

Abstract
We report here a hitherto undescribed form of cell migration. When a suspension of human keratinocytes is plated on a fibrin matrix, single cells invade the matrix and progress through it as rounded cells by dissolving the fibrin and thereby creating tunnels. These tunnels are cylindrical or helical, the latter being the result of constant change in the path of cellular advance around the helical axis. Helical tunnel formation is strongly promoted by epidermal growth factor. The rate of migration of the cell through the track of a helical tunnel (up to 2.1 mm per day) is about 7-fold greater than through a cylindrical tunnel. Pericellular fibrinolysis leading to tunnel formation depends on the presence of plasminogen in the medium and its conversion to plasmin by a cellular activator. Formation of tunnels requires that plasminogen activator be localized on the advancing surface of the keratinocyte; we propose that the tunnel is cylindrical when the site of release of plasmin is located at a fixed point on the cell surface and helical when the site of release precesses.
Keywords: helical tunnels, fibrinolysis, epidermal growth factor
Cell locomotion is key to many developmental processes, including embryo implantation, morphogenesis, tissue renewal and remodeling, wound healing, and metastasis (1). In living tissues, cells are either attached to a basement membrane or embedded in an extracellular matrix whose composition varies and can include fibronectin, vitronectin, laminins, and collagens (2). Cells adhere to matrix proteins through a repertoire of membrane-bound receptors including integrins, cadherins, selectins, syndecans, and members of the Ig superfamily (3, 4). Cell locomotion thus necessitates cycles of release from the matrix and of attachment. The leading cell edge detaches first from the matrix, reorganizes its cytoskeleton, and extends cytoplasmic protrusions (5, 6). While the lamellipodia and filopodia extend forward onto the matrix, the trailing cell edge in turn detaches from the matrix and retracts, allowing the cell to propel itself (7).
Detachment of the cell from the substratum involves pericellular proteolysis in which serine and matrix metalloproteinases (MMPs) play a crucial role (8). During wound repair, migrating cells such as neutrophils and macrophages, smooth muscle cells, endothelial cells, and keratinocytes, invade and degrade fibrin clots in the form of a three-dimensional lattice. A fibrin clot is the result of the activation of the clotting cascade, the last step of which is the conversion of plasma fibrinogen to fibrin by thrombin (9). The fibrin is then covalently crosslinked by factor XIII of the clotting cascade, a plasma transglutaminase that also crosslinks plasma fibronectin and vitronectin to fibrin. Cell locomotion through a clot necessitates proteolysis of fibrin and involves a cascade of events to which plasmin is usually central. However, there is evidence that pericellular fibrinolysis may also occur independently of the plasminogen cascade through matrix metalloproteinases (membrane type-1 MMP) (10). To form plasmin, circulating plasminogen binds to specific cell surface receptors, where it is converted into plasmin by cleavage of a single peptide bond by the serine protease, urokinase-type plasminogen activator (uPA). However, pro-uPA is converted into active uPA only after it has bound to high-affinity cell surface receptors (uPAR), tethered to the plasma membrane via a glycosylphosphatidylinositol anchor. Plasmin that diffuses away from the plasma membrane is rapidly inactivated by specific plasminogen activator inhibitors (PAIs), most importantly PAI-1 and -2. This provides means of regulating the process of plasminogen activation (11). uPAR that is present on the surface of a variety of cell types, including keratinocytes (12), is also directly involved in cell binding to vitronectin (13). Moreover, in complexes with transmembrane adaptor, integrins, and tyrosin-kinases, uPAR may be involved in signal transduction (11).
By far the most commonly studied form of cell locomotion is that of cells attached and spread out on glass or plastic surfaces (14–16). Sophisticated methods have been applied to the study of cell movement, such as coating the surface of a culture dish with supracolloidal gold particles and detailed analyses of the phagokinetic tracks of 3T3 cells (17). This approach has been used to study the locomotion of various cell types, including keratinocytes, on different extracellular matrix proteins (18, 19). But apart from epithelial cells attached to a basement membrane, there would appear to be few sites in the body where a surface would limit migration to two dimensions. The locomotory system may depend on the differentiated properties of each cell type; for example, the movements by which a leukocyte emerges from a capillary do not resemble the movement of cells on artificial surfaces (20).
We describe here an original migration by means of which human keratinocytes pass through a fibrin matrix. Locomoting keratinocytes dig cylindrical and helical tunnels into fibrin matrices while advancing as rounded cells. This pattern of migration is very efficient and completely different from that described for other cell types. It is a form of locomotion that seems adapted to a keratinocyte deprived of its basement membrane by wounding and obliged to rapidly repair the wound in an environment of fibrin.
Experimental Procedures
Fibrin Gel Matrix.
Fibrin matrices were made from a freeze-dried surgical fibrinogen prepared from human plasma obtained from blood donors (Biocol-Human Thrombin, LFB, Les Ullis, France) as described (21). The protein composition of the product has been reported in detail (22). It contains clottable fibrinogen (95–130 mg ml−1), fibrinonectin (4–13 mg ml−1), factor XIIIa (10–25 units ml−1), and traces of other plasma proteins. The freeze-dried fibrinogen was reconstituted with twice the volume of sterile distilled water recommended by the manufacturer. It was then mixed (vol/vol) with isotonic sodium chloride solution containing human thrombin (5 NIH units ml−1) and evenly distributed into culture dishes before clotting occurred. The fibrin clot resulting from the thrombin-induced activation of fibrinogen was covalently crosslinked by the plasma transglutaminase factor XIIIa present in the fibrinogen preparation. Under these conditions, the fibrin matrix was transparent and suitable for microscopy of cells migrating through the matrix. The final fibrinogen concentration in the matrix (30 mg ml−1) was 8–10 times higher than in plasma.
Cell Culture.
Human keratinocytes originating from foreskin of newborns were cultivated as described (23). The medium was supplemented with epidermal growth factor (EGF) (10 ng ml−1) at the first feeding. Human fibroblasts were also obtained from the foreskin of a newborn but were cultivated in DMEM containing 10% FBS. Human melanocytes, skeletal muscle myoblasts, endothelial cells, and mammary epithelial cells were from Clonetics (San Diego) and were cultivated according to the manufacturer's guidelines.
Plasminogen and PAI.
Plasminogen from human plasma and hrPAI-1 were from Calbiochem. The first was 95% pure and the second was 98% pure, as determined by SDS/PAGE.
Migration Assay.
Cells were dissociated with trypsin and plated at low density (103 cells per cm2) onto the fibrin matrix in complete keratinocyte medium containing human recombinant EGF (hrEGF) (10 ng ml−1) and 10% FBS, but in the absence of irradiated 3T3 cells. Cultures were incubated for 24 h at 37°C and then fixed in 3.7% formaldehyde. The cells and tunnels were then examined under an inverted microscope equipped with phase-contrast optics (Zeiss Axiovert 35). Individual tunnels were analyzed either on photographs or on prints of images obtained with a television camera (AVC-D7CE, Sony, Tokyo).
Isolation of Single Cells.
The fibrin matrix surrounding an individual cell was cut with a disposable 2.0-mm sterile skin biopsy punch (Stiefel, France) under an inverted microscope. The small fibrin cylinder containing the single cell was then carefully transferred to a Petri dish containing irradiated 3T3 cells. The cell was grown into a colony. Individual clones were passaged after 7 days of cultivation and further cultivated (24) or fixed after 12 days of cultivation and stained with 1% Rhodamine.
Results
Entry of Keratinocytes into the Fibrin Matrix and the Formation of Cylindrical Tunnels.
Human epidermal keratinocytes or dermal fibroblasts obtained from the foreskin of newborns were plated at 103 cells/cm2 on transparent fibrin matrices and cultivated for 24 h in medium supplemented with 10% FBS and hrEGF (10 ng ml−1). Fibroblasts were able to attach and spread on the surface of the fibrin matrix but did not enter or appear to degrade it (Fig. 1A). They were mostly nonmotile. The behavior of keratinocytes was quite different: some, like fibroblasts, attached to the fibrin, spread out on the surface, and were nonmotile (Fig. 1B). Others degraded a little of the fibrin matrix locally but remained on the surface, moving only a short distance (Fig. 1C). Sometimes, a motile cell invaded the fibrin matrix but the motility lacked any translational component (Fig. 1D). More commonly, while retaining its spherical shape, a keratinocyte was able to advance over a considerable distance through the matrix, leaving behind the fibrin-free tunnel that it had created (Fig. 1E). The migrating cell was always located at the blind (forward) end of the tunnel, indicating that the cell was responsible for the destruction of the fibrin matrix and that the tunnel is a static record of the cell's migration during the 24-h period before fixation. The mean length of cylindrical tunnels was 365 μm, and the mean diameter was 19.0 μm, slightly larger than a cell diameter (Figs. 1 and 3).
Figure 1.
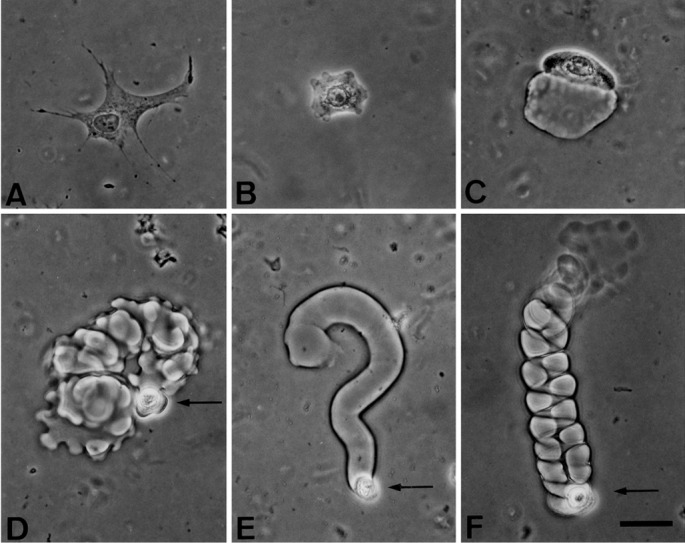
Motility and migration of human keratinocytes after inoculation onto a fibrin matrix. Cells inoculated onto fibrin matrices (on average 100 μm thick) were fed with culture medium supplemented with 10% FBS and hrEGF (10 ng ml−1). Cultures were examined and fixed 24 h later. (A) Attached and spread but nonmotile human diploid fibroblast (strain AFF11, culture XIII); (B–F) human diploid keratinocytes (strain YF29, culture V); (B) nonmotile keratinocyte; (C) keratinocyte that has penetrated into the matrix and moved a short distance; (D) highly migratory keratinocyte that has invaded the matrix; (E) cylindrical tunnel formed by a keratinocyte; (F) right-handed helical tunnel formed by a keratinocyte. Note the narrow wall of fibrin between the coils of the helix. Arrows point to single cells at the forward end of their tunnel. (Phase contrast, bar = 50 μm.)
Figure 3.

Schematic representation of a helical tunnel. (A) Left-handed helix; (B) right-handed helix. The long arrow indicates the axis of the helix: a, direction of rotation; b, helical pitch; c, diameter of the helix; and d, length of the helix. The dark circle indicates the position of the cell.
Helical Tunnels.
A more striking form of tunnel was helical (Fig. 1F and 2 A and B). As the cell advanced, it appeared to precess around the translational axis, creating a helical tunnel through the fibrin. The mean diameter of the cell track within the helix (19.8 μm) was not significantly different from that of the cylindrical tunnels (19.4 μm), but the overall diameter of the helix was 37.5 μm. Both right- and left-handed helices could be observed in different tunnels in the same gel. However, once a tunnel was begun, its helix usually retained the same handedness and the same direction of advance (Fig. 3).
Figure 2.
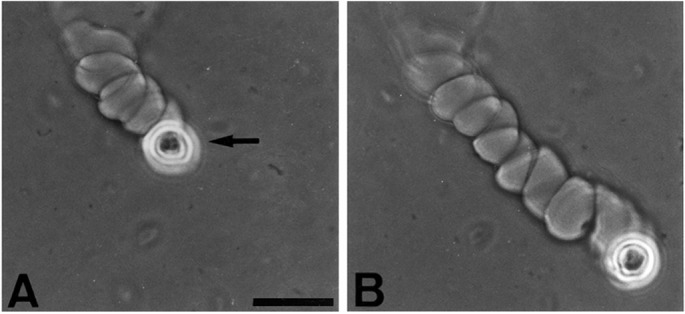
Helical migration rate of a human keratinocyte. A human keratinocyte (strain YF29, culture V) was placed on a fibrin matrix as described in Fig. 1. It was photographed 8 h after plating (A) and 5 h later (B). Note the site of invasion of the matrix and the subsequent migration along a right-handed helix, whereas the helical axis remained nearly linear. By 13 h, the cell made 11 complete rotations around the helix axis and the helical path extended at a rate of 96 μm/hr for a total of 1,243 μm. Arrow points to the cell. (Phase contrast, bar = 50 μm.)
Effect of EGF on the Type of Tunnel.
EGF is known to promote migration of keratinocytes (25), as well as fibroblasts (26). In our experiments, the presence of EGF had marked effects on keratinocyte migration and tunnel formation through the fibrin matrix. Using subconfluent cultures for inoculation, we found that the proportion of cells undergoing surface movement increased by a factor of 2-fold or less in the presence of 10 ng/ml (Table 1). The proportion of cells forming cylindrical tunnels increased somewhat more than 2-fold, but the proportion forming helical tunnels increased 6-fold to about one-quarter of all cells inoculated. A more detailed study of the effects of EGF on helical migration showed that the formation of helical tunnels increased progressively at concentrations greater than 0.1 ng/ml (Fig. 4A). The number of helical turns per tunnel also increased progressively (Fig. 4B). On the other hand, the pitch of the helices (length of tunnel divided by number of turns) decreased slightly (Fig. 4C). The overall length of the helix therefore increased by a factor slightly less than the number of turns (Fig. 4D).
Table 1.
EGF increases helical tunnel formation more strikingly than other forms of keratinocyte motility
| Presence of EGF, 10 ng/ml | Total cells | Surface cells
|
Tunnel-forming cells
|
||
|---|---|---|---|---|---|
| Motile* | Cylindrical | Helical | |||
| Expt. 1, n.c. | − | 235 | 63 (27) | 4 (1.7) | 10 (4.3) |
| + | 398 | 214 (54) | 16 (4.0) | 98 (24.6) | |
| Expt. 2, n.c. | − | 260 | 126 (49) | 4 (1.5) | 13 (5.0) |
| + | 327 | 183 (56) | 11 (3.4) | 87 (27) | |
| Expt. 3, c. | − | 204 | 28 (14) | 0 | 0 |
| + | 187 | 66 (35) | 3 (1.6) | 9 (4.8) | |
All experiments were performed on keratinocyte strain YF29. Inoculated cells: n.c., nonconfluent, rapidly growing; c., confluent, slowly growing. Expt., experiment.
Over one cell diameter in 24 h. Numbers in parentheses give percentage of total cells.
Figure 4.

Effect of EGF on number and properties of tunnels. Human keratinocytes were plated on fibrin matrix and cultivated in medium containing hrEGF at different concentrations. After 24 h, cultures were fixed, and the number of helical tunnels was counted and expressed as percent of total cells attached to the fibrin (A). The properties of the tunnels, including the number of turns (B), helical pitch (C), and the length of the track composing the helix (D), were estimated from a total of 30 helical tunnels.
The relation between the lengths of cylindrical and helical tunnels was also examined. In 24 h, the mean length of the cell track through a helical tunnel in the presence of EGF at 10 ng/ml was found to be 1,837 μm, compared with 365 μm for cylindrical tunnels, an increase of 5-fold. However, the mean rate of translational advance of the cell (overall length of the helix) was only 296 μm, or slightly less than that through cylindrical tunnels (365 μm).
Although not studied in detail, transforming growth factor (TGF)-α and the hepatocyte growth factor/scatter factor appeared able to promote tunnel formation, both cylindrical and helical. Fibroblast growth factor (FGF)-7 was slightly active, but FGF-1 and -2 had no effect on keratinocyte migration. TGF-β1 and TGF-β2 reduced somewhat the number of cylindrical tunnels but completely prevented the formation of helical tunnels. This shows that the process that moves the cell around a helical axis is more sensitive to inhibition than the translational component.
Tunnel formation, both cylindrical and helical, was a property of human keratinocytes isolated from different body sites, i.e., foreskin, axilla, groin, sole, face, thigh, and outer epithelial sheath of hair follicles (a total of 16 different strains) and from humans of different ages (from newborn to 78 years). As expected, corneal epithelial keratinocytes also formed tunnels, but melanocytes, skeletal muscle myoblasts, and vascular endothelial cell types, like fibroblasts, did not. Mammary epithelial cells, a cell type related in origin to keratinocytes, were the only nonkeratinocyte cell type found to make tunnels, both cylindrical and helical. However, these tunnels occurred less frequently and were shorter than those made by keratinocytes.
Immortalized and neoplastic keratinocytes such as GMA cells (27), SCC 9 and 13, and A431 cells had either drastically reduced or no ability to migrate and to form tunnels, even in presence of EGF.
The Axis of Helical Tunnels Is Straighter than That of Cylindrical Tunnels.
The existence of both cylindrical and helical tunnels raises the question of the significance of the two patterns of keratinocyte migration. It seemed possible that a helical tunnel enables the cell to maintain a straighter path of migration. Measurements were therefore made on the rectilinearity of the two types of tunnel.
As mentioned above, the cell in a helical tunnel advances along its track about seven times faster than a cell in a cylindrical tunnel (Table 2, column 1). The average overall length of the helical tunnels is nevertheless a little shorter than that of the cylindrical tunnels (column 2). However, when the rectilinear displacement of the cell during a 24-h period was measured, it was found to be greater when the tunnel was helical (column 3). Correspondingly, the movement that does not contribute to rectilinear displacement is reduced (column 4). The “wasted” distance traveled without contributing to rectilinear displacement, expressed as a fraction of the total length of the tunnel, is therefore 2.5 times greater for cylindrical tunnels than for helical tunnels (column 5).
Table 2.
Rectilinear displacement of a cell moving through cyclindrical and helical tunnels
| Type of tunnel | Track length, μm (column 1) | Total tunnel length, μm (column 2) | Rectilinear displacement of the cell, μm (column 3) | (column 4) column 2 minus column 3 | Curvature of tunnels, column 4 divided by column 2 |
|---|---|---|---|---|---|
| Cylindrical | 322 | 322 | 222 | 100 | 0.31 |
| Helical | 2,156 | 277 | 241 | 36 | 0.13 |
Measurements were made on photographs of 20 tunnels of each type, generated during a period of 24 h. Calculation of curvature of tunnels shows that helical tunnels are straighter than cylindrical tunnels.
The Cytoskeleton in Relation to Tunnel Formation.
Like cells in suspension culture, keratinocytes migrating through tunnels maintained their spherical shape under phase-contrast microscopy (Fig. 2 A and B). Electron microscopy showed that their keratin network was concentrated in a cap adjacent to the nucleus and that numerous microspikes were present. The cells made only occasional contacts with the fibrin matrix, and at these sites the tunnel wall appeared condensed (data not shown). In the presence of colchicine (1 μg ml−1 or cytochalasin I (1 μg ml−1), formation of helical tunnels was abolished (data not shown). This result indicates that the microtubules and microfilaments are both necessary for movement of keratinocytes through helical tunnels.
Fibrinolysis Is Essential for Tunnel Formation.
Cellular advance through tunnels was diminished by the presence of the protease inhibitor aprotinin at concentrations in excess of 5 international Kallikrein inhibiting units (KIU)/ml and completely suppressed by 500 KI units/ml (Fig. 5A). Because aprotinin is known to inhibit plasmin, we suspected that tunnel formation would depend on fibrinolysis. Tunnel formation was suppressed by removal of serum from the culture medium and restored in large part by the addition of 95% pure plasminogen. The dependence of migration on plasminogen activation was confirmed by the observation that tunnel formation in the presence of serum was eliminated by the addition of 98% pure PAI-1. Two such experiments are shown in Fig. 5B.
Figure 5.

Effect of plasmin inhibitors on keratinocyte migration. Human keratinocytes were plated on fibrin matrix and cultivated in medium containing 10% serum in the absence of EGF. After 24 h, the cultures were fixed, and the number of motile cells was counted. (A) Effect of aprotinin (Antagosan, Hoechst Pharmaceuticals). All evidence of motility disappeared at concentrations of 500 kallikrein units/ml. (B) Effect of PAI-1. All motility disappeared at a concentration of 10–15 μg/ml.
Similar experiments were performed by the addition of plasminogen and its inhibitor (Table 3). In the absence of any serum or added plasminogen, some motility and tunnel formation were detected. We attribute these to residual plasminogen in the fibrinogen preparation used to make the matrices. The addition of plasminogen increased the number of cylindrical tunnels by 4-fold and the number of helical tunnels by 7-fold. The addition of PAI-1 reduced the number of both to one-half.
Table 3.
Effect of plasminogen and PAI-1 on tunnel formation
| Total motile cells | Cylindrical tunnels | Helical tunnels | |
|---|---|---|---|
| No addition | 49 (15.4) | 11 (3.4) | 2 (0.6) |
| + plasminogen (0.5 units/ml) | 197 (71.9) | 46 (16.8) | 14 (5.1) |
| + plasminogen (0.5 units/ml) + PAI-1 (0.5 μg/ml) | 57 (20.9) | 21 (7.7) | 7 (2.6) |
Human keratinocytes were plated on fibrin matrix and cultivated in serum-free medium in the absence of EGF. Plasminogen and PAI-1 were added to the medium at the indicated concentration. After 24 hours, cultures were fixed, and the number of tunnels was counted. Numbers in parentheses are percentage of adherent cells.
These results indicate that the fibrinolysis necessary for tunnel formation by keratinocytes depends on cellular activation of plasminogen. The effects of EGF on tunnel elongation might be related to the ability of this growth factor to promote synthesis of uPAR (28).
Retention of Clonogenicity of Cells After Their Migration Through Tunnels.
When surface cultures of keratinocytes are allowed to reach confluence and become stratified, they lose colony-forming efficiency. Typically, in our experiments, the colony-forming efficiency dropped from 59 to 7%. In contrast, the migration of a keratinocyte through helical tunnels in a fibrin matrix did not impair its capacity to divide and to initiate a colony (Fig. 6). To establish that migration did not affect colony-forming efficiency, a total of 375 cells located at the ends of individual helical tracks were isolated under an inverted microscope and plated onto a feeder layer of lethally irradiated 3T3 cells (24). Thirty-five percent of the keratinocytes formed colonies. Similarly, 264 sessile keratinocytes, as shown in Fig. 1B, were isolated and cultivated; 48% of them were able to form colonies. The proportion of the colonies that aborted was the same whether the founding keratinocytes were nonmotile or motile (50 and 44%, respectively). Cells isolated from colonies initiated by nonmotile keratinocytes could invade fibrin and make tunnels. It may be concluded that motility and nonmotility are not properties of different keratinocyte populations but are reversible states of the same population.
Figure 6.
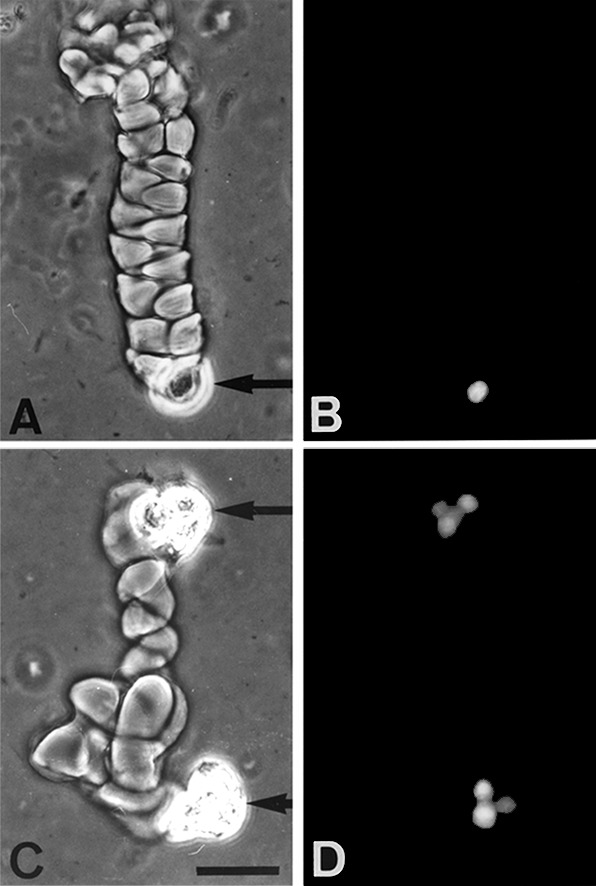
Multiplication of keratinocytes migrating through helical tunnels. Human keratinocytes (strain YF29, culture V) were plated on a fibrin gel matrix as described in Fig. 1, in the absence of EGF. After 68 h, the culture was fixed and stained with the nuclear dye Hoechst 33342. (A) A helical tunnel; arrow indicates the advancing cell; (B) the same helix after Hoechst 33342 staining. Note the presence of a single cell at the end of the tunnel. (C) Two helical tracks extending in opposite directions by presumed progeny of a single cell division in a fibrin matrix. (D) The same tracks after Hoechst 33342 staining. Note the presence of a three-cell colony at one end of the track and a four-cell colony at the other. Two consecutive divisions have occurred in each half of the helix. (Phase contrast, bar = 50 μm.)
These experiments also showed that clonogenic keratinocytes do not undergo terminal differentiation when they are maintained in a fibrin matrix as they do when suspended in medium containing methylcellulose (29, 30). Thus the interaction of the cells with fibrin or some other component of the matrix, possibly fibronectin (31), fosters the preservation of growth potential.
Discussion
The locomotion of keratinocytes in a fibrin scaffold by cylindrical or helical tunnel formation is a striking example of locomotion as a physically integrated process, coordinated both spatially and temporally (1).
Proteolysis is required for locomotion through tunnel formation as it is for surface locomotion. It has been known for a long time that fibrinolysis is linked to the migration of different cell types (32–35). Human keratinocytes produce urokinase (uPA) and its receptor (uPAR) (12, 36, 37), as well as tissue-type plasminogen activators (38). In cell culture, uPA is produced by migrating keratinocytes located at the edge of a growing keratinocyte colony (39). uPA is not found in normal epidermis but appears in epidermal outgrowths after wounding and in keratinocytes migrating under a wound clot (37, 40).
Mechanism of Cellular Advance Through a Tunnel.
The tunnel that the cell leaves behind is its kinetic track, analogous to the two-dimensional phagokinetic tracks of 3T3 cells studied earlier (17). The keratinocyte makes the tunnel by digesting the fibrin at the blind end of the tunnel while advancing into the newly created space. It is not clear how the keratinocyte adheres to the fibrin matrix of the tunnel wall while the interior of the tunnel is being dissolved. It seems possible that the cell maintains adhesion points that are protected from fibrinolysis.
A cylindrical tunnel is formed by a cell whose track does not change in a periodic manner during its advance, whereas a helical tunnel is formed by a cell whose track suggests a rotational process as the cell revolves about the axis of the helix (precession). This form of movement may be related to the so-called circus movement described long ago (41). The tunnel may be viewed as the result of a chemical/mechanical propeller that combines the release of plasminogen activator at a localized site on the advancing surface of the cell with mechanical forces that propel the cell by acting on the fibrin wall of the tunnel. These forces depend on the same cytoskeletal system as other forms of movement, namely microfilaments and microtubules. The rate of advance of the keratinocyte through a cylindrical fibrin tunnel was ≈322 μm/day, similar to the rate at which 3T3 cells migrate over plastic surfaces (≈250 μm/day) (17). However, the rate of migration of a keratinocyte through the track of a helical tunnel is about seven times greater.
The production of tunnels through a fibrin matrix by keratinocytes has disclosed that this cell type has migrational properties not generally possessed by other cell types. The significance of these properties with respect to the functions of the keratinocyte can easily be seen. The formation of helical tunnels is the most interesting, because it would seem to require that the site of release of plasminogen activator at the surface of the cell precesses around the axis of the helix. The effects of this property are: (i) a great increase in the rate of advance of the cell through the track; (ii) no increase (in fact, a slight decrease) in the total rate of extension of the tunnel; (iii) a straighter tunnel with a greater rectilinear displacement of the cell from its starting point. An orientation of the helical tunnel at the time of its formation is therefore better preserved over time than that of a cylindrical tunnel.
The ability of the mammary epithelial cell to make cylindrical and helical tunnels shows that an epithelial cell type other than the keratinocyte (although developmentally related to it) possesses the necessary machinery. However, the differences between the tunnels of these cells and those of the keratinocyte suggest the function of this form of migration may not be identical in the two cell types.
Keratinocyte Movement Requiring Fibrinolysis Is Essential in Wound Healing.
The ability of a keratinocyte to form a tunnel through a fibrin matrix permits a particular form of cell migration that may be adapted to the healing of a wound covered with fibrin. Indeed, keratinocytes move 20 times faster by tunnel formation into a fibrin matrix than when attached on native collagen I, the main matrix component of the dermis (19). That suggests that locomoting keratinocytes may preferentially use the plasmin(ogen) proteolytic system during wound closure. However, keratinocytes migrate at the interface of the collagen-rich wound bed and of the fibrin clot, and consequently they may also use the matrix metalloproteinase, especially because MT1-metalloproteinase, which acts as a pericellular fibrinolysin (10), is made by the keratinocytes located at the wound edge (42). Hence, migration of keratinocytes in a wound may result from the synergistic action of the matrix metalloproteinase and plasmin(ogen), especially because they are both up-regulated by EGF (19, 28).
Ablation of the gene for plasminogen or uPA/tissue-type plasminogen activators (tPA) in mice produces a number of abnormalities, including defects in wound healing and in keratinocyte migration (43–45). Additional ablation of the gene for fibrinogen relieves the defect in epidermal wound healing and in the migration of keratinocytes (46). When fibrinogen is available, the inability of the cells to lyse the resulting fibrin by means of uPA/tPA is therefore the cause of the defect in wound healing. Of the two PAs, uPA alone is sufficient to permit normal wound healing (44).
The gene for the cellular receptor for uPA (uPAR) is most strongly activated in keratinocytes at the front of epithelial outgrowths of skin wounds (12). That would explain why the activation of plasminogen would be confined to the vicinity of the tunnel-making cell, but absence of this receptor has not been found to produce any abnormality in wound-healing experiments (44). The confinement of the fibrolytic process to the end of the tunnel may also be explained by the observation that plasminogen is bound to the surface of the keratinocyte and is activated there to form plasmin (47). One of the unexpected features of tunnel formation by keratinocytes is that neoplastic or transformed keratinocytes have diminished or no ability to make tunnels. That is surprising because tumors have enhanced abilities to activate PA and to carry out fibrinolysis (48), which is particularly important for metastasis (49). Transformed keratinocytes may be deficient in some other aspect of the locomotion process that impedes or prevents their ability to make tunnels, possibly through the enhanced production of PAI-1 (50). However, high levels of PAI-I have been linked to enhanced metastasis and poor survival (51, 52).
Concluding Remarks.
Our observations demonstrate that a spherical keratinocyte can propel itself through fibrin by a three-dimensional locomotory mechanism other than that used for surface locomotion. Additional studies are needed to identify possible relations to cell rolling and “swimming”, the nature of the cell–fibrin contacts, and, if any, the repertoire of integrins involved (53).
Acknowledgments
We dedicate this paper to H. Green, whose comments and criticisms were invaluable. We are grateful to G. Albrecht-Buehler and E. Reich for critical reading of the manuscript. This work was supported by grants to Y.B. from Institut National de la Santé et de la Recherche Médicale (CRI 9602), the Association pour la Recherche sur le Cancer, the Ligue Nationale contre le Cancer, Mutuelle Générale de l'Education Nationale, and from the Centre Régional de Transfusion Sanguine. V.R. was supported by the Laboratoire Français du Fractionnement et des Biotechnologies.
Abbreviations
- uPA
urokinase-type plasminogen activator
- uPAR
high-affinity cell surface receptors
- PAI
plasminogen activator inhibitor
- EGF
epidermal growth factor
- hrEGF
human recombinant EGF
References
- 1.Lauffenburger D A, Horwitz A F. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 2.Gumbiner B M. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- 3.Hynes R O, Lander A D. Cell. 1992;68:303–322. doi: 10.1016/0092-8674(92)90472-o. [DOI] [PubMed] [Google Scholar]
- 4.Woods A, Couchman J R. Trends Cell Biol. 1998;8:189–192. doi: 10.1016/s0962-8924(98)01244-6. [DOI] [PubMed] [Google Scholar]
- 5.Stossel T P. Science. 1993;260:1086–1094. doi: 10.1126/science.8493552. [DOI] [PubMed] [Google Scholar]
- 6.Galbraith C G, Sheetz M P. Curr Opin Cell Biol. 1998;10:566–571. doi: 10.1016/s0955-0674(98)80030-6. [DOI] [PubMed] [Google Scholar]
- 7.Lee J, Ishihara A, Jacobson K. Trends Cell Biol. 1993;3:366–370. doi: 10.1016/0962-8924(93)90084-e. [DOI] [PubMed] [Google Scholar]
- 8.Murphy G, Gavrilovic J. Curr Opin Cell Biol. 1999;11:614–621. doi: 10.1016/s0955-0674(99)00022-8. [DOI] [PubMed] [Google Scholar]
- 9.Blasi F. BioEssays. 1993;15:105–111. doi: 10.1002/bies.950150206. [DOI] [PubMed] [Google Scholar]
- 10.Hiraoka N, Allen E, Apel I J, Gyetko M R, Weiss S J. Cell. 1998;95:365–377. doi: 10.1016/s0092-8674(00)81768-7. [DOI] [PubMed] [Google Scholar]
- 11.Chapman H A. Curr Opin Cell Biol. 1997;9:714–724. doi: 10.1016/s0955-0674(97)80126-3. [DOI] [PubMed] [Google Scholar]
- 12.Rømer J, Lund L R, Eriksen J, Pyke C, Kristensen P, Danø K. J Invest Dermatol. 1994;102:519–522. doi: 10.1111/1523-1747.ep12373187. [DOI] [PubMed] [Google Scholar]
- 13.Wei Y, Waltz D A, Rao N, Drummond R J, Rosenberg S, Chapman H A. J Biol Chem. 1994;269:32380–32388. [PubMed] [Google Scholar]
- 14.Bray D. In: Cell Movements. Bray D, editor. New York: Garland; 1992. pp. 3–72. [Google Scholar]
- 15.Anderson K I, Wang Y-L, Small J V. J Cell Biol. 1996;134:1209–1218. doi: 10.1083/jcb.134.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oliver T, Dembo M, Jacobson K. J Cell Biol. 1999;145:589–604. doi: 10.1083/jcb.145.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Albrecht-Buehler G. Cell. 1977;11:395–404. doi: 10.1016/0092-8674(77)90057-5. [DOI] [PubMed] [Google Scholar]
- 18.Sarret Y, Woodley D T, Grigsby K, Wynn K, O'Keefe E J. J Invest Dermatol. 1992;98:12–16. doi: 10.1111/1523-1747.ep12493517. [DOI] [PubMed] [Google Scholar]
- 19.Pilcher B K, Dumin J A, Sudbeck B D, Krane S M, Welgus H G, Parks W C. J Cell Biol. 1997;137:1445–1457. doi: 10.1083/jcb.137.6.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S, Springer T A. J Cell Biol. 1999;144:185–200. doi: 10.1083/jcb.144.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ronfard V, Rives J-M, Neveux Y, Carsin H, Barrandon Y. Transplantation. 2000;70:1588–1598. doi: 10.1097/00007890-200012150-00009. [DOI] [PubMed] [Google Scholar]
- 22.Burnouf-Radosevich M, Burnouf T, Huart J J. Vox Sang. 1990;58:77–84. doi: 10.1111/j.1423-0410.1990.tb02066.x. [DOI] [PubMed] [Google Scholar]
- 23.Rochat A, Kobayashi K, Barrandon Y. Cell. 1994;76:1063–1073. doi: 10.1016/0092-8674(94)90383-2. [DOI] [PubMed] [Google Scholar]
- 24.Barrandon Y, Green H. Proc Natl Acad Sci USA. 1987;84:2302–2306. doi: 10.1073/pnas.84.8.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrandon Y, Green H. Cell. 1987;50:1131–1137. doi: 10.1016/0092-8674(87)90179-6. [DOI] [PubMed] [Google Scholar]
- 26.Ware M F, Wells A, Lauffenburger D A. J Cell Sci. 1998;111:2423–2432. doi: 10.1242/jcs.111.16.2423. [DOI] [PubMed] [Google Scholar]
- 27.Barrandon Y, Morgan J R, Mulligan R C, Green H. Proc Natl Acad Sci USA. 1989;86:4102–4106. doi: 10.1073/pnas.86.11.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lund L R, Rømer J, Rønne E, Ellis V, Blasi F, Danø K. EMBO J. 1991;10:3399–3407. doi: 10.1002/j.1460-2075.1991.tb04904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green H. Cell. 1977;11:405–416. doi: 10.1016/0092-8674(77)90058-7. [DOI] [PubMed] [Google Scholar]
- 30.Hotchin N A, Gandarillas A, Watt F M. J Cell Biol. 1995;128:1209–1219. doi: 10.1083/jcb.128.6.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams J C, Watt F M. Nature (London) 1989;340:307–309. doi: 10.1038/340307a0. [DOI] [PubMed] [Google Scholar]
- 32.Ossowski L, Quigley J P, Reich E. In: Proteases and Biological Control. Reich E, Rifkin D B, Shaw E, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1975. pp. 901–913. [Google Scholar]
- 33.Strickland S, Reich E. Cell. 1976;9:231–240. doi: 10.1016/0092-8674(76)90114-8. [DOI] [PubMed] [Google Scholar]
- 34.Bode V C, Dziadek M A. Dev Biol. 1979;73:272–289. doi: 10.1016/0012-1606(79)90067-8. [DOI] [PubMed] [Google Scholar]
- 35.Valinsky J E, Reich E, Le Douarin N M. Cell. 1981;25:471–476. doi: 10.1016/0092-8674(81)90065-9. [DOI] [PubMed] [Google Scholar]
- 36.Morioka S, Lazarus G S, Baird J L, Jensen P J. J Invest Dermatol. 1987;88:418–423. doi: 10.1111/1523-1747.ep12469754. [DOI] [PubMed] [Google Scholar]
- 37.Rømer J, Lund L R, Eriksen J, Ralfkiaer E, Zeheb R, Gelehrter T D, Dano K, Kristensen P. J Invest Dermatol. 1991;97:803–811. doi: 10.1111/1523-1747.ep12486833. [DOI] [PubMed] [Google Scholar]
- 38.Chen C-S, Lyons-Giordano B, Lazarus G S, Jensen P J. J Cell Sci. 1993;106:45–53. doi: 10.1242/jcs.106.1.45. [DOI] [PubMed] [Google Scholar]
- 39.Jensen P J, John M, Baird J. Exp Cell Res. 1990;187:162–169. doi: 10.1016/0014-4827(90)90131-s. [DOI] [PubMed] [Google Scholar]
- 40.Grøndahl-Hansen J, Lund L R, Ralfkiaer E, Ottevanger V, Danø K. J Invest Dermatol. 1988;90:790–795. doi: 10.1111/1523-1747.ep12461511. [DOI] [PubMed] [Google Scholar]
- 41.Holtfreter J. J Morphol. 1946;79:27–62. doi: 10.1002/jmor.1050790103. [DOI] [PubMed] [Google Scholar]
- 42.Saarialho-Kere U K, Kovacs S O, Pentland A P, Olerud J E, Welgus H G, Parks W C. J Clin Invest. 1993;92:2858–2866. doi: 10.1172/JCI116906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord J J, Collen D, Mulligan R C. Nature (London) 1994;368:419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- 44.Bugge T H, Flick M J, Danton M J S, Daugherty C C, Rømer J, Danø K, Carmeliet P, Collen D, Degen J L. Proc Natl Acad Sci USA. 1996;93:5899–5904. doi: 10.1073/pnas.93.12.5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rømer J, Bugge T H, Pyke C, Lund L R, Flick M J, Degen J L, Danø K. Nat Med. 1996;2:287–292. doi: 10.1038/nm0396-287. [DOI] [PubMed] [Google Scholar]
- 46.Bugge T H, Kombrinck K W, Flick M J, Daugherty C C, Danton M J S, Degen J L. Cell. 1996;87:709–719. doi: 10.1016/s0092-8674(00)81390-2. [DOI] [PubMed] [Google Scholar]
- 47.Reinartz J, Batrla R, Boukamp P, Fusenig N, Kramer M D. Exp Cell Res. 1993;208:197–208. doi: 10.1006/excr.1993.1238. [DOI] [PubMed] [Google Scholar]
- 48.Unkeless J, Danø K, Kellerman G M, Reich E. J Biol Chem. 1974;10:4295–4305. [PubMed] [Google Scholar]
- 49.Ossowski L, Reich E. Cell. 1983;35:611–619. doi: 10.1016/0092-8674(83)90093-4. [DOI] [PubMed] [Google Scholar]
- 50.Deng G, Curriden S A, Wang S, Rosenberg S, Loskutoff D J. J Cell Biol. 1996;134:1563–1571. doi: 10.1083/jcb.134.6.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bajou K, Noël A, Gerard R D, Masson V, Brunner N, Holst-Hansen C, Skobe M, Fusenig N E, Carmeliet P, Collen D, Foidart J M. Nat Med. 1998;4:923–928. doi: 10.1038/nm0898-923. [DOI] [PubMed] [Google Scholar]
- 52.Johnsen M, Lund L R, Rømer J, Almholt K, Danø K. Curr Opin Cell Biol. 1998;10:667–671. doi: 10.1016/s0955-0674(98)80044-6. [DOI] [PubMed] [Google Scholar]
- 53.Palecek S P, Loftus J C, Ginsberg M H, Lauffenburger D A, Horwitz A F. Nature (London) 1997;385:537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]