

Abstract
PDZRhoGEF (PRG) belongs to a small family of RhoA-specific nucleotide exchange factors that mediates signaling through select G-protein-coupled receptors via Gα12/13 and activates RhoA by catalyzing the exchange of GDP to GTP. PRG is a multidomain protein composed of PDZ, regulators of G-protein signaling-like (RGSL), Dbl-homology (DH), and pleckstrin-homology (PH) domains. It is autoinhibited in cytosol and is believed to undergo a conformational rearrangement and translocation to the membrane for full activation, although the molecular details of the regulation mechanism are not clear. It has been shown recently that the main autoregulatory elements of PDZRhoGEF, the autoinhibitory “activation box” and the “GEF switch,” which is required for full activation, are located directly upstream of the catalytic DH domain and its RhoA binding surface, emphasizing the functional role of the RGSL-DH linker. Here, using a combination of biophysical and biochemical methods, we show that the mechanism of PRG regulation is yet more complex and may involve an additional autoinhibitory element in the form of a molten globule region within the linker between RGSL and DH domains. We propose a novel, two-tier model of autoinhibition where the activation box and the molten globule region act synergistically to impair the ability of RhoA to bind to the catalytic DH-PH tandem. The molten globule region and the activation box become less ordered in the PRG-RhoA complex and dissociate from the RhoA-binding site, which may constitute a critical step leading to PRG activation.
Keywords: Guanine Nucleotide Exchange Factor (GEF), Intrinsically Disordered Proteins, Protein Dynamics, Protein Structure, X-ray Scattering
Introduction
The Rho family of the monomeric GTPases, comprising 22 human proteins classified into 6 subfamilies, function in cell regulation as molecular switches, cycling between the biologically inactive GDP-bound state and biologically active GTP-bound form (1). Of all the Rho proteins, RhoA is one of the few most ubiquitous and physiologically critical, exerting control over cytoskeleton, gene expression, cell morphogenesis, and regulation of enzymatic activities (1–4). The activation of RhoA and other Rho proteins requires dedicated guanine nucleotide exchange factors (or GEFs),4 which catalyze the exchange of GTP for GDP on the GTPase. Most (although not all) such GEFs belong to the Dbl-homology family in which the catalytic function is served by a tandem of two domains: the Dbl-homology (DH) domain and the assisting pleckstrin-homology (PH) domain (5, 6). The mechanism by which the DH-PH tandem accomplishes nucleotide exchange is relatively well understood; in general terms, GEF binds to and transiently stabilizes nucleotide-free GTPase, which upon release from the complex preferentially binds GTP, which is 10-fold more abundant in the cell than GDP (5, 7).
For the cell to respond to specific stimuli with RhoA activation, the GEFs are autoinhibited in their native state and assume an active form only in response to such stimulation. Because many GEFs are multidomain proteins, the autoinhibition phenomenon is thought to arise from a supramodular, “closed” architecture in which other functional domains interact with the DH-PH tandem, occluding the functional surfaces and consequently interfering with RhoA binding (8). In this model upstream stimulation of specific receptors initiates cascades of intermolecular interactions that eventually involve these other functional domains within GEFs, so that they become engaged in complexes and dislodged from the DH-PH tandem, thereby relieving autoinhibition and allowing for the nucleotide exchange reaction.
Although many Rho GTPases appear to be activated through cascades involving tyrosine kinase receptors, RhoA is also activated downstream of certain physiologically important G-protein-coupled receptors (9, 10). In this case stimulation of the target G-protein-coupled receptors results in the interaction with and dissociation of a specific trimeric G-protein, releasing a free Gα subunit in the GTP-bound state. A small subfamily of the Dbl-like GEFs comprises three proteins that contain the so-called RGSL (regulator of G-protein signaling like) domain that interacts with Gα and thereby, presumably, activates the DH-PH tandem, located ∼200 amino acids downstream of the RGSL domain. The three GEFs that allow for this type of coupling of G-protein-coupled receptors with Rho GTPases are p115 (11), PDZRhoGEF (or PRG) (12), and leukemia-associated Rho-GEF (or LARG) (13).
We are particularly interested in the PDZRhoGEF exchange factor, which has been demonstrated to function in smooth muscle and mediate agonist-stimulated Ca2+ sensitization, resulting in enhanced contractility of smooth muscle at constant concentrations of intracellular Ca2+ (14, 15). This cascade is initiated by the stimulation of G-protein-coupled receptors coupled to Gα12/13 (15). Like LARG, but unlike p115, PRG contains an additional PDZ domain at the N terminus, implicated in interactions with the C-terminal tails of select receptors (16–18). The PDZ domains are followed by a ∼150-residue-long linker (L1) that appears to be disordered based on in silico sequence analysis. The portion common to all three GEFs includes the RGSL domain followed by another linker (L2) ∼200 residues in length, also presumed to be disordered, the DH-PH tandem, and a C-terminal-located unique fragment containing some coiled-coil elements that may mediate homo- and/or heterodimerization.
A key question in this field is how exactly the RGSL-containing GEFs are activated at the level of the molecular structure. The crystal structures of the RGSL domains of PRG and p115 are known (19, 20) as are those of the DH-PH tandems of all three GEFs either alone or in complex with nucleotide free RhoA (21–23). Moreover, the structure of the complex of the RGSL domain with the activated Gα13 is also known (24). However, little is understood about the possible interactions and communication between the individual domains in the context of the intact GEFs.
Our recent study showed that in solution a fragment of PRG encompassing residues 37–1081 (all functional domains with the two interweaving linkers) shows only 18% of the activity of the isolated DH-PH tandem (25). This autoinhibition was relieved only partly (35% of the full activity) by the truncation removing the RGSL domain but leaving the linker sequence (L2) in place. Interestingly, removal of most of the linker up to residue 671 had a marginal effect. Thus, a key autoinhibitory element is contained between residues 672 and 712. Very similar results were obtained recently for the p115 GEF (23, 26). We also found that this fragment, although lacking defined secondary structure, appears to interact in solution with the DH domain, and its presence also affects the conformation of residues 712–724 (25). Interestingly, an earlier structural and biochemical study of the DH-PH tandem of LARG revealed that this latter N-terminal fragment of the DH domain is critical for interaction with RhoA, dubbing it the GEF switch (22). The motif is conserved in PRG and p115, suggesting a common feature. A recent study of p115 showed that the presence of a fragment upstream of the GEF switch motif exerts an influence on the conformation of the switch that becomes disordered, limiting the ability of the DH domain to engage RhoA in a productive fashion (23). Although significantly advancing our knowledge, these studies still fail to explain how the RGSL domain may initiate the signal that relieves the autoinhibition.
Here we present a detailed study of the structure of PRG using a combination of biophysical techniques including circular dichroism (CD), dynamic light scattering, small-angle x-ray scattering (SAXS), and x-ray crystallography. We show conclusively that in its native form PRG does not form any supramodular assemblies, so that there are no direct interactions between the PDZ or RGSL domain and the DH-PH tandem. Moreover, binding of RhoA does not involve any globular domains other than the DH-PH tandem. However, we present evidence suggesting that both linkers of PRG form relatively compact, molten globule-like assemblies. The molten globule within the L2 linker may sterically interfere with RhoA binding and, therefore, be a part of the autoinhibitory mechanism. These features are likely to be found in all three RGSL-domain-containing GEFs.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Truncated fragments of human PRG (Fig. 1) were subcloned into pDEST15 vector (Invitrogen) with a GST tag followed by a tobacco etch virus cleavage site, target sequence, and a C-terminal noncleavable His8 tag (25). Cell pellets were resuspended in buffer A (50 mm Tris, pH 7.5, 500 mm NaCl, 50 mm (NH4)2 SO4, 5 mm 2-mercaptoethanol) and lysed using EmulsiFlex C3 homogenizer (Avestin, Ottawa, ON, Canada). The cell lysate was clarified by centrifugation at 35,000 × g for 30 min at 4 °C and loaded onto nickel-nitrilotriacetic acid column pre-equilibrated with buffer A. After binding to the resin for 1 h with gentle rocking of the column, protein was eluted with 200 mm imidazole in Buffer A. Eluted fractions were applied on a glutathione-Sepharose 4B column (Amersham Biosciences) pre-equilibrated with buffer B (50 mm Tris, pH 7.8, 200 mm NaCl, 50 mm (NH4)2 SO4, 1 mm EDTA, 5 mm DTT). Protein was bound to the resin at 4 °C for 1 h and eluted with 20 mm glutathione in buffer B. Fractions containing fusion proteins were digested overnight with recombinant tobacco etch virus protease in buffer B and subsequently purified using Superdex-200 size exclusion column (Amersham Biosciences) pre-equilibrated with buffer C (20 mm Tris, pH 7.8, 200 mm NaCl, 50 mm (NH4)2 SO4, 1 mm EDTA, 5 mm DTT, 5% glycerol). For experiments involving free PRG fragments, fractions were pooled, concentrated, and re-applied on Superdex-200 gel filtration column (Amersham Biosciences) pre-equilibrated with buffer C to remove any aggregated protein.
FIGURE 1.

Schematic representation of multidomain PRG fragments used in this study. FL, full-length, L1, PDZ-RGSL linker; L2, RGSL-DH linker; AB, activation box; GS, GEF switch.
The expression and purification of non-prenylated human RhoA (residues 1–181) were performed as described previously (27). The PRG-RhoA complex was formed by mixing both proteins in 1:2 molar ratio and dialyzing overnight at 4 °C against buffer C. To remove the fraction of unbound RhoA, the PRG-RhoA complex was purified on Superdex-200 size exclusion chromatography column (Amersham Biosciences) in buffer C. The formation of a stable complex was verified by SDS-PAGE.
Far-UV CD Measurements
The far-UV CD spectra were recorded using an AVIV 410 spectrometer (AVIV Biomedical, Lakewood, NJ) at 25 °C. The concentration of PRG 37–490 and PRG 277–1081 in buffer C was 0.4 mg/ml, and a cell with an optical path length of 1 mm was used. The mean residue ellipticity [θ] (deg × cm2 × dmol−1) was calculated using the following equation,
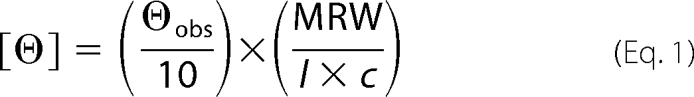 |
where θobs is the observed ellipticity, MRW is the mean residue molecular weight, l is the path length in cm, and c is the protein concentration in g/ml. The spectra were recorded in a 185–260-nm range, and data spanning 200–240 nm were used for deconvolution using a neural network algorithm program SOMCD (28).
Hydrogen/Deuterium Exchange Mass Spectrometry Measurements
Hydrogen/deuterium exchange mass spectrometry measurements were performed by the ExSAR company (Monmouth Junction, NJ). Hydrogen/deuterium isotopic exchange in the PRG 37–1081 fragment was initiated by mixing 10 μl of the protein solution (∼1.3 mg/ml) with 10 μl of deuterated buffer D (50 mm citrate, pH 6.0) at 23 °C. The mixture was incubated for 30, 100, 300, or 1000 s at 23 °C to allow for exchange of backbone amide hydrogens for deuterium atoms. Quenching was accomplished by adding 30 μl of cold buffer E (0.8% formic acid, pH 2.3, 1.6 m guanidine HCl). The deuterium-labeled protein was proteolysed by passing through an immobilized pepsin column at 200 μl/min equilibrated in buffer F (0.05% trifluoroacetic acid). The resulting peptic fragments were loaded onto a reverse-phase trap column and desalted with buffer F at 200 μl/min for 3 min followed by separation by a C18 HPLC (Magic C18; Michrom BioResources, Inc., Auburn, CA) with a linear gradient of 13–40% buffer G (95% acetonitrile, 5% H2O, 0.0025% trifluoroacetic acid) for 23 min and detected by a Finnegan LCQ mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Fully deuterated PRG sample was prepared by incubating a mixture of 22 μl of PRG with 2 μl of 100 mm Tris(2-carboxyethyl)phosphine in D2O at 60 °C for 3 h and processed as described above. Non-deuterated PRG was used as a negative control. The deuteration level for each reporter peptide, D%, was calculated by the following equation,
 |
where mon is the centroid value of an ion after on-exchange experiment, mn is the centroid value of an ion after non-deuterated experiment, and mf is the centroid value of an ion after fully deuterated experiment.
Dynamic and Static Light Scattering Measurements
The hydrodynamic radius (Rh) and dispersity of PRG fragments were measured using a DynaPro Titan instrument equipped with a Temperature Controlled MicroSampler operating at a wavelength of 830.9 nm (Wyatt Technology, Santa Barbara, CA). Briefly, protein samples in buffer C were concentrated to 0.5, 1.0, 2.0, and 3 mg/ml and spun at 21,000 × g for 30 min at 4 °C to remove dust and particulate material before application into a 12-μl quartz microsampling cell. All measurements were performed at 10 °C, and a minimum of 100 individual Autocorelation Functions were averaged and analyzed using the DYNAMICS software package (Wyatt Technology). The Rh was determined from the translational diffusion constant, Dt, using the Stokes-Einstein relation,
 |
where k is the Boltzmann's constant, T is temperature (K), η is solvent viscosity, and Rh is the hydrodynamic radius. Sample polydispersity, Pd, was analyzed using a regularization algorithm implemented into a DYNAMICS program that attempts to fit the distribution of exponentials to ACF to obtain an approximation of the real distribution of Rh. Percent polydispersity, %Pd, was calculated using the Equation 4,
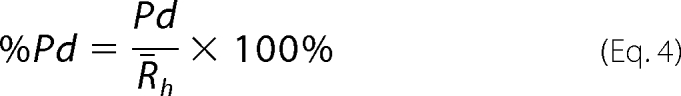 |
where Pd is sample polydispersity, and R̄h is the mean hydrodynamic radius
Static light scattering (SLS) measurements of PRG 37–1081 and its complex with RhoA were performed on a Viscotek TDA 305 instrument with triple detector array (Malvern Instruments, Westborough, MA). Protein samples were concentrated to ∼1.5 mg/ml and passed through a Superdex-200 10/30 size exclusion column (Amersham Biosciences) before SLS analysis.
Analytical Size Exclusion Chromatography
The ASEC of PRG fragments was performed on an ÄKTA FPLC protein purification system equipped with a Superdex-200 10/30 GL size exclusion chromatography column (Amersham Biosciences). Briefly, the protein in buffer C was concentrated to ∼1.5 mg/ml, spun at 21,000 × g for 30 min at 4 °C to remove particulate material and applied on a gel filtration column. The column was calibrated with a kit for the 12–200-kDa range (Sigma).
SAXS Data Collection and Reduction
SAXS data were collected in several experimental sessions on the EMBL X33 beamline of the storage ring DORIS III (DESY, Hamburg) (29) using a robotic sample changer (30). Different PRG constructs (including mutants) with and without RhoA were measured at 10 °C in a concentration range from 1 to 10 mg/ml. The data were recorded using a Pilatus 1 m pixel detector (DECTRIS) at a sample detector distance of 2.7 m and wavelength of 1.5 Å, covering the range of momentum transfer 0.012 < s < 0.6 Å−1 (here, s = 4π sinθ/λ, where 2θ is the scattering angle). A standard data collection time of 2 min was used for all samples and divided into eight 15-s frames to assess and correct for radiation damage. The frames were processed automatically (31), yielding radially averaged curves of normalized intensity versus the momentum transfer. The scattering from the buffer, recorded before and after each sample, was averaged for the background subtraction.
The data analysis was done using PRIMUS (32). The forward scattering I(0) and the radii of gyration, Rg, were evaluated using the Guinier approximation (33) assuming that at very small angles (s < 1.3/Rg) the intensity is represented as I(s) = I(0) exp (−(sRg)2/3). These parameters were also computed from the entire scattering patterns using the program GNOM (34), that provides the distance distribution functions, P(r), and the maximum particle dimensions, Dmax. The expected molecular weight (Mr) of the solute was estimated by comparison of the forward scattering with that from reference solutions of bovine serum albumin (Mr = 66 kDa). The excluded volume of the hydrated particle (the Porod volume, vp) was computed using the Porod invariant (35).
Shape Reconstruction
The low resolution shapes of various PRG constructs were reconstructed ab initio from the scattering data using DAMMIN (36) and DAMMIF (37). These programs represent the particle by an assembly of densely packed spheres and employ simulated annealing to construct a compact interconnected model fitting the experimental data to minimize the discrepancy,
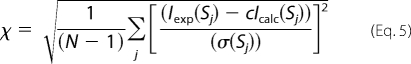 |
where N is the number of experimental points, c is a scaling factor, and Iexp(s), Icalc(s), and σ(sj) are the experimental intensity, the calculated intensity, and experimental error at the momentum transfer sj, respectively. Alternative, higher resolution ab initio models were constructed using GASBOR (38), which models the particle in solution as a protein-like assembly of dummy residues and represents the internal structure more accurately than DAMMIN or DAMMIF. Multiple DAMMIN, DAMMIF, and GASBOR calculations were performed to assess the stability of resulting solutions. 10–20 independent reconstructions were performed, and the models were averaged with the program DAMAVER (39).
Combined ab Initio and Rigid Body Modeling
High resolution crystal structures of different domains (PDZ, RGSL, DH-PH, and RhoA) were used for a combined ab initio/rigid-body modeling with BUNCH (40). The rigid-body models were generated for the PRG constructs (including mutants) in the presence and absence of bound RhoA (Table 1). Starting from a random domain arrangement, BUNCH uses simulated annealing to guide the translations and rotations of domains to minimize the discrepancy, χ, between the experimental and calculated data (Equation 5) while maintaining chain connectivity without steric clashes. Missing linkers between individual subunits are modeled using dummy residues starting from a random initial configuration generated by PRE_BUNCH. For different constructs, either a single scattering curve of the particular construct was fitted or multiple curves containing deletion mutants were fitted simultaneously. The relative positions of DH and PH domains were fixed, as it has been previously shown that they form a rigid unit in solution (41). For PRG-RhoA complexes, DH, PH, and RhoA were grouped into a single rigid body. Multiple BUNCH runs were performed, yielding stable and consistent rigid body models for each sample.
TABLE 1.
Summary of analytical size exclusion, dynamic light scattering, and SLS measurements
| Mr | Mr ASEC | Mr SLS | Rh (MAD) | % Pd (MAD)a | |
|---|---|---|---|---|---|
| kDa | kDa | kDa | nm | ||
| PDZ-L1-RGSL | 49 | 145 | 5.1 (0.23) | 11.5 (3.30) | |
| N-DH-PH | 48.7 | 4.1 (0.18) | 10.5 (3.50) | ||
| N-DH-PH /RhoA | 69.3 | 4.3 (0.07) | 13.5 (2.35) | ||
| N-DH-PH 4R | 49.3 | 4.4 (0.31) | 13.8 (5.99) | ||
| N-DH-PH 4R/RhoA | 69.9 | 4.4 (0.18) | 12.6 (2.66) | ||
| RGSL-L2-DH-PH | 93.2 | 250 | 6.3 (0.20) | 14.9 (3.84) | |
| RGSL-L2-DH-PH /RhoA | 114.6 | 260 | 6.0 (0.05) | 13.1 (7.55) | |
| RGSL-L2-DH-PH 4R | 92.8 | 7.0 (0.35) | 23.8 (4.95) | ||
| RGSL-L2-DH-PH 4R/RhoA | 115.2 | 6.2 (0.20) | 12.6 (5.05) | ||
| PDZ-L1-RGSL-L2-DH-PH | 118.2 | 420 | 115 | 7.7 (0.22) | 19.9 (2.78) |
| PDZ-L1-RGSL-L2-DH-PH /RhoA | 139.6 | 450 | 137 | 7.2 (0.32) | 18.1 (4.35) |
| PDZ-L1-RGSL-L2-DH-PH 4R | 118.8 | 390 | 7.8 (1.16) | 19.0 (6.05) | |
| PDZ-L1-RGSL-L2-DH-PH 4R/RhoA | 140.2 | 420 | 7.0 (0.45) | 9.74 (1.28) |
a Mr, Mr ASEC, and Mr SLS are molecular weight, molecular weight determined with analytical size exclusion, and molecular weight determined by static light scattering, respectively. Rh and % Pd are the hydrodynamic radius and percent polydispersity, respectively, determined in each case from 5–10 independent measurements. MAD is mean absolute deviation.
Modeling Ensembles of Structures
To assess the degree of the dynamics and conformational heterogeneity of the various constructs, the SAXS data were analyzed using the Ensemble Optimization Method (EOM) (42) which assumes coexistence of a range of conformations in solution to fit the experimental SAXS data. In the first step RANCH was used to generate a pool of 10,000 models with random arrangement of high resolution structures of individual domains connected by modeled linkers. For PRG-RhoA complexes, the DH-PH tandem and RhoA were used as a single rigid body, similarly to the aforementioned BUNCH analysis. The theoretical scattering curve was then calculated for each model by CRYSOL (43). In the second step, a genetic algorithm (GAJOE) selected subsets of ∼20 protein models. The average theoretical scattering was calculated for each subset and fitted to experimental SAXS data. The subset best-fitting experimental data are reported as a final solution. A dozen independent EOM runs were performed, and the obtained subsets were analyzed to yield the Rg distributions in the optimal ensembles. To group EOM models into extended and compact conformations, the average Dmax was calculated for each ensemble. Models with Dmax values above the average were classified as extended, and models with Dmax values below the average were assigned to the compact class.
Crystallization and X-ray Structure Determination of RhoA Complex
The complex of the wild-type N-DH-PH with RhoA was prepared as described above and screened using the JCSG+ suite crystallization matrix (Qiagen, Valencia, CA) using either the screen solution or 1.5 m NaCl in the reservoir (44) and yielded diffraction-quality crystals directly from the initial setup. The best crystals of N-DH-PH/RhoA were grown in 0.15 m dl-malic acid, pH = 7.0, 20% w/v PEG 3350. Crystals exhibit P21 symmetry and unit cells close to that of the DH-PH/RhoA structure (21). Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-BM beamline at the Advanced Photon Source, Argonne National Laboratory. The structure was determined using the molecular replacement pipeline BALBES (45) and refined with PHENIX (46) to final R and Rfree values of 21.1 and 27.1%, respectively.
RESULTS
Expression and Preliminary Characterization of Multidomain Fragments of PRG
The full-length human PRG is made up of a single polypeptide chain comprising 1522 amino acids (12). It contains a short, putatively disordered N-terminal fragment of 36 residues followed by a catalytically functional module consisting of PDZ, RGSL, and DH-PH domains. Downstream of this module there is a C-terminal domain of unknown structure and function that may mediate in vivo homo- and heterodimerization of the RGSL-family GEFs (47, 48) but is not important for the catalytic activity. As previously described (25), we were able to overexpress in Escherichia coli and purify to homogeneity several multidomain fragments of human PRG (Fig. 1). These included: 1) the PDZ-L1-RGSL fragment (residues 37–490), which comprises the PDZ domain, the ∼180 residue long linker (L1), followed by the RGSL domain; 2) the RGSL-L2-DH-PH fragment (residues 277–1081), which includes the RGSL domain, the L2 linker ∼220 residues in length, and the DH-PH tandem; 3) the N-DH-PH fragment (residues 672–1081), which includes the catalytically active DH-PH tandem along with the N-terminal extension thought to exert a regulatory function; 4) the intact functional module of PDZ-L1-RGSL-L2-DH-PH (residues 37–1081). Furthermore, we generated variants of both RGSL-L2-DH-PH and PDZ-L1-RGSL-L2-DH-PH incorporating the activating charge reversal mutations D706R, E708R, E710R, and D712R (4R). Finally, we were able to purify complexes of those variants containing the DH-PH tandem, both wild-type and with activating mutations, with the human recombinant RhoA overexpressed in the non-prenylated form in E. coli (27).
One of the characteristics of many recombinant proteins that hinders the in vitro biochemical and biophysical assays is irreversible aggregation induced by improper expression, purification, or handling procedures (49). Therefore, to evaluate possible supramodular architecture, we needed to confirm that all PRG variants exist in solution in monodisperse and monomeric state. Dynamic light scattering is a method of choice to accurately determine the stability of a target protein, its hydrodynamic radius (Rh), and consequently the overall conformation (50, 51). The low polydispersity values of the samples tested indicate that they are relatively stable and of quality sufficient to proceed with structural analysis (Table 1). Comparison of hydrodynamic radii of PRG variants with Rh, calculated for theoretical models of globular, branched, and linear polymers, suggests that PRG is a relatively extended molecule (supplemental Table S1).
The ASEC elution profiles for PDZ-L1-RGSL-L2-DH-PH, PDZ-L1-RGSL, and RGSL-L2-DH-PH show apparent molecular masses of ∼ 420, 250, and 145 kDa, respectively (Table 1). These results are consistent with extended, rather than globular conformations, but we could not exclude the possibility of oligomerization. However, SLS measurements of the PDZ-L1-RGSL-L2-DH-PH fragment as well as that of its complex with RhoA were consistent with monomeric species in solution (Table 1). Given that the PDZ-L1-RGSL-L2-DH-PH fragment contains all the elements that could potentially be responsible for oligomerization, the shorter fragments are also expected to be monomeric.
Secondary Structure Content and Dynamics of Interdomain Linkers
Our first aim was to determine whether the linker regions that connect the PDZ and RGSL domains (L1) and RGSL with the DH domain (L2) contain any detectable secondary structure elements. Although both linkers are predicted to be intrinsically disordered (52), secondary structure prediction (53) suggests the presence of α-helical segments amounting to ∼26 and ∼20% of the sequence in L1 and L2, respectively. Circular dichroism measurements of PDZ-L1-RGSL and RGSL-L2-DH-PH fragments revealed that the content of the secondary structure within L1 and L2 linkers is actually significantly higher than expected (Fig. 2). Assuming that the linkers are completely unstructured, and taking into account the known structures of PDZ and RGSL domains, the α-helical content of PDZ-L1-RGSL and RGSL-L2-DH-PH fragments should be ∼35 and ∼45%, respectively (these values would increase by ∼5%, if we include the predicted putative α-helical content). However, the experimentally determined α-helical content for the PDZ-L1-RGSL fragment is 79% (±7%), and that of the RGSL-L2-DH-PH fragment is 57% (±5%). The β-structure content in PDZ-L1-RGSL is <5%, whereas for the RGSL-L2-DH-PH it is roughly 13% (±3%) or ∼8% higher than if the L2 linker was completely unstructured. Thus, we conclude that at any point in time the ensemble of PRG molecules in solution contains a portion with L1 and L2 linkers with significant content of secondary structure elements, most probably transient α-helices.
FIGURE 2.

Results of far-UV CD measurements. A, shown are spectra of PDZ-L1-RGSL (solid triangles) and RGSL-L2-DH-PH (solid circles) in normalized molar ellipticity units. B, shown is percent content of secondary structure elements of PDZ-L1-RGSL and RGSL-L2-DH-PH. Percent values assuming completely unfolded linker, including secondary structure predictions and experimental data are shown as striped, white, and black bars, respectively. MRE, mean residue ellipticity.
Next we used hydrogen-deuterium exchange mass spectrometry in an effort to analyze the dynamics of the PDZ-L1-RGSL-L2-DH-PH fragment. This method allows for the monitoring of the D/H exchange rate, which is significantly suppressed within stably folded domains as a consequence of reduced solvent accessibility (54). The data revealed that both linkers are highly solvent-accessible, with low levels of protection from hydrogen/deuterium exchange (supplemental Fig. S1A). These results are not inconsistent with the CD data because high solvent accessibility does not necessarily imply a lack of transiently structured elements. For example, the PDZ domain (residues 37–123), a fully structured, small globular module, undergoes nearly full D/H exchange after even the shortest quenching times (supplemental Fig. S1B). The hydrogen/deuterium exchange mass spectrometry data are consistent with the notion that PRG is highly dynamic in solution and that the L1 and L2 linkers are accessible for D/H exchange and, thus, not sequestered between the globular domains.
The Conformation of PRG in Solution
Our next objective was to determine whether the four globular domains within the functional fragment of PRG form a supramodular entity in solution. Because the three- and four-domain PRG constructs proved recalcitrant to characterization by crystallographic methods, we turned to SAXS, which can probe the structure in a broad range of macromolecular sizes under near native conditions (55) and different levels of flexibility, from relatively rigid molecules with flexible hinge regions (56) to natively unfolded proteins, like Tau protein (57, 58).
The experimental SAXS data are presented in Fig. 3 and supplemental Fig. S2A. Except the four-domain construct at the highest concentration, the samples did not show any concentration-dependent aggregation. Therefore, the scattering data extrapolated to infinite dilution were used for analysis and modeling. First, we calculated the overall parameters from SAXS data, i.e. the radius of gyration (Rg), maximum size (Dmax), and the excluded volume (vp) (Table 2). The Rg values of most constructs were significantly higher than those of globular proteins of similar Mr but smaller than the Rg values of unfolded proteins. For example, PDZ-L1-RGSL construct (49 kDa) has the Rg of 5.8 ± 0.2, whereas folded bovine serum albumin (BSA, 66 kDa) and natively unfolded Tau protein (45 kDa) (58) have Rg values of ∼3.1 ± 0.2 and 6.6 ± 0.3 nm, respectively (Table 2). The Rg/Rh ratio provides information about the flexibility of the macromolecule. For rigid structures, Rg is close to Rh (Rg/Rh ranging from ∼1.1 for anisometric polymers to ∼0.8 for a solid sphere). For flexible systems Rg may significantly exceed Rh, such that Rg/Rh reaches 1.5 for a random coil (59). For most of the PRG constructs, Rh is slightly higher than Rg, confirming the limited flexibility of the samples. On the other hand, for the well folded N-DH-PH constructs, the Rg/Rh ratio was ∼0.7, which clearly points to their rigidity and compactness (see Tables 1 and 2).
FIGURE 3.

Scattering profiles for truncated (A) and four-domain PRG fragments (B). Experimental data, fit to ab initio, rigid body, and EOM models are shown as open circles, yellow-dotted lines, blue solid lines, and red dashed lines, respectively. Experimental SAXS profiles were appropriately displaced along the logarithmic axis for better visualization and overlaid with corresponding fits.
TABLE 2.
Overall structural parameters of PRG variants and their complexes with RhoA obtained by SAXS
| Rg | Dmax | vp | χs | χg | χrb | χfa | |
|---|---|---|---|---|---|---|---|
| nm | nm | nm3 | |||||
| PDZ-L1-RGSL | 5.8 | 21.5 | 118 | 0.932 | 1.1 | 1.36 | 0.80 |
| N-DH-PH | 2.95 | 9.8 | 84 | 1.00 | 1.08 | 1.02 | 0.84 |
| N-DH-PH /RhoA | 2.8 | 9.1 | 120 | 1.14 | 1.28 | 1.00 | 0.71 |
| N-DH-PH 4R | 2.87 | 9.2 | 84 | 0.92 | 1.26 | 1.10 | 0.79 |
| N-DH-PH 4R/RhoA | 2.8 | 9.2 | 124 | 0.92 | 1.13 | 0.95 | 0.80 |
| RGSL-L2-DH-PH | 6.5 | 22 | 215 | 0.8 | 0.85 | 0.8 | 0.72 |
| RGSL-L2-DH-PH /RhoA | 5.85 | 20 | 240 | 0.87 | 1.17 | 1.0 | 0.95 |
| RGSL-L2-DH-PH 4R | 7.3 | 27.5 | 233 | 0.90 | 0.97 | 1.1 | 0.78 |
| RGSL-L2-DH-PH 4R/RhoA | 6.44 | 25 | 270 | 0.77 | 0.8 | 0.83 | 0.74 |
| PDZ-L1-RGSL-L2-DH-PH | 7.7 | 28 | 295 | 0.89 | 1.09 | 1.00 | 0.97 |
| PDZ-L1-RGSL-L2-DH-PH /RhoA | 6.6 | 24.5 | 240 | 0.76 | 0.88 | 0.72 | 0.69 |
| PDZ-L1-RGSL-L2-DH-PH 4R | 8.1 | 30 | 325 | 0.79 | 0.79 | 0.82 | 0.72 |
| PDZ-L1-RGSL-L2-DH-PH 4R/RhoA | 7.6 | 27 | 330 | 0.79 | 0.80 | 0.71 | 0.684 |
a Rg, Dmax, and vp are radius of gyration, maximum size, and excluded volume, respectively. χs, χg, χrb, and χf are discrepancies between the experimental data and computed scattering curves from DAMMIN, GASBOR, BUNCH, and EOM models, respectively.
Further evidence for the limited flexibility of the studied constructs is provided by the distance distribution functions, P(r) (see Figs. 5 and supplemental Fig. S2B). For PDZ-L1-RGSL as well as for the wild type and the 4R mutant of RGSL-L2-DH-PH, the function yielded large Dmax values and shoulders at higher distances, representing weak interdomain correlation peaks. These features are typical for flexible multidomain proteins with extended conformations (see Fig. 5) (60). The same is true for the P(r) functions of the wild type and the 4R mutant of PDZ-L1-RGSL-L2-DH-PH, both free and in complex with RhoA. It is noteworthy that the interdomain correlation peaks are even more reduced in the wild-type and the 4R mutant of PDZ-L1-RGSL-L2-DH-PH, as compared with RGSL-L2-DH-PH constructs, suggesting yet higher flexibility in the constructs with two linkers. The shapes of P(r) functions of N-DH-PH and its 4R variant agree well with the model of a two-domain DH-PH tandem and reflect the transition toward a more globular assembly in the RhoA complexes (supplemental Fig. S2B).
FIGURE 5.

Rigid body and ab initio models of PRG variants. A, RGSL-L2-DH-PH. B, PDZ-L1-RGSL-L2-DH-PH. C, PDZ-L1-RGSL. The left panels in A and B show models of the wild-type PRG +/− RhoA, and the center panels show models of the 4R mutant of PRG +/− RhoA. The right panel shows distance distribution function, P(r), of the wild-type and 4R mutant of PRG variants in isolation (solid and dashed black line, respectively) and RhoA complexes (solid and dashed red line, respectively). Ab initio models and folded domains of PRG and RhoA are depicted in surface representation (gray, ab initio; orange, PDZ; green, RGSL; blue, DH; purple, PH; red, RhoA), and linker regions are represented as yellow spheres.
To qualitatively assess the compactness and flexibility of PRG fragments, we used the Kratky plot (I(s) × s2 as a function of s) (Fig. 4 and supplemental Fig. S2C). With the exception of the compact, well folded N-DH-PH (supplemental S2C), all PRG variants show the Kratky plot shapes between those for a globular BSA and for a natively unfolded Tau (58), further confirming limited intramolecular flexibility of the PRG constructs. Qualitatively, the RGSL-L2-DH-PH fragment appears to be more compact than PDZ-L1-RGSL, although the PDZ-L1-RGSL-L2-DH-PH fragment, which includes both L1 and L2 linkers, seems to be most flexible. This conclusion agrees well with the analysis of P(r) functions for different constructs as well as with the light scattering and ASEC results (Table 1 and supplemental S1).
FIGURE 4.

Comparison of Kratky plots of PDZ-L1-RGSL (solid triangle), RGSL-L2-DH-PH (green solid square), and PDZ-L1-RGSL-L2-DH-PH (solid circle), with bovine serum albumin (open circle), and protein tau (open triangle).
The shapes obtained ab initio from the SAXS data using DAMMIN (36), particularly for the four-domain constructs, showed extendedness and excluded volumes somewhat larger than expected for the proteins of the corresponding Mr (Table 2). The excluded volume increase can be attributed to the flexibility of the constructs, making the protein cover effectively larger spatial volumes.
The program GASBOR (38) was then used to generate alternative ab initio models with predefined numbers of dummy residues for each construct and, therefore, consistent with the expected volumes. The envelopes generated by GASBOR (gray surface in Fig. 5) are in general compatible with the DAMMIN models being highly extended and not showing clear domain boundaries. These envelopes are also consistent with the flexibility of the constructs, supposedly representing an average over the sets of existing conformations.
To obtain a more detailed structural picture of PRG, we performed combined ab initio/rigid body modeling using the available high resolution structures of individual domains and experimental SAXS data using the program BUNCH (40). The SAXS-based modeling is most informative when one simultaneously uses the scattering patterns from several deletion mutants (40). Therefore, we used either single or multiple scattering curves for combined ab initio/rigid body modeling (Table 2, Figs. 3 and 5). Inspection of the rigid body models generated by BUNCH reveals that both linker regions seem to contain partially folded, compact elements, and therefore, cannot be completely unstructured (Fig. 5). It is conceivable that these relatively compact pseudo-domains have the characteristics of molten globules, a state in which there are stable secondary structure elements but only a few tertiary contacts (61). The χ values of the rigid body models, however, are not perfect, especially in the case of PDZ-L1-RGSL (Fig. 3, Table 2), which can be explained by conformational heterogeneity of the studied samples.
We then compared the ab initio models generated by GASBOR with the corresponding rigid body models (Fig. 5). The rigid body models of all multidomain PRG fragments agree relatively well with the ab initio molecular envelopes at low resolution. There are, however, discrepancies whereby the individual domains (e.g. the PH domain) stick out of the envelope. This can be rationalized taking into account the fact that the ab initio calculated envelopes represent the averaged structural heterogeneity of the protein rather than single conformers, and these results further suggest that the protein adopts multiple conformations.
Overall, these observations are consistent with the fact that the SAXS curves are a result of a dynamic equilibrium of co-existing conformations in solution. To obtain an insight into the range of conformations sampled by the various PRG variants and to quantitatively describe flexibility, we used the EOM technique (42). This approach allows for identification of an ensemble of conformers, for which the collective simulated scattering curve provides the best fit to the experimental data (see “Experimental Procedures”). The EOM provided good fits to the experimental data for all constructs (Table 2, Fig. 3). The analysis of the Rg distributions and the models generated by EOM strongly suggests that the linker conformations within the ensemble of the molecules may range from highly extended to compact, in which case the flanking globular domains are located in close proximity to each other (Fig. 6). It is very important to note that the experimental data cannot be satisfactorily fitted by single models with either compact or extended conformations, as evidenced by the poor χ values of such fits. When an ensemble of conformers is used instead, the fit is significantly improved (Table 2, Fig. 6). Moreover, the experimental data can be rather well fitted even if only few representative EOM models, containing one compact and one extended conformation, are used for simulation. This strongly suggests that the conformation of PRG in solution is a heterogeneous mixture of compact and extended states.
FIGURE 6.

The radius of gyration distribution for the EOM models of PDZ-L1-RGSL (A), RGSL-L2-DH-PH (B), and PDZ-L1-RGSL-L2-DH-PH (C). The distribution for the pools of models is shown by black and red solid lines for the isolated PRG and the RhoA complex, respectively, whereas the distribution for the selected ensemble of models is represented by black and red dashed lines for the isolated PRG and the RhoA complex, respectively. The representative conformations of the respective EOM models are shown in the insets. Folded domains of PRG are depicted in surface representation (orange, PDZ; green, RGSL; blue, DH; purple, PH; red, RhoA), and linker regions are represented as yellow spheres. χe, χc, and χE are discrepancies to the representative extended, compact, and ensemble of models, respectively.
It is noteworthy that the molten globules are also present in EOM models, but their exact location within the linker and the level of compactness vary. Although single PDB models generated by EOM do not represent the biologically relevant species, they adhere to the geometrical and chemical restrains such as bond lengths and dihedral angles. Therefore, visual inspection of the individual models selected by EOM provides a way to assess the peculiarities of the local structure. The fact that the EOM models fit well the experimental data when molten globule-like moieties are present within the linker regions suggests that a single BUNCH model representing the “intermediate” conformation may be a reasonable approximation of the PRG structure.
Structural Consequences of Activating Mutations and RhoA Binding
Given the activating character of the four charge reversal mutations within the activation box upstream of the DH domain (residues 672–711), we used SAXS to explore the impact of these mutations on the conformation of RGSL-L2-DH-PH and PDZ-L1-RGSL-L2-DH-PH. Rigid body models do not indicate any significant changes to the overall architecture (Fig. 5). However, the putative molten globule within the L2 linker appears to be less compact in the mutated variants, perhaps providing less of a steric hindrance when RhoA is bound (Fig. 7). Moreover, the variants carrying the mutations seem to be more dynamic than the wild-type variants, as evidenced by the increase of the radius of gyration (Table 2). Interestingly, the analysis of Dmax values of the EOM models for the wild-type proteins showed that ∼60% were classified as compact, whereas for the 4R mutant only 40% were in that group, suggesting higher dynamics in the latter (data not shown).
FIGURE 7.

Conformational changes within the molten globule region of wild-type and 4R mutant of N-DH-PH (A), RGSL-L2-DH-PH (B), and PDZ-L1-RGSL-L2-DH-PH (C). Structures of wild-type and 4R mutant of PRG were superimposed using residues 714–932 of the DH domain. DH domain is shown as a blue schematic; wild-type and 4R mutant linkers are represented by orange and light blue spheres, respectively. A putative position of RhoA is shown in semi-transparent surface representation. The PH domain was omitted for clarity.
We also investigated the solution structures of complexes of various fragments of PRG with RhoA. In general terms, the binding of RhoA does not alter the scattering curves in any critical way (Fig. 3). However, the wild-type and the constitutively active mutants of RGSL-L2-DH-PH and PDZ-L1-RGSL-L2-DH-PH become more compact upon binding RhoA (Tables 1 and 2). We previously reported that the binding of RhoA to the DH-PH tandem induces virtually no structural changes in the latter (41), and thus, any difference in the present SAXS data must stem from the rearrangements of domains and linkers upstream of the DH-PH tandem. In all models generated for RhoA complexes, the L2 linker is displaced from the RhoA-binding site on the DH domain, and the molten globule region upstream of the DH domain becomes less ordered than in an isolated protein. Consistent with the data for the isolated 4R mutants, the Rg values for RhoA complexes are larger than for the wild-type PRG-RhoA (Table 2), suggesting increased disorder within the L2 linker.
Given the importance of the activation box in the regulation of catalytic activity, we wondered if the 672–711 segment becomes ordered upon binding of RhoA and if the charge reversal mutations cause visible structural consequences. To this end we crystallized the autoinhibited 672–1081 variant (N-DH-PH) in complex with RhoA (data statistics are provided in Table 3). As was the case with the crystal structure of the complex of isolated DH-PH domain with RhoA (21), there is interpretable electron density starting with residue Gln-714, indicating that the 672–713 fragment is completely disordered. In both cases the GEF switch, i.e. residues 714–730, is ordered and engages RhoA (Fig. 8). Its conformation is virtually identical to that observed in the known crystal structures of RGSL-containing GEFs regardless of the RhoA presence (Fig. 8A, inset). The same result was obtained with the 672–711 segment harboring the 4R mutations (not shown).
TABLE 3.
Crystallographic statistics
| N-DH-PH/RhoA | |
|---|---|
| Data collectiona | |
| Resolution range (Å) | 50.0–2.85 (2.90–2.85) |
| Space group | P1211 |
| Unit-cell parameters (Å, °) | a = 84.4, b = 92.4, c = 113.6, β = 118.6 |
| Observations | |
| Unique | 38,049 |
| Total | 137,404 |
| Completeness (%) | 91.6 (58.8) |
| Redundancy | 3.6 |
| Rmergeb | 0.087 (0.33) |
| 〈I/σ(I)〉 | 12.55 (1.98) |
| Refinementa | |
| Resolution range (Å) | 48.3-2.85 |
| No. of reflectionsc | 38,028 |
| R factord | 0.211 |
| Rfreec | 0.271 |
| No. of atoms | |
| Protein, non-H | 8,658 |
| Nonprotein | 39 |
| Root mean square deviations | |
| Lengths (Å) | 0.009 |
| Angles (°) | 1.278 |
| Overall B factor (Å2) | 52.8 |
a Values in parentheses correspond to the last resolution shell.
b Rmerge = ΣhklΣi|Ii(hkl) − 〈I(hkl)|/ΣhklΣiIi(hkl)〉.
c % of the reflections were reserved for calculation of Rfree.
d R factor = Σhkl‖Fobs| − |Fcalc‖/Σhkl|Fobs|.
FIGURE 8.

The GEF switch region of PRG. A, a fragment of PRG encompassing the GEF switch (residues 714–730) is shown as blue sticks, whereas the part of an α1 helix of the DH domain is represented as a blue ribbon. The RhoA switch 1 is depicted as a green stick/ribbon diagram. An omit electron density map calculated with the GEF switch removed from the model using FFT program within the CCP4 suite (72) is contoured at 1.9 σ and shown as green mesh. The inset shows superposition of the GEF switch region of PRG, LARG (PDB code 1X86), and p115 (PDB code 3ODO) shown as blue, green, and orange ribbons, respectively. αΝ1 and αΝ2 are the short α-helical extensions of the DH domain, and N and C are the N and C termini. B, the sequence alignment of three RGSL-GEFs shows the location and sequence homology of the activation box and the GEF switch. The negatively charged residues of the activation box are shown in red, and the residues from two short a-helical extensions of the DH domain are colored green.
DISCUSSION
Our results show conclusively that the functional fragment of PRG encompassing the four globular domains (PDZ, RGSL, DH, and PH) does not have a simple supramodular architecture in which the domains would interact directly to form a globular moiety. We also show evidence that both interdomain linkers, L1 and L2, are partly ordered and may contain modules similar to molten globules. It is, therefore, clear that PRG and probably the other two members of the RGSL subfamily of GEFs are not autoinhibited by the expected canonical mechanism (8) in which PDZ and RGSL domains might inhibit the catalytic activity of the DH-PH tandem by steric hindrance.
In view of the results reported here and in two previous papers from our group (25, 41) as well as data from other laboratories (23, 26), it is necessary to revise a model of PRG autoinhibition and activation. First, as has been demonstrated by our recent study, most of the autoinhibition is traced to the short fragment upstream of the DH domain, encompassing residues 672–711 (25). This negatively charged region seems to interact with positively charged patches on DH domain surface (i.e. Arg-867 and Arg-868), which are involved in interactions with RhoA, and affects the conformation of the GEF switch, which is immediately downstream. The fact that the autoinhibited N-DH-PH fragment assumes in the complex with RhoA a structure that is identical to that described for the isolated DH-PH tandem shows that the autoinhibition is not caused by permanent steric impairment but by a shift of the equilibrium between the catalytically competent and impaired species. The recent crystallographic study of the p115-isolated DH-PH tandem containing disordered N-terminal extension that includes the activation box (23) is consistent with the notion that in the native GEF the GEF switch is intrinsically less ordered due to the presence of the adjacent L2 linker. This is likely to be the main source of autoinhibition in this GEF family (Fig. 9, inset).
FIGURE 9.

Model of regulation of PDZRhoGEF. In a resting state, in cytosol, activity of PRG is inhibited by steric and electrostatic interference with RhoA binding caused by the activation box and the molten globule region within the linker. Anchoring at the membrane and interaction with Gα subunit might be one of the primary forces activating PRG and results in conformational changes within the linker, which consequently displaces inhibitory elements from RhoA-binding site. The inset shows three main regulatory structural elements of PDZRhoGEF include the GEF switch (green schematic representation), the activation box (red line), and the molten globule (red semi-circle). The DH, PH, and RhoA are shown in a surface representation and are colored blue, purple and gray, respectively. The surface patch on the DH domain, which is involved in RhoA binding, and is affected by the presence of the L2 linker is colored yellow. Two arginine residues within this patch (Arg-867 and -868), most probably involved in electrostatic interactions with negatively charged activation box, are shown in red. The switch 1 region of RhoA is shown in orange. MG, molten globule; AB, activation box; GS, GEF switch.
The activation mechanism of PRG appears to be much more subtle and complex than originally believed. It seems that the regulatory function of the PDZ domains, as suggested by several studies (16–18), is at best indirect. The removal of the PDZ domain from PRG has essentially no effect on the catalytic activity (25), and so any activating effects of PDZ-mediated interactions is likely to stem from the recruitment of PRG to the membrane. Likewise, the RGSL domain clearly has no direct interaction with DH-PH, and when its covalent link to the latter is severed, its presence has no effect on the catalytic process (25). Identical observations are reported for p115 (23, 26). Nevertheless, the removal of RGSL domain causes partial relief of autoinhibition. Our present data suggest that although the RGSL domain is not in direct contact with the DH-PH tandem, it may still be physically in its proximity due to the relatively compact structure of the L2 linker. Thus, interactions with Gα12/13 may lead to structural perturbations transmitted through the L2 linker.
Interestingly, a recent SAXS structure determination of the L2-DH-PH fragment of p115 also suggests that the linker contains at least some ordered elements (23). This investigation did not explore that structure of a RGSL-L2-DH-PH, and it is possible that such a variant would show an even higher degree of compactness in the L2 region.
As pointed out earlier, amino acid composition and bioinformatics analysis suggest that the L2 linker is intrinsically disordered. It has been shown that such intrinsically disordered proteins may regulate many enzymatic, transcriptional, and signal transduction processes (62, 63). The presence and functional role of molten globules is well documented in intrinsically disordered proteins (63). For example, the yeast P2β ribosomal stalk protein exists in a partially disordered state (molten globule) under native conditions and adopts a tighter conformation after dimerization or binding to ribosome (64). Importantly, disordered segments linking structured modules in multidomain proteins have also been shown to be important for the regulation of activity. The human transcriptional coactivator p300, for example, contains a disordered, 60-residue loop within its histone acetyltransferase (HAT) domain (65), which has an autocatalytic activity and regulates p300 via autoacetylation of several lysine residues contained within this region. In addition, another regulatory domain, the cell-cycle regulatory domain-1 (CRD1), has been identified within the presumably disordered linker between cysteine/histidine-rich domain and bromodomain (66). The CRD1 domain utilizes two SUMO modification sites to repress the transcription process (67).
In conclusion, our structural investigation of the PRG in solution suggests that the autoinhibition/activation cycle of PRG is complex and does not conform to a simple mechanistic paradigm of closed-open supramodular equilibrium. Clearly, the autoinhibitory activity resides in the 672–712 fragment in such a way that it disturbs the catalytically competent conformation of the GEF switch. The system is dynamic so that in solution assays utilizing non-prenylated RhoA, it is leaky and shows 20-fold enhancement of nucleotide exchange on the GTPase. However, in vivo, PRG is unlikely to encounter sufficient concentration of free RhoA in the cytosol because GDP-bound, prenylated RhoA is sequestered in the complex with RhoGDI (68). Translocation of PRG to the membrane, mediated either by the PDZ domain or the RGSL domain, can by itself enhance the apparent rate of nucleotide exchange due to increased local concentrations of both PRG- and membrane-bound RhoA. Moreover, the RGSL domain, but not the PDZ domain, may exert a regulatory function on the GEF switch through the L2 linker fragment (Fig. 9).
This model is significantly more complex than the mechanistic schemes proposed for some other Dbl-family GEFs (e.g. Asef, Vav1, or p63RhoGEF) (69–71). Nevertheless, one should bear in mind that distinct, conformational changes are only a part of any in vivo process that involves a plethora of molecules as well as dynamic partitioning between the membrane bound and cytosolic fractions.
Supplementary Material
Acknowledgments
Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM086457. This work was also supported by Human Frontier Science Program Research Grant RGP 55/2006.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1 and S2.
The atomic coordinates and structure factors (code 3T06) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- GEF
- guanine nucleotide exchange factor
- ASEC
- analytical size exclusion chromatography
- DH
- Dbl-homology
- PDZ
- PSD-95/Disc-large/ZO-1
- PH
- pleckstrin-homology
- RGSL
- regulators of G-protein signaling-like
- SAXS
- small angle X-ray scattering
- EOM
- Ensemble Optimization Method
- PRG
- PDZRhoGEF
- LARG
- leukemia-associated Rho-GEF
- SLS
- static light scattering.
REFERENCES
- 1. Etienne-Manneville S., Hall A. (2002) Nature 420, 629–635 [DOI] [PubMed] [Google Scholar]
- 2. Coso O. A., Chiariello M., Yu J. C., Teramoto H., Crespo P., Xu N., Miki T., Gutkind J. S. (1995) Cell 81, 1137–1146 [DOI] [PubMed] [Google Scholar]
- 3. Luo L. (2000) Nat. Rev. Neurosci. 1, 173–180 [DOI] [PubMed] [Google Scholar]
- 4. Ishizaki T., Maekawa M., Fujisawa K., Okawa K., Iwamatsu A., Fujita A., Watanabe N., Saito Y., Kakizuka A., Morii N., Narumiya S. (1996) EMBO J. 15, 1885–1893 [PMC free article] [PubMed] [Google Scholar]
- 5. Rossman K. L., Der C. J., Sondek J. (2005) Nat. Rev. Mol. Cell Biol. 6, 167–180 [DOI] [PubMed] [Google Scholar]
- 6. Zheng Y. (2001) Trends Biochem. Sci. 26, 724–732 [DOI] [PubMed] [Google Scholar]
- 7. Snyder J. T., Worthylake D. K., Rossman K. L., Betts L., Pruitt W. M., Siderovski D. P., Der C. J., Sondek J. (2002) Nat. Struct. Biol. 9, 468–475 [DOI] [PubMed] [Google Scholar]
- 8. Erickson J. W., Cerione R. A. (2004) Biochemistry 43, 837–842 [DOI] [PubMed] [Google Scholar]
- 9. Fukuhara S., Chikumi H., Gutkind J. S. (2001) Oncogene 20, 1661–1668 [DOI] [PubMed] [Google Scholar]
- 10. Bhattacharya M., Babwah A. V., Ferguson S. S. G. (2004) Biochem. Soc. Trans. 32, 1040–1044 [DOI] [PubMed] [Google Scholar]
- 11. Kozasa T., Jiang X., Hart M. J., Sternweis P. M., Singer W. D., Gilman A. G., Bollag G., Sternweis P. C. (1998) Science 280, 2109–2111 [DOI] [PubMed] [Google Scholar]
- 12. Fukuhara S., Murga C., Zohar M., Igishi T., Gutkind J. S. (1999) J. Biol. Chem. 274, 5868–5879 [DOI] [PubMed] [Google Scholar]
- 13. Fukuhara S., Chikumi H., Gutkind J. S. (2000) FEBS Lett. 485, 183–188 [DOI] [PubMed] [Google Scholar]
- 14. Gong M. C., Iizuka K., Nixon G., Browne J. P., Hall A., Eccleston J. F., Sugai M., Kobayashi S., Somlyo A. V., Somlyo A. P. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 1340–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Somlyo A. P., Somlyo A. V. (2003) Physiol. Rev. 83, 1325–1358 [DOI] [PubMed] [Google Scholar]
- 16. Swiercz J. M., Kuner R., Behrens J., Offermanns S. (2002) Neuron 35, 51–63 [DOI] [PubMed] [Google Scholar]
- 17. Taya S., Inagaki N., Sengiku H., Makino H., Iwamatsu A., Urakawa I., Nagao K., Kataoka S., Kaibuchi K. (2001) J. Cell Biol. 155, 809–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yamada T., Ohoka Y., Kogo M., Inagaki S. (2005) J. Biol. Chem. 280, 19358–19363 [DOI] [PubMed] [Google Scholar]
- 19. Longenecker K. L., Lewis M. E., Chikumi H., Gutkind J. S., Derewenda Z. S. (2001) Structure 9, 559–569 [DOI] [PubMed] [Google Scholar]
- 20. Chen Z., Wells C. D., Sternweis P. C., Sprang S. R. (2001) Nat. Struct. Biol. 8, 805–809 [DOI] [PubMed] [Google Scholar]
- 21. Derewenda U., Oleksy A., Stevenson A. S., Korczynska J., Dauter Z., Somlyo A. P., Otlewski J., Somlyo A. V., Derewenda Z. S. (2004) Structure 12, 1955–1965 [DOI] [PubMed] [Google Scholar]
- 22. Kristelly R., Gao G., Tesmer J. J. (2004) J. Biol. Chem. 279, 47352–47362 [DOI] [PubMed] [Google Scholar]
- 23. Chen Z., Guo L., Sprang S. R., Sternweis P. C. (2011) Protein Sci. 20, 107–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Z., Singer W. D., Sternweis P. C., Sprang S. R. (2005) Nat. Struct. Mol. Biol. 12, 191–197 [DOI] [PubMed] [Google Scholar]
- 25. Zheng M., Cierpicki T., Momotani K., Artamonov M. V., Derewenda U., Bushweller J. H., Somlyo A. V., Derewenda Z. S. (2009) BMC Struct. Biol. 9, 36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jaiswal M., Gremer L., Dvorsky R., Haeusler L. C., Cirstea I. C., Uhlenbrock K., Ahmadian M. R. (2011) J. Biol. Chem. 286, 18202–18212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oleksy A., Barton H., Devedjiev Y., Purdy M., Derewenda U., Otlewski J., Derewenda Z. S. (2004) Acta Crystallogr. D. Biol. Crystallogr. 60, 740–742 [DOI] [PubMed] [Google Scholar]
- 28. Unneberg P., Merelo J. J., Chacón P., Morán F. (2001) Proteins 42, 460–470 [DOI] [PubMed] [Google Scholar]
- 29. Roessle M. W., Klaering R., Ristau U., Robrahn B., Jahn D., Gehrmann T., Konarev P., Round A., Fiedler S., Hermes C., Svergun D. (2007) J. Appl. Cryst. 40, 5190–5194 [Google Scholar]
- 30. Round A. R., Franke D., Moritz S., Huchler R., Fritsche D., Malthan M., Klaering R., Svergun D. I., Roessle M. (2008) J. Appl. Crystallogr. 41, 913–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Petoukhov M. V., Konarev P. V., Kikhney A. G., Svergun D. I. (2007) J. Appl. Crystallogr. 40, S223–S228 [Google Scholar]
- 32. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., J., Svergun D. I. (2003) J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 33. Guinier A. (1939) Annu. Phys. (Paris) 12, 161–237 [Google Scholar]
- 34. Svergun D. I. (1992) J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 35. Porod G. (1982) in General Theory; Small-angle X-ray Scattering (Glatter O., Kratky O. eds) pp. 17–51, Academic Press; London [Google Scholar]
- 36. Svergun D. I. (1999) Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Franke D., Svergun D. I. (2009) J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Svergun D. I., Petoukhov M. V., Koch M. H. J. (2001) Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Volkov V. V., Svergun D. I. (2003) J. Appl. Crystallogr. 36, 860–864 [Google Scholar]
- 40. Petoukhov M. V., Svergun D. I. (2005) Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cierpicki T., Bielnicki J., Zheng M., Gruszczyk J., Kasterka M., Petoukhov M., Zhang A., Fernandez E. J., Svergun D. I., Derewenda U., Bushweller J. H., Derewenda Z. S. (2009) Protein Sci. 18, 2067–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bernadó P., Mylonas E., Petoukhov M. V., Blackledge M., Svergun D. I. (2007) J. Am. Chem. Soc. 129, 5656–5664 [DOI] [PubMed] [Google Scholar]
- 43. Svergun D., Barberato C., Koch M. H. J. (1995) J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 44. Newman J. (2005) Acta Crystallogr. D. Biol. Crystallogr. 61, 490–493 [DOI] [PubMed] [Google Scholar]
- 45. Long F., Vagin A. A., Young P., Murshudov G. N. (2008) Acta Crystallogr. D. Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zwart P. H., Afonine P. V., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., McKee E., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Storoni L. C., Terwilliger T. C., Adams P. D. (2008) Methods Mol. Biol. 426, 419–435 [DOI] [PubMed] [Google Scholar]
- 47. Chikumi H., Fukuhara S., Gutkind J. S. (2002) J. Biol. Chem. 277, 12463–12473 [DOI] [PubMed] [Google Scholar]
- 48. Chikumi H., Barac A., Behbahani B., Gao Y., Teramoto H., Zheng Y., Gutkind J. S. (2004) Oncogene 23, 233–240 [DOI] [PubMed] [Google Scholar]
- 49. Bondos S. E., Bicknell A. (2003) Anal. Biochem. 316, 223–231 [DOI] [PubMed] [Google Scholar]
- 50. Jachimska B., Wasilewska M., Adamczyk Z. (2008) Langmuir 24, 6866–6872 [DOI] [PubMed] [Google Scholar]
- 51. Harding S. E. (1995) Biophys. Chem. 55, 69–93 [DOI] [PubMed] [Google Scholar]
- 52. Romero P., Obradovic Z., Kissinger C., Villafranca J. E., Dunker A. K. (1997) in International Conference on Neural Networks, June 9–12, 1997, Houston, TX, Vol. 1, pp. 90–95 [Google Scholar]
- 53. Cole C., Barber J. D., Barton G. J. (2008) Nucleic Acids Res. 36, 197–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Woods V. L., Jr., Hamuro Y. (2001) J. Cell. Biochem. Suppl. 37, 89–98 [DOI] [PubMed] [Google Scholar]
- 55. Mertens H. D., Svergun D. I. (2010) J. Struct. Biol 172, 128–141 [DOI] [PubMed] [Google Scholar]
- 56. Verstraete K., Vandriessche G., Januar M., Elegheert J., Shkumatov A. V., Desfosses A., Van Craenenbroeck K., Svergun D. I., Gutsche I., Vergauwen B., Savvides S. N. (2011) Blood 118, 60–68 [DOI] [PubMed] [Google Scholar]
- 57. Mylonas E., Hascher A., Bernadó P., Blackledge M., Mandelkow E., Svergun D. I. (2008) Biochemistry 47, 10345–10353 [DOI] [PubMed] [Google Scholar]
- 58. Shkumatov A. V., Chinnathambi S., Mandelkow E., Svergun D. I. (2011) Proteins 79, 2122–2131 [DOI] [PubMed] [Google Scholar]
- 59. Rubinstein M., Colby R. H. (2003) Polymer Physics, 1st Ed Oxford University Press, Oxford, UK [Google Scholar]
- 60. Bernadó P. (2010) Eur. Biophys. J. 39, 769–780 [DOI] [PubMed] [Google Scholar]
- 61. Ptitsyn O. B., Uversky V. N. (1994) FEBS Lett. 341, 15–18 [DOI] [PubMed] [Google Scholar]
- 62. Papoian G. A. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 14237–14238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dyson H. J., Wright P. E. (2005) Nat. Rev. Mol. Cell Biol. 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 64. Zurdo J., Sanz J. M., González C., Rico M., Ballesta J. P. G. (1997) Biochemistry 36, 9625–9635 [DOI] [PubMed] [Google Scholar]
- 65. Thompson P. R., Wang D., Wang L., Fulco M., Pediconi N., Zhang D., An W., Ge Q., Roeder R. G., Wong J., Levrero M., Sartorelli V., Cotter R. J., Cole P. A. (2004) Nat. Struct. Mol. Biol. 11, 308–315 [DOI] [PubMed] [Google Scholar]
- 66. Snowden A. W., Anderson L. A., Webster G. A., Perkins N. D. (2000) Mol. Cell. Biol. 20, 2676–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Girdwood D., Bumpass D., Vaughan O. A., Thain A., Anderson L. A., Snowden A. W., Garcia-Wilson E., Perkins N. D., Hay R. T. (2003) Mol. Cell 11, 1043–1054 [DOI] [PubMed] [Google Scholar]
- 68. Longenecker K., Read P., Derewenda U., Dauter Z., Liu X., Garrard S., Walker L., Somlyo A. V., Nakamoto R. K., Somlyo A. P., Derewenda Z. S. (1999) Acta Crystallogr. D. Biol. Crystallogr. 55, 1503–1515 [DOI] [PubMed] [Google Scholar]
- 69. Murayama K., Shirouzu M., Kawasaki Y., Kato-Murayama M., Hanawa-Suetsugu K., Sakamoto A., Katsura Y., Suenaga A., Toyama M., Terada T., Taiji M., Akiyama T., Yokoyama S. (2007) J. Biol. Chem. 282, 4238–4242 [DOI] [PubMed] [Google Scholar]
- 70. Yu B., Martins I. R., Li P., Amarasinghe G. K., Umetani J., Fernandez-Zapico M. E., Billadeau D. D., Machius M., Tomchick D. R., Rosen M. K. (2010) Cell 140, 246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shankaranarayanan A., Boguth C. A., Lutz S., Vettel C., Uhlemann F., Aittaleb M., Wieland T., Tesmer J. J. G. (2010) Cell. Signal. 22, 1114–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Collaborative Computational Project, number 4 (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 15299374 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.