty vector (LXSN).
To determine what functions of the ectopic hTERT were required for induction of TGF-β resistance, two different hTERT mutants were introduced into 184 HMEC at passage 12. One mutant has inactivating amino acid substitutions in the reverse transcriptase domain (17), and the other is a wild-type hTERT with a carboxyl-terminal hemagglutinin epitope tag that shows in vitro, but not in vivo, telomerase activity (24). These constructs induced either no or very low levels of TGF-β resistance (Table 1), suggesting that the reverse transcriptase domain must be intact and hTERT must be capable of forming a complex with in vivo activity to confer TGF-β resistance.
Although the hTERT-transduced HMEC were no longer sensitive to TGF-β-induced growth inhibition, like fully immortal 184A1 and 184B5, as well as normal finite lifespan HMEC (16), they were still capable of responding to TGF-β with differentiated functions. Assays for plasminogen activator inhibitor-1 induction in response to TGF-β exposure showed a 2- to 4-fold induction (data not shown).
hTERT Transduction of Finite Lifespan and Conditionally Immortal HMEC Leads to Telomere Elongation.
Exogenously introduced hTERT caused a rapid increase in mean TRF length in postselection and conditionally immortal HMEC (Fig. 4). Although the initial TRF values in these cultures varied from ≈2.5 kb (184A1 at passage 22) to ≈6.5 kb (184 HMEC at passage 11), all of the hTERT-transduced cultures displayed mean TRF values of ≈10–12 kb within a few passages of infection. In this respect, the immortal HMEC resulting from hTERT transduction of both the finite lifespan and conditionally immortal HMEC differed from the fully immortal HMEC lines that resulted from overcoming senescence and undergoing conversion (Fig. 4 B and C). The lines that overcame senescence and then reactivated endogenous telomerase as part of the conversion process displayed short faint TRFs during conversion, followed by short, regulated mean TRF values of ≈3–7 kb in the fully immortal populations (10, 11). These data also indicate that the acquisition of TGF-β resistance after expression of telomerase activity is not correlated with telomere length.
Figure 4.

Ectopic hTERT causes telomeres to lengthen in normal and conditionally immortal HMEC, whereas telomeres become critically short in immortal HMEC undergoing conversion. (A) Postselection 184 HMEC; hTERT-184(11p) shows rapid telomere lengthening whereas LXSN-184(11p) telomeres continue to shorten to a mean TRF of ≈5 kb at senescence. Both (B) conditional immortal 184A1 transduced at passage 12 and (C) conditional immortal 184A1 transduced at passage 22 with hTERT show rapid telomere lengthening, whereas LSXN-184A1 undergoing conversion show continued telomere erosion to faint critically short mean TRF of ≈2 kb, followed by stabilization.
hTERT Transduction into Preselection Finite Lifespan HMEC Expressing p16 Does Not Confer TGF-β Resistance.
Most finite lifespan HMEC grown in either MCBD 170 or MM cease proliferation after 15–25 PD with high levels of p16 expression (15). To examine the effect of hTERT in these preselection HMEC, 184 HMEC growing in MM were transduced at passage 3 (≈18 PD) with the hTERT gene or control LXSN vector (see Fig. 1C). All hTERT-transduced cultures showed telomerase activity when assayed at passage 4; control cultures had no detectable activity (data not shown).
Typical for MM-grown HMEC, control cultures maintained heterogeneous growth through passage 4, with areas of small actively growing cells interspersed among nonproliferative cells with a flattened, vacuolated morphology. LXSN-184(3p) exhibited complete growth arrest by passage 5. hTERT-184(3p) initially showed growth and morphologies similar to control cells. Of four independently infected dishes, three ceased proliferation by passage 5. hTERT-184(3p) from one dish has maintained growth through passage 41. These hTERT-184(3p) HMEC were examined for growth capacity ± TGF-β, p16 expression, telomerase activity, and TRF length. Similar to hTERT-transduced postselection and 184A1 HMEC, the mean TRF length of hTERT-184(3p) cultures quickly elongated to 10–12 kb (data not shown). There was a gradual decrease in overall p16 protein levels from passages 4–29, corresponding to a decreasing number of p16(+) cells (Fig. 5 A and B). p16(+) cells had the large flat morphology of senescent HMEC, whereas the p16(−) cells had the small refractile morphology characteristic of proliferating HMEC.
Figure 5.
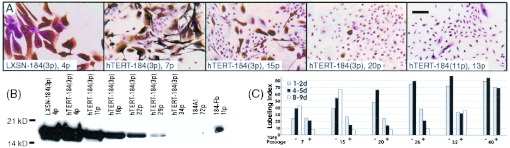
Rare hTERT immortalization of preselection 184 HMEC is associated with gradual down-regulation of p16 and acquisition of TGF-β resistance. (A) Decreasing p16 expression (brown precipitate) in individual cells with increasing passage in the rare preselection HMEC that gained immortality after hTERT transduction at passage 3 [hTERT-184(3p)]. p16(−) postselection HMEC transduced with hTERT [184-TERT(11p)] are shown for comparison. (Bar = 100 μm.) (B) Western blot analysis showing decreasing p16 expression with increasing passage in hTERT-184(3p). (C) Increasing ability to grow in TGF-β correlates with decreasing p16 expression in hTERT-184(3p). Growth was assayed as described in Methods; LI was determined from five separate fields at the indicated days postseeding.
hTERT-184(3p) was assayed for growth in mass cultures ± TGF-β from passages 7 to 40 (Fig. 5C). Cultures at passages 17 and 20 also were assayed to detect rare TGF-β-resistant cells. From passages 7 to 20, increasing time in TGF-β resulted in severe growth inhibition, with <10% LI after 9 days in TGF-β compared with 40–65% LI in the non-TGF-β-exposed cultures. However, by passage 20 rare cells capable of very slow growth in TGF-β could be detected. This first indication of continuous growth capacity in TGF-β corresponded to the passage level where most cells no longer expressed p16. Thereafter, the population showed an increasing number of cells capable of progressively better growth in TGF-β.
Discussion
Our studies on HMEC immortalized by potentially pathologically relevant means (chemical carcinogens, overexpression of the potential breast cancer oncogene ZNF217) have uncovered previously undescribed steps involved in the acquisition and maintenance of an indefinite lifespan (10, 25). Based on these studies, we have postulated that senescence may be overcome first by loss of p16 expression, and second by circumventing a stringent constraint that normally prevents continued growth with very short telomeres. However, p53(+) HMEC that have overcome even this second senescence constraint are only conditionally immortal and must still undergo a conversion process to reactivate telomerase and attain uniform unlimited growth. We suggest that telomerase reactivation in these HMEC during conversion is due to an inherent epigenetic mechanism encountered when continuous proliferation reduces telomeres to a critically short length. Furthermore, we have proposed that acquiring uniform immortal potential (i.e., overcoming stringent senescence and undergoing conversion) may be a key rate-limiting step in malignant progression of cells from long-lived animals such as humans (26). This conversion step has not been reported in human epithelial cells immortalized by viral oncogenes. Viral oncogene-mediated immortalization produces cells, lacking functional p53, that rapidly transform from finite lifespan extended life cultures to full immortality after a short period of crisis.
Our previous studies noted a strict correlation between the reactivation of telomerase during conversion and gaining the capacity to maintain growth in TGF-β (10). The current work demonstrates that expression of hTERT is sufficient to confer TGF-β resistance to HMEC lacking p16 expression. Exogenous introduction of hTERT into postselection finite lifespan and early passage conditionally immortal HMEC (which lack hTERT and p16 expression), produced resistance to TGF-β-induced growth inhibition. Curiously, the hTERT-transduced postselection HMEC acquired TGF-β resistance rapidly, whereas the hTERT-transduced conditional immortal 184A1 line showed an increasing number of cells with progressively better growth in TGF-β over the course of 10–20 passages— a pattern similar to that seen in conditional immortal 184A1 after reactivation of endogenous telomerase during conversion.
The mechanism responsible for hTERT inducing either rapid or gradual TGF-β resistance remains to be elucidated. The experiments using hTERT mutants indicate that in addition to being catalytically active telomerase must be capable of telomere maintenance in vivo to confer TGF-β resistance. However, our data also indicate that there is no correlation between telomere length and TGF-β resistance. The incremental acquisition of TGF-β resistance in conditionally immortal HMEC suggests that the effect of hTERT is likely to be indirect, possibly involving cumulative changes in chromatin structure and/or soluble factors. hTERT expression might indirectly change the abundance, modification, and/or spatial arrangements of signaling molecules involved in TGF-β growth inhibition through altering telomere association with nuclear matrix, or affecting the activities of telomere-associated proteins. Understanding how hTERT expression affects TGF-β resistance will require more detailed examination of the components and interactions of the active telomerase complex and the TGF-β signaling pathway.
hTERT rapidly conferred uniform immortal potential to postselection HMEC and preconversion conditional immortal 184A1 at passage 12 (mean TRF >3 kb) that were growing well when transduced. The mean TRF of hTERT-184A1(12p) never declined to <3 kb and thus the culture never encountered the slow growth phase of conversion, nor expressed high p57 levels. In contrast, hTERT transduction did not confer significant growth advantage, nor did it affect the high p57 levels in conditional immortal 184A1 at passage 22 (mean TRF <2.5 kb) that had already begun the slow growth phase of conversion. Better understanding of how the critically short telomeres in cells that have overcome senescence lead to the altered gene expression occurring during conversion may be necessary to explain this result. Possibly, once telomeres decline to the length (< 3-kb TRF) that triggers high p57 expression and slow growth, a series of alterations is set into motion, which then must proceed irreversibly, resolving only once full immortality is attained. hTERT expression before that length maintains longer telomeres and precludes activation of the entire conversion-associated phenotype of slow-growth and high p57 expression. Because hTERT transduction of 184A1 at both passages 12 and 22 produced similar kinetics of acquiring TGF-β resistance, but different effects on growth without TGF-β, the processes of attaining uniform good growth in the absence and presence of TGF-β appear to be separable.
Consistent with previous reports (27), we show that hTERT does not immortalize p16(+) HMEC and additionally show that it does not induce TGF-β resistance in these cells. In the one instance where hTERT-transduced preselection HMEC became immortal and gradually gained TGF-β resistance, p16 expression was lost. Rare loss of p16 associated with hTERT immortalization also has been reported for hTERT-transduced human keratinocytes (28). Immortal human epithelial cells commonly exhibit loss of p16 expression (12).
Malignant progression in human carcinomas is commonly associated with acquiring the ability to maintain growth in TGF-β (2). In some cases, this acquisition can be attributed to loss of functional TGF-β receptors or other mutations, but in most instances, no mutations are detected. An obligate gain of TGF-β resistance as a consequence of telomerase reactivation could explain why this phenotype is common to carcinoma cells. However, the very gradual nature of conversion-induced telomerase reactivation and TGF-β resistance, along with the potential growth advantage provided by the loss of TGF-β inhibition during carcinogenesis, also could promote selection of the observed mutation-associated mechanisms of TGF-β resistance. Our data showing that hTERT can induce TGF-β resistance suggest that immortality could be more than a passive facilitator of malignant progression. However, although TGF-β resistance may be a tumor-promoting property for immortal epithelial cells, it is not a malignant property per se; normal mesenchymal cells may be TGF-β responsive but not growth inhibited by TGF-β.
More significant in determining whether immortality intrinsically predisposes cells to malignancy may be the pathway by which telomerase becomes activated. In the potentially physiologically relevant model systems of HMEC immortalization we have developed, gaining immortality and reactivating telomerase required overcoming stringent senescence and undergoing a conversion process. Immortal cells produced by this path would (i) incur the as-yet-unknown defect(s) required to overcome senescence; (ii) possess extremely short telomeres, which can give rise to chromosomal aberrations, for an extended period; and (iii) exhibit the conversion-associated prolonged period of slow heterogeneous growth and high p57 levels. In vivo, such conditions might preferentially select for growth of rare aberrant cells that have acquired malignancy-promoting advantages such as AIG, growth factor independence, or angiogenesis. Although we do not yet know whether conversion occurs in in vivo breast carcinogenesis, several aspects of conversion do model what is seen in in vivo cancers, e.g., an extended period of heterogeneous growth, TGF-β resistance in the absence of specific mutations, and the generation of immortal cells with short regulated TRF lengths. Ectopically expressed hTERT produced immortalization of postselection HMEC, and full immortalization of conditionally immortal HMEC with mean TRF >3 kb, via bypassing senescence and conversion. Thus several molecular processes that could significantly impact malignant progression did not occur during these immortalization procedures.
We propose that telomerase activation during human carcinogenesis may involve an obligate stage of very short telomeres, as part of overcoming senescence and undergoing conversion. During this time, chromosomal aberrations may occur, gene expression is altered, and malignant progression may be promoted. Systems of immortalization that increase telomerase expression directly, bypassing senescence and/or conversion, cannot model these behaviors. Thus, testing of therapeutic interventions targeted to these potentially rate-limiting steps in immortalization and tumorigenicity may not be possible in such systems. Our results suggest that the extent to which in vitro culture systems of carcinogenesis model the steps, and order of steps, of in vivo human malignant progression could have significant clinical implications. On the other hand, whereas the hTERT-immortalized HMEC show some changes in signal transduction pathways, hTERT transduction may still generate the least deviant immortal human cells.
Supplementary Material
Acknowledgments
We thank Drs. Robert Weinberg and Jim Koh for reagents. This work was supported by National Institutes of Health Grant CA-24844 and the U.S. Department of Energy under Contract DE-AC03–76SF00098.
Abbreviations
- AIG
anchorage independent growth
- CFE
colony-forming efficiency
- HMEC
human mammary epithelial cells
- LI
labeling index
- PD
population doubling
- TGF-β
transforming growth factor type β
- TRF
terminal restriction fragment
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Kim N W, Piatyszek M A, Prowse K R, Harley C B, West M D, Ho P L C, Coviello G M, Wright W E, Weinrich S L, Shay J W. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 2.Fynan T M, Reiss M. Crit Rev Oncog. 1993;4:493–540. [PubMed] [Google Scholar]
- 3.Bodnar A G, Ouellette M, Frolkis M, Holt S E, Chiu C-P, Morin G B, Harley C B, Shay J W, Lichtsteiner S, Wright W E. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 4.Morales C P, Holt S E, Ouellette M, Kaur J, Yan Y, Wilson K S, White M A, Wright W E, Shay J W. Nat Gen. 1999;21:115–118. doi: 10.1038/5063. [DOI] [PubMed] [Google Scholar]
- 5.Jiang W-R, Jimenez G, Chang E, Frolkis M, Kusler B, Sage J, Beeche M, Bodnar A G, Wahl G M, Tlsty T D, Chiu C-P. Nat Gen. 1999;21:111–114. doi: 10.1038/5056. [DOI] [PubMed] [Google Scholar]
- 6.Wei S, Wei W, Sedivy J M. Cancer Res. 1999;59:1539–1543. [PubMed] [Google Scholar]
- 7.Hahn W C, Counter C M, Lundberg A S, Beijersbergen R L, Brooks M W, Weinberg R A. Nature (London) 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Xie L Y, Allan S, Beach D, Hannon G J. Genes Dev. 1998;12:1769–1774. doi: 10.1101/gad.12.12.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stampfer M R, Bartley J C. Proc Natl Acad Sci USA. 1985;82:2394–2398. doi: 10.1073/pnas.82.8.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stampfer M R, Bodnar A, Garbe J, Wong M, Pan A, Villeponteau B, Yaswen P. Mol Biol Cell. 1997;8:2391–2405. doi: 10.1091/mbc.8.12.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garbe J, Wong M, Wigington D, Yaswen P, Stampfer M R. Oncogene. 1999;18:2169–2180. doi: 10.1038/sj.onc.1202523. [DOI] [PubMed] [Google Scholar]
- 12.Reddel R R. Carcinogenesis. 2000;21:477–484. doi: 10.1093/carcin/21.3.477. [DOI] [PubMed] [Google Scholar]
- 13.Hammond S L, Ham R G, Stampfer M R. Proc Natl Acad Sci USA. 1984;81:5435–5439. doi: 10.1073/pnas.81.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stampfer M R. J Tissue Culture Methods. 1985;9:107–116. [Google Scholar]
- 15.Brenner A J, Stampfer M R, Aldaz C M. Oncogene. 1998;17:199–205. doi: 10.1038/sj.onc.1201919. [DOI] [PubMed] [Google Scholar]
- 16.Stampfer M R, Yaswen P, Alhadeff M, Hosoda J. J Cell Physiol. 1993;155:210–221. doi: 10.1002/jcp.1041550127. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura T M, Morin G B, Chapman K B, Weinrich S L, Andrews W H, Lingner J, Harley C B, Cech T R. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- 18.Meyerson M, Counter C M, Eaton E N, Ellisen L W, Steiner P, Caddle S D, Ziaugra L, Beijersbergen R L, Davidoff M J, Liu Q, et al. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 19.Stampfer M R, Pan C H, Hosoda J, Bartholomew J, Mendelsohn J, Yaswen P. Exp Cell Res. 1993;208:175–188. doi: 10.1006/excr.1993.1236. [DOI] [PubMed] [Google Scholar]
- 20.Allsopp R C, Vaziri H, Patterson C, Goldstein S, Younglai E V, Futcher A B, Greider C W, Harley C B. Proc Natl Acad Sci USA. 1992;89:10114–10118. doi: 10.1073/pnas.89.21.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brenner A J, Aldaz C M. Cancer Res. 1995;55:2892–2895. [PubMed] [Google Scholar]
- 22.Nijjar T, Wigington D, Garbe J C, Waha A, Stampfer M R, Yaswen P. Cancer Res. 1999;59:5112–5118. [PubMed] [Google Scholar]
- 23.Hosobuchi M, Stampfer M R. In Vitro. 1989;25:705–712. doi: 10.1007/BF02623723. [DOI] [PubMed] [Google Scholar]
- 24.Counter C M, Hahn W C, Wei W, Caddle S D, Beijersbergen R L, Lansdorp P M, Sedivy J M, Weinberg R A. Proc Natl Acad Sci USA. 1998;95:14723–14728. doi: 10.1073/pnas.95.25.14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nonet G, Stampfer M R, Chin K, Gray J W, Collins C C, Yaswen P. Cancer Res. 2001;61:1250–1254. [PubMed] [Google Scholar]
- 26.Stampfer M R, Yaswen P. J Mam Gland Biol Neo. 2000;5:365–378. doi: 10.1023/a:1009525827514. [DOI] [PubMed] [Google Scholar]
- 27.Kiyono T, Foster S A, Koop J J, McDougall J K, Galloway D A, Klingelhutz A J. Nature (London) 1998;396:84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- 28.Dickson M A, Hahn W C, Ino Y, Ronfard V, Wu J Y, Weinberg R A, Louis D N, Li F P, Rheinwald J G. Mol Cell Biol. 2000;20:1436–1447. doi: 10.1128/mcb.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}