

Abstract
Dps (DNA protection during starvation) enzymes are a major class of dodecameric proteins that bacteria use to detoxify their cytosol through the uptake of reactive iron species. In the stationary growth phase of bacteria, Dps enzymes are primarily used to protect DNA by biocrystallization. To characterize the wild type Dps protein from Microbacterium arborescens that displays additional catalytic functions (amide hydrolysis and synthesis), we determined the crystal structure to a resolution of 2.05 Å at low iron content. The structure shows a single iron at the ferroxidase center coordinated by an oxo atom, one water molecule, and three ligating residues. An iron-enriched protein structure was obtained at 2 Å and shows the stepwise uptake of two hexahydrated iron atoms moving along channels at the 3-fold axis before a restriction site inside the channels requires removal of the hydration sphere. Supporting biochemical data provide insight into the regulation of this acylamino acid hydrolase. Moreover, the peroxidase activity of the protein was determined. The influence of iron and siderophores on the expression of acylamino acid hydrolase was monitored during several stages of cell growth. Altogether our data provide an interesting view of an unusual Dps-like enzyme evolutionarily located apart from the large Dps sequence clusters.
Keywords: Crystal Structure, Crystallography, DNA-binding Protein, Enzyme Purification, Iron, Iron Metabolism, Metalloproteins, Multifunctional Protein
Introduction
Iron is an essential element for cells as the cofactor in numerous enzymes. The element plays a fundamental role in cell respiration, for example as a component of cytochromes and iron-sulfur proteins. In addition, many processes such as photosynthesis, nitrogen fixation, and methanogenesis are strictly iron-dependent (1). Remarkably, iron of the two redox states differs in solubility: the reduced ferrous Fe2+ form is soluble at 10−1 m, whereas the oxidized ferric Fe3+ species is highly insoluble at physiological pH.
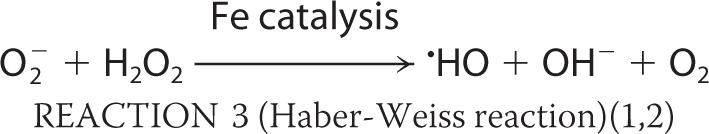
Bacteria regulate iron homeostasis using iron-sensing repressors such as Fur in Escherichia coli (1–3). Fur mainly regulates the synthesis and transport of siderophores, low molecular weight compounds with a high affinity for Fe3+ that are secreted into the external medium (1). The poor bioavailability of iron often limits bacterial growth (1). During respiration, reactive oxygen species (ROS)3 such as hyperoxide anions (O2−) and hydrogen peroxide (H2O2) are formed as byproducts. O2− can destroy the iron-sulfur clusters of many important enzymes; this in turn causes the release of Fe2+ (see Reaction 1 below). Intracellular free Fe2+ from these sources can then react with H2O2 to produce ROS; the most important features of ROS are their hydroxyl radicals, which react with unsaturated lipids, protein side chains, and most destructively DNA (Reaction 2). The Haber-Weiss reaction is able to take place (Reaction 3) as is the catalyzed reduction of H2O2 (e.g. by Dps enzymes; Reaction 4).
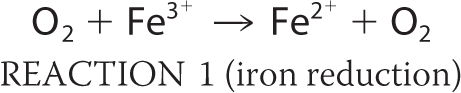 |
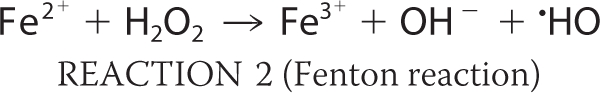 |
 |
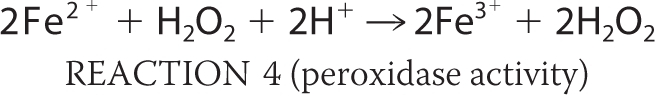 |
Bacteria are protected against H2O2 and ROS by different enzymes such as superoxide dismutases, superoxide reductases, catalases, and Dps proteins (DNA protection during starvation) (4). Dps proteins belong to the superfamily of the ferritin fold but, unlike the 24-mer ferritins, in prokaryotes they occur only as 12-meric complexes with a slightly variable architecture (5). They are active in Fe2+ uptake, oxidation, storage, and H2O2 destruction (1, 6, 7) and also provide physical protection by binding DNA and forming ordered material as shown for E. coli (8, 9), Bacillus cereus (10), and the marine cyanobacterium Trichodesmium erythraeum (11). DNA binding, another function of Dps, is important in the stationary growth phase when Dps is the most abundant protein in E. coli cells (12).
The regulation of Dps proteins is manyfold: most Dps enzymes are induced under stress conditions such as hyperosmotic stress (13–15), temperature shocks (13, 15, 16), oxidative stress (e.g. H2O2) (17–20), nutrient starvation (13, 15, 21), and the presence of ethanol (15, 16). Although Dps proteins are not essential for cell viability under normal growth conditions, it was shown that Dps deletion mutants of some bacteria are susceptible to oxidative stress (22–24), acids, bases, or UV radiation (25). Dps proteins can be induced by increasing iron concentrations in the medium (18) and may be up-regulated during iron starvation (19, 26).
Twenty-eight structures of Dps enzymes have been solved by x-ray crystallography to date, all of which express the same dodecameric quaternary architecture (status of Protein Data Bank as of May 2011). Monomeric Dps proteins express essentially the same fold as ferritin comprising a four-helix bundle (4). Although the 24-mer ferritin superfamily of proteins shows 432-point symmetry, all Dps dodecamers express tetrahedral 23-point symmetry (27). The 23-point symmetry is accompanied by three 2-fold and four 3-fold symmetry axes leading to a high internal symmetry.
Iron storage of Fe2+ through biomineralization as insoluble Fe2O3 in Dps enzymes requires three steps. 1) Iron uptake proceeds via pores embedded in the protein shell. 2) Twelve ferroxidase centers (FOCs) catalyze the oxidation of Fe2+ to Fe3+ by oxygen species. 3) Iron storage as insoluble Fe3+ oxide begins at nucleation centers, which are distributed in the inner sphere of the protein cage (27). Different uptake routes occur for distinct enzymatic species. Dps proteins from E. coli and Bacillus subtilis are proposed to take up iron through four pores located along the 3-fold axis of the complex (28, 29). In contrast, in the DpsA from Halobacterium salinarum, uptake is regulated via 12 channels guiding iron straight to the FOCs (27). Notably, these centers in addition to the quaternary structure are the only conserved fingerprints of Dps sequences.
The Gram-negative bacterium Microbacterium arborescens was isolated from the gut of Spodoptera exigua. Microorganisms living in the midgut have to compete with ROS and antimicrobial peptides (31). Some Dps proteins are known to act as peroxidases and detoxify radicals (6). M. arborescens uses the protein amino acid hydrolase (AAH), a Dps homolog, to both hydrolyze and synthesize N-acylamino acids (31). This class of amino acid conjugates with fatty acids plays an important role in the ability of plants to recognize herbivory (32) and induces the emission of volatile organic compounds (33). Moreover, depending on the iron content of the medium, the Dps protein from M. arborescens displays different amounts of stored iron oxides as determined by inductively coupled plasma optical emission spectrometry (31).
To structurally and functionally characterize Dps proteins from M. arborescens, we set out to crystallize the wild type enzyme and also determined its structure at increased iron concentrations. For the first time, methods from structural biology were used to analyze the iron uptake pathway of this type of Dps enzyme. Our studies reveal iron-hexaquo clusters along the 3-fold axis of the complex and prove this uptake pathway by high resolution x-ray crystallography. Moreover, in this work the activity of AAH as a peroxidase was assayed, and kinetic data were determined. The regulation of the protein through small molecules was monitored.
EXPERIMENTAL PROCEDURES
Western Blot Analysis
M. arborescens strain Se14 was cultivated in BHI medium (Roth). Cells were grown aerobically in a shaker at 200 rpm at 37 °C. To investigate the effect of free iron and iron in small molecule complexes on AAH expression, one of the following compounds was added to the growth medium: FeSO4·7H2O, ferrioxamine B (as desferrioxamine mesylate salt; Sigma), ferrioxamine E (Fluka), bathophenanthroline disulfonic acid (Sigma), 2,2′-dipyridyl (Fisher-Scientific), hydrogen peroxide (Fluka), sodium chloride (Roth), and sorbitol (Fluka). The concentrations used for the experiments are given under “Results.” For the short term experiments, the cultures were grown in BHI medium until exponential growth (7 h) followed by treatment with one of the above mentioned compounds. For the long term experiments, overnight cultures were diluted 1:1000, and samples were taken at distinct time points between 5 and 24 h. For each sample, the A600 was measured, and cells were harvested by centrifugation and resuspended in SDS loading buffer so that samples contained a similar cell number. The samples were separated by electrophoresis using a 12% SDS-polyacrylamide gel and transferred onto a PVDF membrane (Roth) using a semidry transfer unit (Hoefer). The membrane was probed with polyclonal antibodies against purified AAH proteins (Seqlab). The ECL system was used to identify the AAH protein. Emitted light was detected by exposing the membrane to x-ray film (AGFA).
Protein Purification
The purification of AAH was described in an earlier study (31). In brief, M. arborescens strain Se14 was grown in BHI medium for 18 h. Cells were harvested by centrifugation and disrupted by ultrasonication in 50 mm Tris-buffer (pH 8). A fractionated ammonium sulfate precipitation was performed with the clear solution: the pellet with 25% saturation was discarded, and the pellet with 65% saturation was dissolved in buffer and desalted using a 5-ml HiTrap desalting column (GE Healthcare). The volume of the extract was reduced by spinning the solution through a VivaSpin concentrator (molecular mass cutoff, 100 kDa; Viva Science), and the remaining solution was loaded onto a 6-ml Resource Q anion exchange column (GE Healthcare). 50 mm Tris-HCl (pH 8) with 1 m NaCl was used for gradient elution of the protein. The final removal of small contaminants was performed by size exclusion chromatography over a Superdex 200 column (GE Healthcare). The activity of the samples was controlled during this procedure using the conjugation activity assay described in Ping et al. (31). Only active fractions were used for further experiments.
Agarose Gel Electrophoresis of DNA
AAH was added to 380 ng of plasmid DNA (pET28a), yielding a final concentration of 6.5 μm, and incubated for 10 min at room temperature. Afterward, 14.3 μm FeSO4 and 8.6% hydrogen peroxide were added and incubated for 10 min. Samples were applied to and run on a 1% agarose gel. Gels were stained with ethidium bromide and scanned by a BioDoc gel analyzer.
Determination of Kinetic Constants of Peroxidase Activity
The peroxidase activity of AAH from M. arborescens was determined by measuring the oxidation of the substrate ortho-phenylenediamine through H2O2 in the presence of the enzyme. ortho-Phenylenediamine was dissolved in buffer (50 mm Tris-HCl (pH 8)) to a concentration of 9.25 × 10−2 m. Ten different concentrations between 0.925 × 10−2 and 9.25 × 10−2 m were created by diluting this stock with the same buffer. 100 μl of every substrate dilution were added to a flat bottom microtiter plate. The purified enzyme was supplemented to final concentrations of 2.1 × 10−8–1.6 × 10−7 m, and 10 μl of 30% H2O2 were added per well. After 15 min of incubation with shaking at 23 °C in the dark, the reaction was stopped by the addition of 100 μl 0.5 m H2SO4, and absorption at 490 nm was measured in a UV/visible plate reader spectrometer (Spectra Max). Data were collected using the software Softmax Pro 2.2.1. The kinetic constants (Km and kcat) for this substrate were estimated by nonlinear regression using least square fit (GraphPad Prism 5 for Windows software). The calculated value represents a mean of 12 biological replicates with three to four technical repeats. The standard error values were calculated and are given in the plots.
Crystallization and Data Collection
An initial screen against 1400 different crystallization conditions was performed using the sitting drop method. Drops were prepared by mixing 400 + 400 nl (protein + reservoir solution) using 96-well plates (Corning) and the Honeybee 961 nanodrop robot system (Genomic Solutions). Drops were screened for crystallization events with an imaging system (Formulatrix, Waltham). Initial crystals from the full screen diffracting to 3.5 Å were obtained in space group P212121 with four dodecamers of the enzyme in the asymmetric unit. These crystals were further refined using the additive screen from Hampton Research at 18 °C by the hanging drop vapor diffusion method against 0.5 ml of the reservoir solution using chemicals that were all from Fluka. Crystal drops were prepared by mixing 1 μl of protein at 11 mg/ml concentration (in 20 mm Tris-HCl, 50 mm NaCl (pH 8)) with 1 μl of reservoir solution and 0.3 μl of additive. Another crystal form was thereby obtained under the same conditions after 30 days using additives such as spermine, maltose, etc. with a size of 150 × 100 × 100 μm. Single crystals were flash frozen in their mother liquor containing 10% PEG 400, and data collection was performed at 100 K. The crystal system is monoclinic P21 with cell constants of a = 87.55 Å, b = 91.93 Å, c = 128.47 Å, and β = 96.1° (for details, see Table 1). This crystal form contained only one dodecamer in the asymmetric unit and diffracted to a resolution limit of 2.05 Å with a solvent content of 53%. A high resolution data set was collected at beamline PXII, Swiss Light Source at an attenuation factor of 70%. Data were recorded on a MarCCD225 image plate system and 200 × 1° frames were collected (1-s exposure), integrated, and scaled with the XDS program package (34). Complexes of the iron-enriched enzyme were prepared using crystals from the same drop as for the wild type enzyme. A 10 mm iron solution was slowly added (over 30 s) to yield a final concentration of ∼1 mm. Crystals were soaked in this solution for another 30–300 s and flash frozen according to the procedure mentioned above.
TABLE 1.
Data collection and refinement statistics
Values in parentheses are for the highest resolution shell. r.m.s., root mean square.
| AAHL | AAHH | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 1.0 | 1.0 |
| Space group | P21 | P21 |
| Cell dimensions | ||
| a, b, c (Å) | 87.53, 91.92, 128.45 | 87.58, 92.05, 128.58 |
| α, β, γ (°) | 90, 96.10, 90 | 90, 96.04, 90 |
| Resolution (Å) | 30-2.05 (2.10-2.05) | 30-2.0 (2.05-2.00) |
| Rsym or Rmerge | 0.08 (0.79) | 0.10 (0.88) |
| I/σI | 7.3 (1.8) | 7.3 (1.50) |
| Completeness (%) | 99.4 (97.9) | 98.0 (96.8) |
| Redundancy | 3.5 (3.3) | 2.05 (2.0) |
| Refinement | ||
| Resolution (Å) | 30-2.05 (2.10-2.05) | 30-2.0 (2.05-2.00) |
| No. reflections | 120,300 | 129,605 |
| Rwork/Rfree | 0.19/0.23 | 0.19/0.23 |
| No. atoms | ||
| Protein | 13,600 | 13,769 |
| Ligand/ion (Fe2+) | 12 | 17 |
| Water | 920 | 707 |
| B-factors | ||
| Protein | 18.7 | 28.0 |
| Ligand/ion | 22.0 | 43.6 |
| Water | 40.5 | 38.7 |
| r.m.s. deviations | ||
| Bond lengths (Å) | 0.026 | 0.018 |
| Bond angles (°) | 1.68 | 1.26 |
| Ramachandran statistics | ||
| No. residues in favored region | 1,734 (97.9%) | 1,764 (98.3%) |
| No. residues in allowed region | 30 (1.2%) | 22 (1.2%) |
| No. residues in outlier region | 12 (0.6%) | 9 (0.5%) |
| Crystallization conditions | 18% PEG 8000, 2% isopropanol, 0.1 m sodium acetate (pH 7.5), 10 mm spermine | 18% PEG 8000, 2% isopropanol, 0.1 m sodium acetate (pH 7.5), 10 mm spermine |
| PDB code | 2YJJ | 2YJK |
Structure Determination and Refinement
The structure of the Dps enzyme was solved by molecular replacement using the Protein Data Bank coordinates of the dodecameric Dps from Agrobacterium tumefaciens as the basis (Protein Data Bank code 1O9R). The dodecameric protein model was placed using the program MOLREP (35) and initially refined in REFMAC (36). The Phenix program suite (37) was used for one round of automatic model rebuilding, which was continued by iterative model building and refinement using the program package Coot (38) and REFMAC (39). A random set of 7% of the data was omitted during the refinement process and marked as a test set for cross-validation. A crystallographic R/Rfree factor for all atoms was determined to be 0.19/0.23 including the entire protein model, water, and iron atoms (see Table 1). Model superposition was performed by the programs top3d or LSQ included in the CCP4 program package (40). Secondary structure elements were defined according to DSSP (52) algorithm. All structure figures were prepared using the program PyMOL (53). Ramachandran statistics were calculated using the Rampage server.
RESULTS
Sequence Cluster Analysis of AAH/Dps-like Sequences
To monitor the sequence variation of Dps proteins, we used the CLANS program to classify pairwise connections between Dps sequences selected through PSI-BLAST searches (41, 42). Protein sequences related to Dps-like AAH proteins were identified and submitted to clustering according to their similarity using the pairwise p values as artificial attractive forces. The CLANS map shown in Fig. 1 divides Dps sequences into several loosely related groups. Most of these groups contain a large number of similar sequences (e.g. Actinobacteria, γ-Proteobacteria, and Bacilli), whereas the closely AAH-related proteins form a smaller subcluster of only seven protein sequences. Dps proteins most similar to AAH are from the Gram-positive bacteria Clavibacter michiganensis, Leifsonia xyli, and Tropheryma whipplei. These strains are implicated in plant (C. michiganensis and L. xyli) and gastrointestinal (T. whipplei) diseases (43). An alignment of sequences derived from this small subcluster indicates a conservation of residues forming the iron uptake channel along the 3-fold axis as well as residues implicated in FOC formation. Interestingly, the C-terminal residues of the sequences are also highly conserved (supplemental Figs. S1 and S2).
FIGURE 1.

Sequence cluster map of Dps proteins. Dps-like sequences were obtained by a CS-BLAST search, and sequences were clustered according to their sequence similarities in the program CLANS. Connecting lines representing higher similarity are black, whereas sequences connected by gray lines show lower sequence similarity. Five large clusters were identified comprising Dps molecules from Bacilli, Enterococci/Streptococci, γ-Proteobacteria, Actinobacteria I, and Actinobacteria II. There are three subclusters diverging from Actinobacteria II: Mobiluncus species, Methylobacterium/Deinococcus, and a cluster comprising Microbacterium/Clavibacter/Tropheryma.
Crystallization and Specific Features of AAH Structures
AAH was purified from M. arborescens cells as described (31). The AAH initial crystals diffracted to 3.5-Å resolution and four dodecamers (48 monomers) in the asymmetric unit (data are not shown). Using small molecule additives on the basis of these conditions led to a transformation into another morphology resulting in crystals of higher quality. The crystal structure of low iron AAH (AAHL) was obtained in space group P21 with a resolution of 2.05 Å and one dodecamer in the asymmetric unit. The same type of crystal was applied in the iron uptake studies; here crystals were soaked in a Fe2+ solution for 30–300 s, and subsequently a data set to 2-Å resolution was collected (AAHH; for details see Table 1). The AAHL structure was solved by molecular replacement based on the coordinates of the dodecameric Dps from A. tumefaciens that shows a sequence identity of 37%. The structurally closest homolog of AAHL/H is the Dps protein from Deinococcus radiodurans with a root mean square deviation of 1.4 Å (for 147 aligned Cα positions). This protein is part of the second small sequence cluster described in the previous paragraph (Fig. 1).
All protein chains except one lack N- and C-terminal residues due to flexibility. Iron atoms in the AAHL structure reflect the endogenous content and were identified through their anomalous signal and placed accordingly. The occupancy chosen for the 12 atoms (one iron per monomer localized at the FOC) was estimated to be 0.5, yielding B-factors during the positional refinement that reflected the temperature factors of the surrounding atoms. Iron-oxo groups and water molecules in the vicinity of every iron were placed according to the distance between iron and peaks occurring in difference maps. Additional iron positions in the iron-enriched AAHH structure were identified by the anomalous contribution of newly appearing atoms. Occupancies at FOCs significantly increased to ∼1, whereas additional positions located in the uptake channels along the 3-fold axis were modeled with an occupancy of ∼0.2.
Interface Formation in AAHL and Conservation of Residues
The structure of the monomeric AAHL enzyme shows the typical ferritin-like fold of a four-helix bundle with the α3 helix oriented perpendicular as a connecting element between helix α2 and α4 (Fig. 2A). Each AAHL/H subunit forms close contacts to five adjacent subunits. The largest protein interface is formed between the two monomers, which are related by 2-fold symmetry and covers approximately 18% of the protein surface (Fig. 2B). The dimer interface, which is formed by 10 hydrogen bonds and four salt bridges, is the most significant building block of the complex. Four additional surface areas connect each monomer to adjacent protein subunits, covering an additional ∼30% of the monomer surface. Together these interfaces contribute to approximately 50% of the total accessible surface, explaining the high stability of proteins belonging to this class. A small area remains uncovered on the inner wall of the protein complex and is thereby not involved in protein-protein contacts (Fig. 2B).
FIGURE 2.

Structure of dimeric AAH protein. A, ribbon model of a dimer subunit of AAH from M. arborescence. The two subunits are color-coded in orange and blue. The pictures on the right and left are related by a rotation of the subunit by 180° around the y axis. The protein consists of five helices, which are marked by numbers (α1–α5) as well as termini (N terminus (NT) and C terminus (CT)). B, ribbon model and surface representation of a dimer subunit of AAH shown along a 2-fold axis. One monomer is represented as a ribbon model; the second is represented as a surface representation. The two views represent the inner and outer views of the dimer. The interfaces between the surface-encoded monomer and five adjacent proteins are marked with colors (orange, dark green, light green, yellow, and magenta). Numbers represent the interface between the individual subunits in Å2. C, surface representation of one monomer and the symmetry-related monomer with conserved residues extracted from a multiple alignment of AAH against all Dps-like proteins marked in blue. Two tryptophan residues (Trp-44 and Trp-153) and four residues involved in the FOC formation are marked by red and green numbers.
BLAST analysis of conserved and surface-exposed residues displayed on this surface led to the identification of six highly and a few less strongly conserved residues, all of which face the dodecameric inner sphere FOCs or protein-protein interfaces (Fig. 2C). Among those, the two tryptophan residues Trp-44 and Trp-153 of the sequence form a part of the hydrophobic surroundings of the residues building the structural basis of FOCs. The three residue patches (two FOCs and Trp-153) are surrounded by residues of lower conservation, whereas residues facing the outer wall of the complex are not significantly conserved. Ferroxidase and peroxidase activity are the archetypical activities of all Dps enzymes examined to date, whereas DNA binding has not been demonstrated for AAH (31).
Influence of Protein Charges
The AAHL described here was isolated under bacterial wild type growth conditions. The iron presaturation of the enzyme was expected from both the yellow color of the protein sample and the data obtained from mass spectrometry. One possible force driving iron uptake is the difference in charge distribution between the outer and inner walls. The AAH monomer carries a total of 23 negative charges (15 Asp and eight Glu residues) and nine positive charges. The negative residues are located at the outer surface (nine per monomer): the iron uptake channels (four per monomer; 12 for the entire channel) and the inner walling including the FOCs (10 per monomer). In total, this distribution together with the positively charged residues (six outside and three inside) results in an overall excess of negative charges inside the cage interior of approximately −4 per subunit (−48 for the entire complex).
Dodecameric Dps proteins are structurally related enzymes that assemble into temperature-stable protein shells via the large interfaces mentioned above. However, the cavity volume of the enzymes can vary by 10–15% between different specimens; e.g. the volume estimated for AAH is 55,000 Å3, whereas the volume estimated for the E. coli or DpsA from H. salinarum homolog is 61,000 Å3. These volumes allow for the uptake of approximately 500 Fe2O3 units. In former studies, we observed a variable iron load of the protein and determined contents between 11 and 197 iron atoms (31).
Iron Distribution in Low and High Iron Structures
In the AAHL structure, 12 iron atoms were identified at the FOCs using anomalous difference map peaks. The coordination geometry is pseudohexameric through interactions with His-43, Asp-70 (OD1 and OD2), Glu-74, a water molecule at a distance of ∼2.8 Å, and a μ-oxo group at a distance of 2.1 Å (Fig. 3A). However, heterogeneity within this arrangement was observed among the 12 subunits of the asymmetric unit presumably due to a difference in iron occupancies. Interestingly, the distance between the entrance provided by the pore into the protein shell and the closest FOC is ∼2 nm.
FIGURE 3.

Structure of FOC and distribution of FOCs in dodecameric complex. A, structure of the FOC with the iron atom in brown and the ligating molecules (water (W) and the oxo atom (oxo)) in blue (bond lengths are given in Å). The iron atom has a pseudohexameric coordination sphere with two residues contributing three bonds (Asp-70 and Glu-74) from one subunit (in brown) and a third residue (His-43) located on the second subunit (marked in blue). B, dodecameric arrangement of AAH with all subunits color-coded by different colors. Small brown dots represent the positions of iron atoms bound in the AAHL structure, all of which are bound at the FOCs. C, schematic representation of the 12 iron atoms localized at FOCs and distances given in nm between these atoms. Distances between the entry of iron into the inner shell and the three nearest neighboring FOCs are marked.
During the initial stages of uptake, hydrated Fe2+ atoms become attracted by the highest negative charge density accumulation of −6 (three pairs of Asp-128 and Glu-129 residues related by threefold symmetry) exposed to the outermost sphere of the AAAL/H complex. This structural feature appears four times on the AAH surface surrounding the 3-fold axis of the protein.
Iron uptake pathways of Dps enzymes have scarcely been discovered, and so far only H. salinarum DpsA protein has shown progressive iron uptake via a ladder of steps. In AAH, Asp-131, Glu-132, Gln-138, and Asp-139 from each subunit are among the AAHH residues that line the four uptake channels. We observed two iron positions in the vicinity of the 3-fold axis. To our surprise, both iron atoms showed a clear hydration shell with the presence of six water molecules; this was unexpected as the channel diameter along the 3-fold axis is only slightly wider, that is 0.7–0.9 nm (Fig. 4, C–F). The geometry of the hexahydrated Fe2+ complex relative to the 3-fold axis was unchanged for both clusters. Notably, although the outermost iron atom (Fig. 4E, Fe1) is observed in all four uptake channels, the second iron located inside the channels (Fig. 4E, Fe2) occurs only in one of four channel entries. It is reasonable to assume that the passage of hydrated iron stays stable until the channel diameter is narrowed to 0.4 nm. The outermost iron-water cluster is coordinated by residue Asp-131 via side chain water interactions and further transferred to Gln-138. The distance between the two iron atoms was determined to be ∼5 Å, which reflects the approximate diameter of a hydrated iron atom (Fig. 4, C and D). Finally, the iron atom is translated further through a narrowing in the channel, and the water is stripped off, whereas the naked atom is likely to be coordinated by Asp-139 residues (Fig. 4, C and D).
FIGURE 4.

Electrostatic distribution of AAH dodecamer and iron cluster uptake. A, surface representation of the dodecamer with negatively charged residues marked in red and positively charged residues marked in blue. The view is of the channel, which runs along the 3-fold axis and shows a significant surplus of surrounding negative charges. B, view into the dodecameric shell from the opposite side relative to A with the pathway of iron along negative charged residues marked in yellow. In A and B, the 3-fold axis is marked by triangles. C, cross-section of the ion channel running along the 3-fold axis. Residues lining this channel are depicted in stick representation. Two iron-water clusters are shown with iron atoms in brown and water molecules in blue. A third, hypothetical iron binding site is presented in dark brown. Vertical distances between residues lining the channel are given in nm. On the right-hand side, an overview of the channel is presented emphasizing the distances between the individual iron-water clusters, the distances between the hypothetical iron atom within the protein shell, and the distance of 1.9 nm to the closest FOC. D, schematic view of the translocation process. The lateral dimension of the iron-water cluster further along the pore decreases, leading to the removal of water molecules at the entry point of the cluster into the inner protein shell. E, experimental verification of the two iron-water complexes (Fe1 and Fe2). A |2Fobs − Fcalc| electron density in the vicinity of the molecules is shown. Residues involved in binding are represented in stick presentation. On the left-hand side, the view along the 3-fold axis is shown, and on the right-hand side, the view of the clusters from the side perspective is given.
AAH Is Induced in Stationary Growth Phase
We set out to study the regulation principles of AAH and asked whether the protein is constitutively formed or whether its expression depends on the microbial growth phase. The bacterium M. arborescens Se14 was cultivated for 24 h, and samples were taken at different time points before being analyzed by Western blotting. As shown in the growth displayed in Fig. 5, during the exponential growth phase, cells did not form any AAH. Only at the beginning of the stationary growth phase after approximately 9 h was the protein significantly induced, and its expression remained high until the end of the experiment.
FIGURE 5.

Growth curve and corresponding time points for Western blot analysis of M. arborescens Se14.
AAH Is Constantly Expressed in Presence of Siderophores
Crystallographic studies demonstrate the ability of the protein to incorporate and store iron. We speculated that the limited availability of iron may influence its expression profiles. Gram-negative bacteria use siderophores to bind and enhance iron uptake into the cell, and many species can use siderophores from different species (1). Because the specifically secreted siderophores of M. arborescens are unknown, we tested the influence of two Streptomyces siderophores, ferrioxamine B (as desferrioxamine mesylate salt) and ferrioxamine E, on the expression of AAH in both the short term and long term. For the short term experiments, cultures were grown until exponential growth (7 h) and then treated as described below. For the long term experiments, treated and untreated cultures were compared over the entire growth curve, and siderophore treatment was shown to increase the production of AAH (Fig. 6A). In the control culture, AAH expression was induced in the stationary growth phase. M. arborescens can use externally provided siderophores to increase iron uptake continuously. In cells, the siderophore-bound Fe3+ is reduced to Fe2+ and released to be used as a building unit for enzymes or stored within the AAH protein. The constantly high Fe2+ concentration in the cell may induce the expression of AAH to remove this reactive metal ion. Short term experiments showed that this induction is very fast: in one, AAH was induced only 15 min after the addition of one siderophore in the exponential growth phase. This is shown in Fig. 6B for ferrioxamine E.
FIGURE 6.

Western blot analysis of M. arborescens Se14. The AAH protein was identified with a specific antibody. Samples were taken at distinct time points, the A600 was measured, and the sample was stored for Western blot analysis. The samples were diluted or concentrated to the same A600 before analysis. The control culture (C) was cultivated in BHI medium without supplementation. A, influence of ferrioxamine E (75 and 300 nm) on the expression of AAH over the whole growth curve. B, influence of ferrioxamine E (150–600 nm) on the expression of AAH in the exponential growth phase. Samples were taken 15, 30, and 45 min after treatment. C, influence of FeSO4 (75–300 μm) on the expression of AAH in the exponential growth phase. Samples were taken 15, 30, and 45 min after treatment. D, influence of FeSO4 (75–300 μm) on the expression of AAH over the whole growth curve. E, influence of Fe2+ on the expression of AAH in the exponential growth phase. The culture was treated with FeSO4 or FeCl3 (300 μm), and samples were taken after 15 and 30 min. F, influence of iron chelators on the expression of AAH. M. arborescens was cultivated for 20 h in the presence of bathophenanthroline disulfonic acid (25–50 μm) and 2,2′-dipyridyl (150–250 μm).
Fe2+ Transiently Induces Expression of AAH
Because siderophores induced the expression of AAH, we wondered whether free iron in the cell culture would as well. Fig. 6C demonstrates that the addition of FeSO4 in the exponential growth phase had the same effect as siderophores. Only 15 min after the addition of iron, AAH expression was visible; however, this effect occurred only transiently. This was further substantiated by M. arborescens cultivation for 24 h in BHI medium enriched by different iron concentrations (Fig. 6D). These long term experiments showed that AAH was not expressed constantly in the exponential growth phase as was the case for the siderophore experiments. Moreover, AAH was shown to be induced only by Fe2+ and then only to remove this reactive iron species from the growth medium. After a while, all Fe2+ was oxidized to Fe3+, and AAH was no longer required. In contrast, Fe3+ had no visible effect on the expression of AAH during the exponential growth phase (Fig. 6E).
Iron Chelators Suppress AAH Expression
To investigate the influence of iron depletion, M. arborescens cells were cultivated in the presence of two iron chelators (25–50 μm bathophenanthroline disulfonic acid and 150–250 μm 2,2′-dipyridyl). In contrast to siderophores, these chelators should not be transported into the bacterial cells. Accordingly, Fig. 6F demonstrates that AAH was not produced in the presence of these iron chelators.
AAH Is Not Induced by H2O2, Hyperosmotic Stress, or Temperature Shifts
The expression of Dps proteins is often regulated by stress conditions such as nutrient starvation, oxidative, and osmotic stress. However, there are big differences between bacterial species. To investigate under which conditions AAH is preferentially formed, M. arborescens Se14 strain was exposed to different conditions known to induce the stress response in related bacteria (13, 15). In contrast to the induction of AAH by increased iron concentrations, no induction of AAH occurred in response to hydrogen peroxide (30 μm–1 mm), osmotic stress (0.3 m NaCl and 0.7 m sorbitol) or temperature shifts to 43–45 °C (data not shown).
AAH Peroxidase Activity
To protect the bacterial cell against ROS and Fe2+, AAH exhibits peroxidase activity. Using an agarose gel, the qualitative effects of hydrogen peroxide, iron, and AAH on DNA are shown in Fig. 7A. Although hydrogen peroxide in the presence of Fe2+ destroyed DNA (lane 5), DNA was preserved by the addition of AAH (lane 6). Upon addition of AAH, the mobility of the DNA remained unchanged, indicating that no co-crystallization or complex formation took place (lane 2). To quantify the peroxidase activity, an assay based on ortho-phenylenediamine was used. The oxidation of the substrate was followed by absorption measurements at 490 nm. A Km of 635 ± 110 mm and a kcat of 143 ± 39 s−1 were determined by a nonlinear fit of the Michaelis-Menten plot of the results (Fig. 7B). The peroxidase activity was shown to be independent from the stored iron in the nucleus.
FIGURE 7.

Peroxidase activity of AAH. A, protection of DNA in the presence of AAH shown by DNA separation in an agarose gel. pET28a vector DNA was exposed to an excess of iron and hydrogen peroxide. B, nonlinear fit of Michaelis-Menten plot of AAH peroxidase activity assayed by oxidation of ortho-phenylenediamine. One exemplary data set with standard errors is shown.
DISCUSSION
In this study, we present the structure of the AAH protein from M. arborescens at low and high iron concentrations alongside supporting biochemical data describing the induction of AAH during the life cycle of the bacterium. Although Dps enzymes in general express three major independent functions, including peroxidase activity, iron uptake/storage, and DNA complexation, the Dps-like AAH protein from M. arborescens is, to our knowledge, the first enzyme to additionally catalyze the synthesis and hydrolysis of N-acylamino acids (31). Initial attempts to characterize AAH by sequence revealed that, although strongly related to previously characterized Dps enzymes by structure (4, 9, 27), AAH appeared in a protein sequence cluster distantly related to most of the Dps proteins characterized to date. We speculate that AAH enzymes of this cluster may have evolved as a result of conditions given by the unique environment of the bacterium.
Our analysis of the protein-protein interfaces led to the identification of an interface along the 2-fold axis that is prominent in all Dps proteins and suggests that this interface is the major determinant for early dimer formation as the initial building block in subsequent Dps assembly.4 However, this observation is in contrast to the molecular mechanism developed for in vitro oligomerization of Dps from Mycobacterium smegmatis that is assumed to be assembled from preformed protein trimers (44). The overall surface covered after dodecamer formation is large (∼50%) but may be explained by the fact that the Dps cage has to be tightly closed to ensure the selective uptake and gradual release of ions over the protein shell. We also searched for putative binding motifs against known catalytic centers. Although we identified esterases with potentially similar active sites, these sites, located in the inner sphere, have not yet been proven to contribute to AAH activity.
The dodecameric structure of the protein is reminiscent of all Dps enzymes investigated so far, although the inner diameter of the protein shell is smaller as has been shown for the DpsA from H. salinarum (27, 45–47). Consequently, the structure motif of all Dps proteins, the FOC, is maintained in AAH, and iron atoms are observed at identical positions (27). However, the uptake channels of iron deviate in enzymes of different classes. Twelve channels were observed in the archaeal system of DpsA from H. salinarum, whereas in AAH or the Dps complex from E. coli, only four channel openings were observed. Although the channels running along the 3-fold axis of many Dps enzyme structures (e.g. E. coli, B. subtilis and Listeria innocua) are presumed to determine the extent of iron uptake, iron atoms have never been proven to enter through those sites. Our results are supported by recent mutational studies of the Dps enzyme from L. innocua in which individual residues along the iron uptake pathway were mutated and their influence on uptake kinetics was studied (28).
The observation of two iron binding sites as hexaquo-iron complexes was initially unexpected but perfectly resembles the biological situation of hydrated ions in solution. Furthermore, the channel diameter of about 0.9 nm as observed for the AAH allows the cluster to move symmetrically along the 3-fold axis. Iron taken up in its hydrated form may diminish the attraction between the protein and the ion species, leading finally to increased uptake rates of iron through the channel. This may be further supported by the central Gln-138 residue, which in contrast to charged residues (Asp-131 and Glu-132) weakly interacts with the iron-hexaquo complex to ensure improved uptake.
The arrangement of this channel in related proteins such as Dps from E. coli and B. subtilis clearly indicates the conservation of residues involved in iron uptake. However, in E. coli, B. subtilis, and L. innocua Dps proteins, the absence of iron atoms in the structures increases the sphere of hydration. Interestingly, a recently published structure of Dps from Streptococcus pyrogenes shows a sodium ion, which was modeled nearby the iron atoms we observed in the vicinity of the AAAH iron uptake channel (48). The position of this ion together with surrounding water molecules is similar to that of the first iron atom observed in the AAH structure and may reflect traces of iron rather than sodium. Notably, the AAH complex and similar enzymes seem to contain a structural mechanism by which the water of iron-hexaquo complexes is stripped off before entering the inner cavity. This mechanism is provided by side chains of conserved aspartate residues located at the end of the ion channel with a distance of only 4 Å between the carboxylate groups. The removal of water from this site may have different origins. A partially “free Fe(II)” atom located inside the cavity might be more reactive and bind more strongly to the FOCs. Moreover, it should be important to provide a selectivity filter against the uncontrolled entry of small molecules to keep these outside the cavity.
The uptake mechanism found in AAH obviously does not resemble the pathway observed for the halophilic DpsA protein from H. salinarum. Here, the non-hydrated ion is taken up due to the small diameter of the individual asymmetric uptake channels. Although most of the known Dps structures follow the uptake routes described for AAH with the FOCs located approximately 2 nm away from the entry point of the channel, in the DpsA protein, the uptake channel immediately discharges into the FOCs, and no additional transfer intermediates are required. The storage of iron was not detected in the AAH and has so far only been described for the DpsA complex. In this complex, the uptake channels along the 3-fold axis do not exist; instead, these channels are blocked by side chains, and a glutamate residue at the inner sphere of the complex is the anchor point for small iron-oxo clusters.
Although AAH is regulated by the concentration of Fe2+, this reactive iron species needs to be removed to prevent oxidative stress. AAH as a Dps protein oxidizes Fe2+ to Fe3+ and at the same time the iron is sequestered and H2O2 is simultaneously reduced to water, thus protecting against oxidative stress. In addition, the Dps protein AAH from M. arborescens is able to hydrolyze and form N-acylamino acids, which may induce defense responses in some plants (31, 33). The formation of N-acylamino acids is an uncommon feature of Dps proteins. Many Dps enzymes are regulated by stress conditions or iron concentrations. AAH is induced in the stationary growth phase as has been shown for other Dps proteins (9, 26, 49, 50). During this phase, bacteria may be exposed to oxidative stress, nutrient starvation, high cell density, and the presence of toxic metabolites. In contrast, the production of AAH is iron-regulated. In the presence of ferrioxamine siderophores from Streptomyces, AAH is constitutively expressed. In contrast, the addition of FeSO4 results in the temporary induction of AAH, whereas FeCl3 has no effect on the expression of AAH consistent with an exclusive regulation by Fe2+. Under normal culture conditions, Fe3+ is prevalent in the growth medium. Supplementing with siderophores facilitates the uptake of Fe3+, which in turn is reduced to Fe2+ in the bacterial cell. On the other hand, Fe2+ causes the Fenton reaction, which is prevented by the Dps proteins. Due to siderophore recycling and continuous Fe3+ import, the expression of AAH lasts for several hours. In contrast, free FeSO4 is oxidized rapidly and causes only a temporary induction of AAH. In the presence of iron chelators, which cannot function as siderophores, iron undergoes complexation, and AAH is not induced. The regulation of AAH via Fe2+ has a protective function by preventing the Fenton reaction and thus the formation of ROS. Astonishingly, H2O2 did not induce AAH. M. arborescens may use other enzymes (e.g. catalases) for its detoxification. Interestingly, most Dps proteins are induced by iron depletion (19, 26, 50). This may be caused by deficiency of the cofactor Fe2+, which allows the Dps protein to reduce H2O2 to water. This indicates that AAH may work not only as a detoxifying enzyme but also as an iron storage protein. This is similar to ferritins, which are likewise induced by an excess of iron (51). Taken together, the regulation of Dps proteins is highly variable, and these enzymes appear to have different tasks in different organisms. In addition, it remains unknown why AAH is also able to hydrolyze and synthesize N-acylamino acids.
Some Dps proteins protect DNA physically by forming Dps-DNA complexes (8–11). Because the molecular weight of these DNA complexes is very high, they cannot enter an agarose gel. In contrast to E. coli Dps, AAH from M. arborescens is not able to bind DNA by forming such a complex, but protection against DNA damage by H2O2 could be shown. The scavenging of hydrogen peroxide was quantitatively measured by the oxidation of o-phenylenediamine. The kinetic constant (Km) was determined by substrate versus velocity-plotted values and showed that the reaction follows Michaelis-Menten kinetics (Km = 635 mm). The presented results reflect in vitro conditions. M. arborescens was isolated from the gut of S. exigua larvae. Because the gut interior is anoxic, this impacts the equilibrium between Fe2+ and Fe3+ and in consequence the amount of ROS. Nevertheless, in Drosophila melanogaster, ROS were shown to be used to prevent bacterial infections in the gut (30, 54). ROS are synthesized by an NADPH oxidase enzyme (dDuox) using the innate immune system of the gut epithelial cells. Thus, AAH could be an instrument for survival under the harsh conditions occurring in the insect gut.
Supplementary Material
Acknowledgments
We are grateful for the technical assistance, maintenance, and operation of the Swiss Light Source beamline PXII. The support of Dr. Franziska Berndt in the crystallization process is greatly appreciated. We thank Emily Wheeler for editorial assistance.
This work was supported by institutional funds from the Max Planck Society, German Science Foundation Deutsche Forschungsgemeinschaft Grant ZE522/3-2, a grant from the Human Frontier Science Program Foundation (to K. Z.), and the Jena School of Microbial Communication.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
The atomic coordinates and structure factors (codes 2YJK and 2YJJ) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
J. Pesek, R. Büchler, and W. Boland, unpublished biochemical data.
- ROS
- reactive oxygen species
- AAH
- amino acid hydrolyase
- AAHL
- low iron AAH structure
- AAHH
- high iron AAH structure
- FOC
- ferroxidase center
- BHI
- brain-heart infusion
- AAHL
- low iron AAH
- AAHH
- high iron AAH.
REFERENCES
- 1. Andrews S. C., Robinson A. K., Rodríguez-Quiñones F. (2003) FEMS Microbiol. Rev. 27, 215–237 [DOI] [PubMed] [Google Scholar]
- 2. Hantke K. (2001) Curr. Opin. Microbiol. 4, 172–177 [DOI] [PubMed] [Google Scholar]
- 3. McHugh J. P., Rodríguez-Quinoñes F., Abdul-Tehrani H., Svistunenko D. A., Poole R. K., Cooper C. E., Andrews S. C. (2003) J. Biol. Chem. 278, 29478–29486 [DOI] [PubMed] [Google Scholar]
- 4. Chiancone E., Ceci P. (2010) Front. Biosci. 15, 122–131 [DOI] [PubMed] [Google Scholar]
- 5. Battista J. R. (1997) Annu. Rev. Microbiol. 51, 203–224 [DOI] [PubMed] [Google Scholar]
- 6. Chiancone E. (2008) Rend. Lincei-Scienze Fis. Nat. 19, 261–270 [Google Scholar]
- 7. Chiancone E., Ceci P., Ilari A., Ribacchi F., Stefanini S. (2003) Iron and Proteins for Iron Storage and Detoxification, Kluwer Academic Publishers, Capri, Italy: [DOI] [PubMed] [Google Scholar]
- 8. Zhao G., Ceci P., Ilari A., Giangiacomo L., Laue T. M., Chiancone E., Chasteen N. D. (2002) J. Biol. Chem. 277, 27689–27696 [DOI] [PubMed] [Google Scholar]
- 9. Almirón M., Link A. J., Furlong D., Kolter R. (1992) Genes Dev. 6, 2646–2654 [DOI] [PubMed] [Google Scholar]
- 10. Wang S. W., Chen C. Y., Tseng J. T., Liang S. H., Chen S. C., Hsieh C., Chen Y. H., Chen C. C. (2009) J. Bacteriol. 191, 4522–4533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castruita M., Saito M., Schottel P. C., Elmegreen L. A., Myneni S., Stiefel E. I., Morel F. M. M. (2006) Appl. Environ. Microbiol. 72, 2918–2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ali Azam T., Iwata A., Nishimura A., Ueda S., Ishihama A. (1999) J. Bacteriol. 181, 6361–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Facey P. D., Hitchings M. D., Saavedra-Garcia P., Fernandez-Martinez L., Dyson P. J., Del Sol R. (2009) Mol. Microbiol. 73, 1186–1202 [DOI] [PubMed] [Google Scholar]
- 14. Weber A., Kögl S. A., Jung K. (2006) J. Bacteriol. 188, 7165–7175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Antelmann H., Engelmann S., Schmid R., Sorokin A., Lapidus A., Hecker M. (1997) J. Bacteriol. 179, 7251–7256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Olsen K. N., Larsen M. H., Gahan C. G., Kallipolitis B., Wolf X. A., Rea R., Hill C., Ingmer H. (2005) Microbiology 151, 925–933 [DOI] [PubMed] [Google Scholar]
- 17. Michán C., Manchado M., Dorado G., Pueyo C. (1999) J. Bacteriol. 181, 2759–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reindel S., Schmidt C. L., Anemüller S., Matzanke B. F. (2006) Biometals 19, 19–29 [DOI] [PubMed] [Google Scholar]
- 19. Wiedenheft B., Mosolf J., Willits D., Yeager M., Dryden K. A., Young M., Douglas T. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 10551–10556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Strand K. R., Sun C., Li T., Jenney F. E., Jr., Schut G. J., Adams M. W. W. (2010) Arch. Microbiol. 192, 447–459 [DOI] [PubMed] [Google Scholar]
- 21. Gupta S., Pandit S. B., Srinivasan N., Chatterji D. (2002) Protein Eng. 15, 503–512 [DOI] [PubMed] [Google Scholar]
- 22. Hong Y., Wang G., Maier R. J. (2006) Free Radic. Res. 40, 597–605 [DOI] [PubMed] [Google Scholar]
- 23. Halsey T. A., Vazquez-Torres A., Gravdahl D. J., Fang F. C., Libby S. J. (2004) Infect. Immun. 72, 1155–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ishikawa T., Mizunoe Y., Kawabata S., Takade A., Harada M., Wai S. N., Yoshida S. (2003) J. Bacteriol. 185, 1010–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nair S., Finkel S. E. (2004) J. Bacteriol. 186, 4192–4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sen A., Dwivedi K., Rice K. A., Bullerjahn G. S. (2000) Arch. Microbiol. 173, 352–357 [DOI] [PubMed] [Google Scholar]
- 27. Zeth K., Offermann S., Essen L. O., Oesterhelt D. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 13780–13785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bellapadrona G., Stefanini S., Zamparelli C., Theil E. C., Chiancone E. (2009) J. Biol. Chem. 284, 19101–19109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Papinutto E., Dundon W. G., Pitulis N., Battistutta R., Montecucco C., Zanotti G. (2002) J. Biol. Chem. 277, 15093–15098 [DOI] [PubMed] [Google Scholar]
- 30. Vallet-Gely I., Lemaitre B., Boccard F. (2008) Nat. Rev. Microbiol. 6, 302–313 [DOI] [PubMed] [Google Scholar]
- 31. Ping L., Büchler R., Mithöfer A., Svatos A., Spiteller D., Dettner K., Gmeiner S., Piel J., Schlott B., Boland W. (2007) Environ. Microbiol. 9, 1572–1583 [DOI] [PubMed] [Google Scholar]
- 32. van Poecke R. M., Dicke M. (2004) Plant Biol. 6, 387–401 [DOI] [PubMed] [Google Scholar]
- 33. Alborn T., Turlings T. C., Jones T. H., Stenhagen G., Loughrin J. H., Tumlinson J. H. (1997) Science 276, 945–949 [Google Scholar]
- 34. Kabsch W. (1993) J. Appl. Crystallogr. 26, 795–800 [Google Scholar]
- 35. Vagin A., Teplyakov A. (2000) Acta Crystallogr. D Biol. Crystallogr. 56, 1622–1624 [DOI] [PubMed] [Google Scholar]
- 36. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 37. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 39. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., Murshudov G. N. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 [DOI] [PubMed] [Google Scholar]
- 40. Bailey S. (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 41. Frickey T., Lupas A. (2004) Bioinformatics 20, 3702–3704 [DOI] [PubMed] [Google Scholar]
- 42. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Balaji V., Sessa G., Smart C. D. (2011) Phytopathology 101, 349–357 [DOI] [PubMed] [Google Scholar]
- 44. Chowdhury R. P., Vijayabaskar M. S., Vishveshwara S., Chatterji D. (2008) Biochemistry 47, 11110–11117 [DOI] [PubMed] [Google Scholar]
- 45. Ceci P., Ilari A., Falvo E., Chiancone E. (2003) J. Biol. Chem. 278, 20319–20326 [DOI] [PubMed] [Google Scholar]
- 46. Grant R. A., Filman D. J., Finkel S. E., Kolter R., Hogle J. M. (1998) Nat. Struct. Biol. 5, 294–303 [DOI] [PubMed] [Google Scholar]
- 47. Ilari A., Latella M. C., Ceci P., Ribacchi F., Su M., Giangiacomo L., Stefanini S., Chasteen N. D., Chiancone E. (2005) Biochemistry 44, 5579–5587 [DOI] [PubMed] [Google Scholar]
- 48. Haikarainen T., Tsou C. C., Wu J. J., Papageorgiou A. C. (2010) J. Biol. Inorg. Chem. 15, 183–194 [DOI] [PubMed] [Google Scholar]
- 49. Nicodème M., Perrin C., Hols P., Bracquart P., Gaillard J. L. (2004) Curr. Microbiol. 48, 51–56 [DOI] [PubMed] [Google Scholar]
- 50. Polidoro M., De Biase D., Montagnini B., Guarrera L., Cavallo S., Valenti P., Stefanini S., Chiancone E. (2002) Gene 296, 121–128 [DOI] [PubMed] [Google Scholar]
- 51. Harrison P. M., Arosio P. (1996) Biochim. Biophys. Acta 1275, 161–203 [DOI] [PubMed] [Google Scholar]
- 52. Kabsch W., Sander C. (1983) Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 53. The PyMol Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC [Google Scholar]
- 54. Ha E. M., Oh C. T., Bae Y. S., Lee W. J. (2005) Science 310, 847–850 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.