

Abstract
The K/BxN serum transfer model of arthritis is critically dependent on FcγR signaling events mediated by Syk kinase. However, the specific cell types in which this signaling is required are not known. We report that deletion of Syk kinase in neutrophils, achieved using sykf/f MRP8-cre+ mice, blocks disease development in serum transfer arthritis. The sykf/f MRP8-cre+ mice display absent joint disease and reduced deposition of pathogenic anti-GPI Abs in the joint (with a reciprocal accumulation of these Abs in the peripheral circulation). Additionally, sykf/f MRP8-cre+ mice manifest poor edema formation within 3 hours following formation of cutaneous immune complexes (Arthus reaction). Together, this suggests that neutrophil-dependent recognition of immune complexes contributes significantly to changes in vascular permeability during the early phases of immune complex disease. Using mixed chimeric mice, containing both wild type and sykf/f MRP8-cre+ neutrophils, we find no impairment in recruitment of Syk-deficient neutrophils to the inflamed joint, but they fail to become primed, demonstrating lower cytokine production following removal from the joint. They also display an increased apoptotic rate compared to wild type cells in the same joint. To our surprise, mast cell-deficient c-kitsh/sh mice developed robust arthritis following serum transfer while c-kitW/Wv mice did not, suggesting that previous conclusions concerning the central role of mast cells in this model may need to be revised. Basophil-deficient mice also responded normally to K/BxN serum transfer. These results demonstrate that Syk-dependent signaling in neutrophils alone is critically required for arthritis development in the serum transfer model.
INTRODUCTION
Neutrophils are the most numerous leukocytes in the peripheral blood, comprising up to 50 percent of the compartment. They are among the first cells recruited at the initiation of inflammatory responses, and they make up the majority of cells found in sites of infection and tissue injury. Neutrophils are able to respond quickly to a large variety of stimuli, including immune-complexes, complement, and pathogen associated molecular patterns. Following activation, neutrophils initiate phagocytosis, generate reactive oxygen species and cytokines, and release pre-formed granules containing inflammatory mediators. Different types of granules are released depending on the strength and type of signal, allowing neutrophils to modulate their responses (1, 2). Neutrophils are therefore able to shape the immune response by affecting the early inflammatory milieu. Indeed, neutrophils are known to be critical for the effective response to multiple infectious organisms (3). Recent discoveries have greatly broadened our knowledge on the functional role of this cell type in pathologic processes besides infection (4). It is noteworthy that neutrophils are responsible for much of the damage to host tissues in some types of autoimmune disorders, such as rheumatoid arthritis (RA) (5-7).
The K/BxN serum transfer mouse model of inflammatory arthritis reproduces many of the pathological hallmarks of human RA. In this model, transient arthritis is induced in naïve mice by transfer of serum from K/BxN transgenic mice containing antibodies against glucose-6 phosphate isomerase (GPI) (8, 9). Clinical swelling and immune cell infiltrates are associated with IgG and C3 deposition along the cartilagenous surface leading to joint damage, including pannus formation and erosion of cartilage and bone (10). Though inflammation peaks between day seven and nine following transfer, disease may persist up to a month with no evidence of extra-articular inflammation (8). Inflammation in the K/BxN serum transfer model requires a variety of cellular mediators, including macrophages (11) and neutrophils (12), but does not require the adaptive immune system, making it a useful model for studying the role of the innate immune system in auto-inflammation (8). In addition to IgG, FcγRs and complement, inflammation depends on the production of leukotriene B4 (LTB4) (13), tumor necrosis factor-α (TNFα), and IL-1β (14). There is some evidence that tissue-resident mast cells are primarily responsible for recognizing immune-complexes in the joint, then initiating disease by inducing neutrophil and monocyte extravasation through release of chemokines (15-17).
Neutrophils are the predominant infiltrating cell in the arthritic joint, and are required for K/BxN serum-induced disease (18). The LTB4/BLT1 (the cellular receptor for LTB4) pathway plays a crucial role in neutrophil recruitment in this model (13, 19). In addition to Fcγ receptors and BLT1, neutrophils express most of the other receptors known to be required for inflammation, including C5aR (20), and thus are able to recognize disease-causing immune-complexes as well as other inflammatory mediators in the peripheral blood and the joint. These data suggest that neutrophils are key mediators of joint inflammation in the K/BxN serum transfer model. However, it is unknown if neutrophils are required for the recognition of immune complexes. Mice expressing the human FcγRIIA receptor only in neutrophils develop both arthritis, following KxB/N serum transfer, and nephritis, in response to injection of nephrotoxic serum (7, 20), demonstrating the ability of neutrophils to initiate antibody-mediated inflammation. However, these mice lack FcRs in all other cell types, perhaps exaggerating the role of neutrophil FcRs. Previous data in the K/BxN model suggests that neutrophils initially respond to the inflammatory signals from other Fcγ-expressing cells such as mast cells (15) and macrophages (11). It is currently unknown how neutrophil Fcγ signaling participates in the initiation and progression of inflammation within the articular space.
Spleen receptor tyrosine kinase (Syk) is required for signaling through Fcγ receptors, integrins, and other scavenger receptors that utilize Immunoreceptor Tyrosine-based Activation Motifs (ITAMs) to initiate intracellular signaling (21-23). Mice deficient in Syk die late in gestation due to dysregulated macrophage inflammation leading to vascular and lymphatic malformations (24, 25). The role of Syk in immune cells has been studied by transfer of syk−/− fetal liver cells into lethally irradiated recipient mice to generate chimeric mice lacking Syk in all hematopoietic lineages. Syk-deficient fetal liver chimeras are completely resistant to disease in the K/BxN serum transfer model, reflecting the critical role of FcγR signaling in inflammatory cells (26). However, the relative importance of Syk-mediated signaling in different cell types for the initiation or progression of disease cannot be determined using chimeric mice. In order to address this question, we have generated strains of Syk conditional mutant mice, which lack Syk in specific myeloid cell lineages. Using these mice, we have recently demonstrated the importance of Syk based signaling in neutrophils alone as being critical for appropriate host defense to Staph aureus infection in vivo (3). We report here for the first time that specific deletion of Syk in neutrophils is sufficient to block the initiation of arthritis in the K/BxN serum transfer model. These results suggest that neutrophils alone are essential for establishment of immune complex-mediated arthritis in this model. Indeed, using mast cell and basophil-deficient animals, we find no requirement for these cells in the K/BxN model. These observations suggest that models for the pathophysiologic processes in immune complex arthritis may need to be revised.
MATERIALS and METHODS
Mice
Sykf/f mice (27) were backcrossed to C57BL/6 (Charles River) for 8 generations, then crossed to MRP8-cre+ (28), Mx-1-cre+ (29), LysM-cre+ (30) or CD11c-cre+ (31) strains for conditional syk deletion. Importantly, the MRP8-cre gene also contains an ires-GFP marker, allowing us to track cells expressing the Cre recombinase by flow cytometry. Induction of Mx-1 expression by injection of polyinosinic:polycytidylic acid (poly I:C) was performed as described (29). Control mice for all experiments included either sykf/+ or syk+/+ with the relevant Cre, or sykf/− or sykf/f without Cre, to both control for Syk and Cre expression in the various cell lineages. NOD/ShiLtJ, C57BL/6J-KitW-sh/BsmJ (c-kitsh/sh) (32), C57BL/6J-KitW-v/J (c-kitW/+), WB/ReJ KitW/J (c-kit+/Wv) mice were purchased from Jackson Laboratories. The c-kitW/+ and c-kit+/Wv were intercrossed to obtain c-kitW/Wv animals (16). NOD/ShiLtJ mice were crossed to C57BL/6J carrying the KRN TCR transgene to obtain K/BxN mice (8). B6.SJL mice carrying the Ly5.1 allele, used for generation of mixed chimeric mice, were purchased from Taconic Farms. Basophil-deficient mice were generated by interbreeding Basoph8 mice (containing a YFP-ires-Cre gene inserted into the mast cell protease 8 gene; a basophil specific marker) with Rosa-flxDTα mice (containing a loxP-flanked diphtheria toxin α-chain gene inserted into the Rosa26 locus) as described (33). Mixed chimeric mice were generated by injecting lethally irradiated B6 recipients with bone marrow cells, as described (34). Briefly, male 8-12 week old B6.SJL CD45.1+ mice were lethally irradiated and injected i.v. with 5 × 106 mixed bone marrow cells. Chimeric animals were used for experiments after 8-10 weeks. The percentage of chimerism was determined by flow cytometry on peripheral blood or inflammatory joint neutrophils using CD45.1 versus CD45.2 mAbs as described (34). Syk−/− chimeras were obtained by reconstituting lethally irradiated wild type mice with Syk−/− fetal liver, as described (26). All animals were kept in a specific pathogen-free facility at University of California-San Francisco (UCSF) and used according to protocols approved by the UCSF committee on animal research.
K/BxN serum transfer and clinical scoring
Arthritogenic serum was collected in Serum Gel Z/1.1 tubes (Seidstadt) and pooled from 8 week old arthritic K/BxN mice. Disease was induced by injecting 200μL of serum i.p. into 6-8 week old recipient mice on days 0 and 2. Clinical scores were assessed on a 0-12 scale (0-3 per paw) as follows: 0, no edema: 1, localized edema or erythema; 2, localized edema and erythema on 3 or more toes and at talocrural joint or over one entire surface of paw; 3, marked edema and erythema over entire paw surface. Mice were scored daily (19).
Histology
Paws were removed from euthanized mice above the tibiotalar and radiocarpal joints, fixed in 10% formalin for 38 hours, and decalcified with Cal-Ex II (Fisher Scientific) for 48 hours prior to paraffin embedding. Sections, stained with hematoxylin and eosin (H&E) by the UCSF Pathology core, were evaluated for neutrophil infiltration, synovial hyperplasia, and cartilage and bone erosion as previously described (19). Sections stained with toluidine blue were evaluated for the presence of blue-staining mast cells in the dermis as described (20). Images were taken on a Leica DMLB microscope at 50X or 200x magnification.
Immunofluorescent staining
Frozen ankle sections were prepared as previously described (19). Briefly, paws were removed from euthanized mice above the tibiotalar and radiocarpal joint and flash frozen in O.C.T (Tissue-Tek) prior to sectioning. Frozen sections were obtained using the CryoJane Tape-Transfer System (Instrumedics). Sections were fixed in cold acetone and stained with fluorescently labeled goat anti-mouse IgG F(ab’)2 (Jackson Immunoresearch Laboratories) (19).
Arthus reaction
The cutaneous Arthus reaction was performed as previously described (35). Briefly, 30μL of 1mg/mL rabbit anti-chicken ovalbumin IgG or control rabbit IgG was injected intradermally (i.d.) on the back of anaesthetized mice, followed by the administration of 0.5% Evans Blue and 2.5mg/mL chicken ovalbumin (OVA, Sigma #5503) in PBS i.v. (200μL per mouse). Skin from the i.d. injection site was collected 3 hours later and placed in N,N-dimethylformamide for 48 hours to extract the Evans Blue. Evans Blue concentration was determined by absorbance at 650nm and edema was evaluated as the ratio of Evans Blue concentration in the tissue to that in the peripheral blood.
Leukocyte isolation
To isolate bone marrow neutrophils, femora, tibiae, and humeri were collected from euthanized 8-10 week old mice and flushed with saline. Collected marrow was homogenized with a 19g needle and filtered through a 70μm filter, followed by hypotonic red blood cell (RBC) lysis. Neutrophils were purified by isolating cells from the Percoll layer of a 62% Percoll gradient, as previously described (34). The resulting neutrophils were at least 85% pure. Splenocytes were isolated by mechanical homogenization of whole spleens through a 70μm filter, followed by hypertonic RBC lysis. Cells sorted by F4/80 and Ly6G surface markers were cytospun and stained by Wright-Giemsa. To obtain peritoneal cells, the peritonea of euthanized mice were lavaged with 5 ml of PBS/2mM EDTA to isolate resident cells. Peripheral blood leukocytes were isolated by collecting 100μL of blood into heparinized saline, followed by hypertonic RBC lysis (3). Synovial fluid leukocytes were isolated by ankle joint aspiration with a 26 gauge needle into PBS/2mM EDTA for staining, flow cytometry, and stimulation. Anesthetized mice were exsanguinated to reduce peripheral blood contamination of joint aspirates.
Leukocyte stimulation
Synovial leukocytes or bone marrow neutrophils were isolated as above, suspended in RMPI + 10% FCS then stimulated for 6 hours at 37°C under CO2 with 1 μg/ml of brefeldin-A (eBiosciences) at 4 × 105 cells/100μL. Following stimulation, cells were placed on ice, surface stained for Ly6G and stained intracellularly for TNFα and IL-6 using the eBiosciences IC fixation kit as described by the manufacturer. Reagents for stimulation were as follows: 10 ng/mL LPS or rabbit IgG immune complexes at 420μg/mL for cytokine stimulation and 10 μg/mL for superoxide production. Immune complexes were generated by incubating rabbit anti-chicken ovalbumin (Cappel #55304) with OVA in saline, in a 10:1 ratio for 2 hours incubation at 37°C, forming a visible insoluble immune complex precipitate (36).
Flow cytometry
For flow cytometry, isolated leukocytes were treated with 1 μg/ml of Fc block (eBioscience), then surface stained with the following anti-mouse fluorescein isothiocyanate (FITC)-, APC-, APC-Cy7-, PE-Cy7-, Biotin -, phycoerythrin (PE)-, PerCP-Cy5.5- or Alexa fluor 647-conjugated specific Abs (3): CD11b (M1/70), CD11c (HL3), CD62L (MLE-14), CD45.1 (A20), CD45.2 (104), Ly6G (1A8), Ly6C (AL-21), SiglecF (29A1.4), NKp46, DX5 (JORO50), CD131, FcεR1α (MAR1), c-Kit (2B8), all from eBiosciences or BD Pharmingen; F4/80 (CI:A3-1; Serotec); 7/4 (Caltag); followed by Streptavidin-Pacific Orange. After final wash, cells were resuspended in staining/wash buffer containing 1 μg/ml propidium iodide (PI, Sigma-Aldrich) and/or Annexin V (BD #555419) for viability staining according to manufacturer instructions. For intracellular Syk and cytokine staining, cells were fixed using the eBiosciences kit as described (3) and stained for TNFα (MP6-XT22) or IL-6 (MP5-20F3) from eBiosciences, and/or Alexa Fluor 488-conugated mouse anti Syk (5F5). Five-color flow cytometry was performed on a Becton Dickinson FACScan and data analyzed with FlowJo software (Tree Star, Inc.). Peripheral blood and synovial neutrophils and monocytes/macrophages were defined as CD11b+ Ly6G+ and CD11b+ Ly6G− respectively (3). Macrophages were gated as CD11b+ and F4/80+ cells (37). Peritoneal mast cells were defined as c-Kit FcεR1 double positive cells.
Cytokine assays
Serum cytokine concentrations were determined using Cytokine Multiplex Kits for the Luminex technology according to the manufacturer’s protocol (Invitrogen).
ELISA for serum anti-GPI
Anti-GPI antibodies were identified by sandwich ELISA. Briefly, serum was incubated on plates coated with rabbit GPI (Sigma), and detected with an HRP-conjugated anti-mouse IgG-Fc antibody and TMB reagent (KPL 50-76-00). The reaction was stopped with acid and read at 450nm on a SpectraMax M5 (Molecular Devices). Results are displayed as arbitrary units and standardized between experiments by normalizing to K/BxN serum (12).
Analysis and statistics
Graphs and statistical test were performed using Prism. Statistical significance was determined by one-way ANOVA unless otherwise indicated, and error bars represent standard error of the mean. *= p<0.05, ** = p< 0.01, and ***= p<.0001.
RESULTS
Syk deletion in neutrophils protects against antibody-mediated arthritis
To determine the requirement for Syk in antibody-mediated arthritis, mice with LoxP flanked Syk alleles (sykf/f) were crossed with cell-specific Cre recombinase (Cre)-expressing strains. Inducing Cre expression under the control of the human MRP8 promoter (sykf/f MRP8-cre+) resulted in nearly complete Syk deletion in neutrophils as measured by intracellular flow cytometry (Fig. 1A-C, Supplemental Fig. 1). Intracellular Syk staining in neutrophils from sykf/f MRP8-cre+ mice overlapped staining in cells from syk−/− fetal liver chimeras, confirming loss of Syk expression. In contrast, peripheral blood 7/4+ Ly6G− monocytes as well as splenic F4/80+ macrophages and DX5+ basophils from sykf/f MRP8-cre+ mice exhibited similar Syk expression to sykf/f littermate controls (Fig 1A, B). The relative levels of Syk in monocytic cell types from Sykf/f MRP8-cre+ mice are similar to those seen in wild type C57BL/6 controls, in contrast to the Syk staining intensity in Sykf/f MRP8-cre+ neutrophils, which is similar to Syk−/− neutrophils (Fig. 1C). Use of intracellular staining for Syk allowed us to confirm that every animal used in the following experiments demonstrated >90% reduction in Syk expression in peripheral blood neutrophils, as previously reported for sykf/− MRP8-cre+mice (3).
Figure 1.
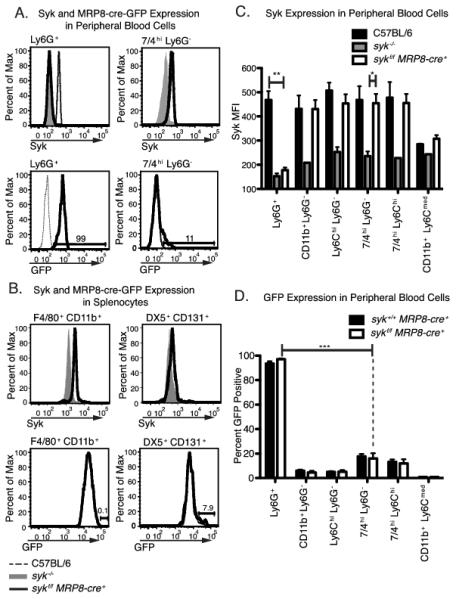
Neutrophil-specific deletion of Syk in sykf/f MRP8-cre+ mice. Peripheral blood and splenic leukocytes from mice of the indicated genotypes were stained for surface markers and Syk for analysis by flow cytometry, as described in the Materials and Methods. Cell populations were either analyzed for Syk or GFP expression. Leukocytes from syk−/− fetal liver chimeras served as a negative control for Syk staining. GFP positivity indicates the current expression of the MRP8-cre+-ires-GFP construct. A, B, Syk and GFP expression in peripheral blood and splenic leukocytes. Histograms are representative of at least 3 separate experiments, with at least 3 mice per genotype per experiment. C, Mean fluorescence intensity of Syk staining in peripheral blood populations in sykf/f MRP8-cre+ mice (n= 8) as compared to C57BL/6 wild type mice and sykf/f cells (n= 2-3). Only the significant differences by one-way ANOVA for Ly6G+ and 7/4hiLy6G− cells are shown. Only staining in Ly6G+ cells was significantly different between sykf/f MRP8-cre+ and wild type controls. D, Percent GFP positive of peripheral blood leukocytes from syk+/+MRP8-cre+ and sykf/f MRP8-cre+ mice (n= 6). Only the significant differences, by one-way ANOVA, between Ly6G+ and 7/4hiLy6G− cells in sykf/f MRP8-cre+mice are shown. GFP positivity in Ly6G+ cells was significantly higher than in all other cell populations analyzed. *=p<0.05, **=p<0.01, *** = p<0.001
The MRP8-cre allele contains an ires-GFP construct that allows us to detect cells expressing Cre (3). While 95- 98 % of Ly6G+ neutrophils expressed GFP, less than 20% of the various monocyte populations were GFP+, in general at a lower intensity, resembling a shoulder on the negative peak (Fig. 1A, D). Since this level of GFP expression did not correlate with significant differences in Syk expression (Fig. 1A, C), it is unclear if this level of potential Cre expression is physiologically relevant. Nevertheless, determining GFP levels allows us a potentially more sensitive approach to follow cellular expression of Cre in the MRP8-cre+ animals. Resident macrophages, basophils, mast cells and some peripheral blood monocyte populations show low levels (5-20%) of GFP expression (Fig 1B, D and Supplemental Fig 1). Though we cannot rule out the effects of this low level of non-neutrophil Cre expression, no other cell population besides neutrophils shows efficient GFP expression and Syk deletion. We have also crossed the MRP8-cre+strain to the ROSA26-YFP reporter strain (38) and have confirmed recombination predominantly in neutrophils, with less than 10% recombination in monocytes/macrophages (C. Abram and C. Lowell, unpublished). We also crossed sykf/f mice to the Mx-1-cre+, LysM-cre+, and CD11c-cre+ strains to afford inducible deletion of Syk in all hematopoietic cells, in myeloid leukocytes only or in CD11c+ dendritic cells, respectively. The specificity and efficiency of Syk deletion in these strains was also monitored by intracellular flow cytometry and was previously described (3, 39).
The multiple sykf/f Cre-expressing strains were treated with serum from K/BxN arthritic mice to determine the importance of Syk expression in various cell types for the initiation and progression of arthritis. Syk deletion in all hematopoietic cells using polyinosinic:polycytidylic acid (poly I:C) treated sykf/f Mx1-cre+ mice (29) resulted in complete protection against antibody-mediated arthritis (Fig. 2A). These results confirm previous findings using Syk-deficient fetal liver chimeras, which lack Syk in all hematopoietic lineages and were completely resistant to serum transfer arthritis (26). Deletion of Syk in myeloid cells (sykf/− LysM-cre+) was also protective (Fig. 2B), reflecting the previously published importance of macrophages and granulocytes in the development of disease (12). However, deletion of Syk in dendritic cells (sykf/f CD11c-cre+) had no effect on the course arthritis (Fig. 2C). This was expected, as dendritic cells have not been shown to contribute to inflammation during the effector phase of arthritis (9, 40).
Figure 2.
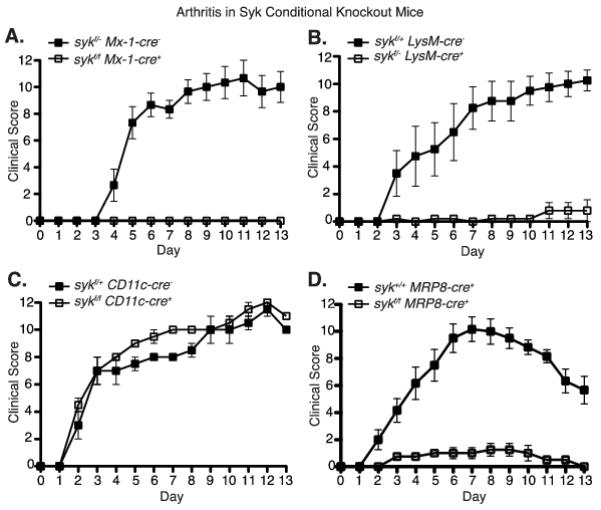
Conditional deletion of Syk in myeloid cells or neutrophils specifically protects mice from K/BxN serum-induced arthritis. Clinical score was recorded daily following K/BxN serum transfer in mice of the indicated genotypes, as described in Materials/Methods. Solid squares represent littermate controls; hollow squares represent Syk conditional knockouts. A, sykf/f Mx-1-cre mice that have Syk deficiency in all hematopoietic cells (n = 3). B, sykf/−LysM-cre mice that have Syk deficiency in myeloid cells (n=4). C, sykf/f CD11c-cre mice that have Syk deficiency in dendritic cells (n=2). D, sykf/f MRP8-cre mice that have Syk deficiency in neutrophils (n=4). Representative plots from 2 - 6 independent experiments per genotype. Time points after day five were significant (p > 0.01, two-way ANOVA) between the conditional knockouts and wild type controls in panels A, B, and D.
Syk deletion in neutrophils (sykf/f MRP8-cre+) completely protected mice from K/BxN serum induced arthritis as compared to control mice (either Sykf/f or Syk+/+ MRP8-cre+) (Fig. 2D, Supplemental Fig. 2A). Histological evaluation of ankle sections from sykf/f MRP8-cre+ mice following K/BxN serum transfer showed no evidence of cellular infiltration, pannus formation, or cartilage and bone erosion, while these features were readily apparent in joints from control sykf/f mice (Fig. 3A, B). Moreover, sykf/f mice developed robust synovial fluid accumulation containing neutrophils and macrophages (Fig. 3B, C) while sykf/f MRP8-cre+ mice showed no joint swelling and no synovial fluid could be collected from these animals. Sykf/f MRP8-cre+ mice also displayed less evidence of systemic inflammation following serum transfer, as demonstrated by lower serum levels of KC, MCP-1and TNFα (Fig. 3D). Thus, the lack of Syk in neutrophils greatly diminished both the systemic and local inflammatory response to articular immune-complexes.
Figure 3.
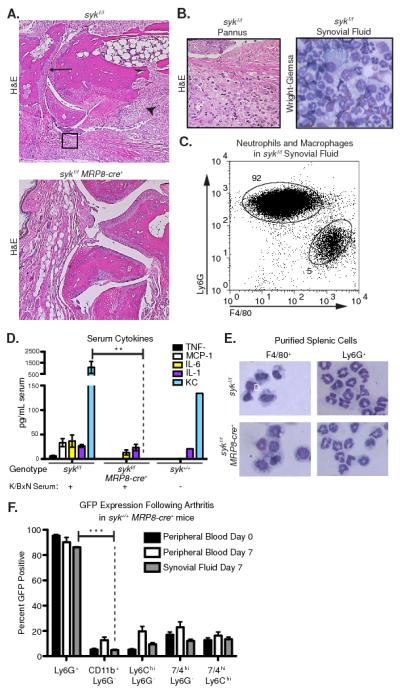
Joint inflammation, synovial neutrophil recruitment and serum cytokine levels are elevated in sykf/f but not sykf/f MRP8-cre+ mice following K/BxN serum injection. A, Representative H&E-stained sections of sykf/f and sykf/f MRP8-cre+ forepaws 7 days after K/BxN serum transfer. Joints from sykf/f animals exhibit pannus formation and leukocyte infiltration (arrowhead), bone erosion (open arrowhead), and narrowing of the synovial space (arrow). B, Higher magnification view of sykf/f hindpaw (left) and Wright-Giemsa stain of isolated synovial fluid leukocytes (right). C, Synovial fluid leukocytes from sykf/f mice at Day 7 were stained with anti-Ly6G and F4/80 to enumerate neutrophils monocytes/macrophages, and analyzed by flow cytometry. D, The levels of the indicated cytokines in the serum of sykf/f and sykf/f MRP8-cre+ mice at 7 days following K/BxN serum transfer or untreated C57BL/6 mice was determined by luminex bead array as described in Materials and Methods. Data are from 4-10 mice per group and representative of two independent experiments. E, Splenocytes from sykf/f and sykf/f MRP8-cre+ mice were stained for F4/80 and Ly6G for sorting by flow cytometry to isolate monocytes/macrophages and neutrophils, respectively. Cytospun cells were then stained by Wright-Giemsa. F, Peripheral blood was collected from syk+/+MRP8-cre+ mice prior to K/BxN serum transfer (day 0, n=12) and day 7 following serum transfer (day 7, n=6). Synovial fluid was aspirated from the fore and hind-paws on day 7 (n=5). Peripheral and synovial leukocytes were stained as indicated and analyzed for GFP expression by flow cytometry. Ly6G+ cells were significantly more GFP positive than all other cell types (p > 0.01). Statistics analyzed by two-way ANOVA. **=p<0.01, *** = p<0.001
Deletion of Syk in the neutrophil lineage in sykf/f MRP8-cre+ mice did not alter myeloid cell development as determined by cell counts (3), expression of myeloid cell markers (Table 1), or morphologic examination (Fig. 3E). Similarly, syk−/− fetal liver chimeric mice demonstrated no alteration in myeloid cell development or marker expression, but of course did have a block in B cell maturation (3). To further address the possibility that Syk deletion was occurring in monocyte/macrophage lineages, we examined GFP expression in the synovial leukocytes of syk+/+ MRP8-cre+ mice following K/BxN serum transfer. As in resting animals, the peripheral blood and joint Ly6G+ neutrophils of arthritic mice showed 90 - 95% expression of GFP, while monocyte/macrophage types ranged from 5-20% (Fig. 3F). This level of GFP expression was low and of unclear significance, especially since monocyte/macrophage cell types make up only 5% of the synovial leukocytes (Fig 3C).
Table 1.
Cell surface marker frequencies in syk+/+ MRP8-cre+ and sykf/f MRP8-cre+ mice
| Peripheral blood (n=6) |
Ly6G+ | CD11b+ Ly6G− |
Ly6Chi Ly6G− |
7/4hi Ly6G− |
| syk+/+ MRP8-cre+ | 10 ± 1 | 11 ± 1 | 7 ± 0.5 | 4 ± 0.5 |
| sykf/f MRP8-cre + | 12 ± 1 | 12 ± 1 | 6 ± 0.3 | 4 ± 0.3 |
| Spleen (n=3) |
F4/80+ CD11b− |
F4/80+ CD11b+ |
CD11c+ CD11b− |
DX5+ CD131+ |
| syk+/+ MRP8-cre+ | 20 ± 1 | 3 ± 0.2 | 26 ± 1 | 0.4 ± 0.02 |
| sykf/f MRP8-cre+ | 19 ± 2 | 4 ± 0.2 | 24 ± 1 | 0.3 ± 0.02 |
| Peritoneum (n=3) |
F4/80+ | F4/80med CD11bmed |
c-kit+ FcεRIα+ |
|
| syk+/+ MRP8-cre+ | 65 ± 9 | 3 ± 1 | 0.7 ± 0.1 | |
| sykf/f MRP8-cre + | 56 ± 1 | 3.5 ± 1 | 1 ± 0.2 |
Data represent the percentages of each cell type, defined by the designated markers, in peripheral blood, spleen and peritoneum of resting wild type MRP8-cre+ versus sykf/f MRP8-cre+ mice. ± SEM.
To rule out the possibility that deletion of syk in non-hematopoietic cells may be contributing to the lack of response in the K/BxN arthritis model, we generated chimeric mice by transferring wild type bone marrow into lethally irradiated sykf/f MRP8-cre+ recipients, then tested these animals for arthritis development. The sykf/f MRP8-cre+ mice with wild type hematopoietic cells responded normally (Supplemental Fig. 2B). These results suggest that Syk signaling in neutrophils is required during the initiation and progression of antibody-mediated arthritis, and that Syk-dependent signaling pathways in other cell types is not sufficient to induce arthritis. As Syk is necessary for Fc receptor signaling in response to immune complexes, we expect that the requirement for Syk in K/BxN serum induced arthritis reflects a nonredundant role of neutrophils in responding to pathogenic immune complexes in both the joint and possibly in the serum.
Loss of Syk affects the kinetics of antibody deposition
The deposition of anti-GPI immune complexes in the joint following K/BxN serum injection is influenced both by rapid (minutes) changes in vascular permeability as well as more long term (hours to days) changes (10, 41). Though antibody accumulation is required for disease, it can occur despite the absence of clinical arthritis. To explore whether neutrophil-specific Syk deficiency affected anti-GPI immune complex deposition, we examined both joints and serum for the presence of anti-GPI Abs. To our surprise, the amount of IgG deposited along the cartilage at day 7 was greatly reduced in sykf/f MRP8-cre+ mice (Fig. 4A). The reduced accumulation of anti-GPI antibodies in the joints of sykf/f MRP8-cre+ mice was not due to a reduced serum half-life of the antibodies, since the sykf/f MRP8-cre+ animals consistently had greater anti-GPI serum titers throughout the course of disease (Fig. 4B). Sykf/f MRP8-cre+ chimeric mice with wild type bone marrow do not maintain this trend (Supplemental Fig. 2C), ruling out an effect from possible syk deletion in inflamed endothelial cells. These results suggested that loss of Syk-dependent signaling in neutrophils resulted in a failure of anti-GPI Abs to exit the vasculature and deposit in joints, potentially due to a failure to induce vascular permeability.
Figure 4.
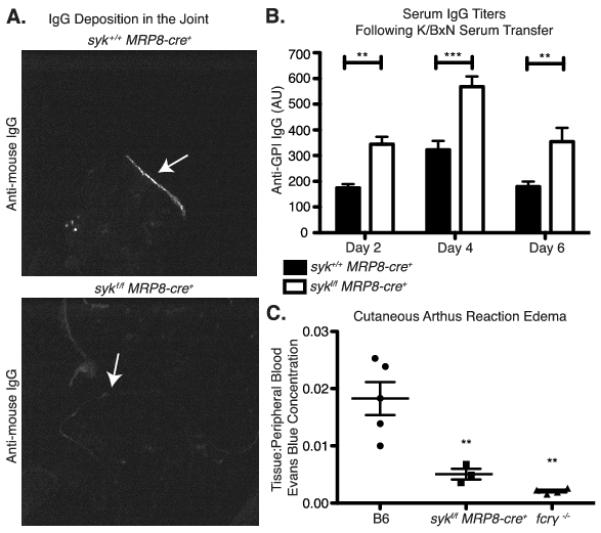
Neutrophil-specific deletion of Syk results in decreased joint antibody deposition following K/BxN serum transfer. A, Cryosections from the forepaws of syk+/+ MRP8-cre+ and sykf/f MRP8-cre+ mice 7 days following K/BxN serum transfer were stained for the Fc portion of IgG. Deposition of IgG lining the cartilage surface is shown by the arrow. B, Serum anti-GPI titers on days 2, 4, and 6 following K/BxN serum transfer in sykf/f and sykf/f MRP8-cre+ mice were determined by ELISA as described in Materials/Methods and analyzed by two-way ANOVA. C, Immune complex induced cutaneous edema was evaluated in syk+/+ MRP8-cre+, wild type, and FcγR−/− mice by Evans Blue extraction three hours following i.v. injection of antigen, as described in Materials and Methods. Results are shown as the ratio of Evans Blue concentration in the tissue to concentration in the peripheral blood (Statistics shown compared to C56BL/6 mice, by one-way ANOVA). **=p<0.01, *** = p<0.001
To evaluate whether neutrophil-specific Syk deficiency affected early events of immune complex-induced vascular permeability, we utilized the cutaneous reverse passive Arthus reaction. Sykf/f MRP8-cre+ mice developed less severe edema at early time points, three hours after immune complex induced inflammation, compared to wild type animals (Fig. 4C). The decrease in edema was not as complete as that seen in FcRγ-deficient mice, suggesting that Fc signaling in other cell types also contributes to increased vascular permeability. A lack of induced vascular permeability in the Sykf/f MRP8-cre+ mice may contribute to the reduced joint deposition of anti-GPI antibodies, which would further protect these animals following K/BxN serum transfer.
Syk-deficient neutrophils migrate to the inflamed joint
The lack of clinical disease in sykf/f MRP8-cre+ mice may reflect both an inability of Syk-deficient neutrophils to respond to joint immune complexes or a block in their ability to migrate into the inflamed joint. To help distinguish these possibilities, we generated mixed chimeric mice to assess the behavior of Syk-deficient neutrophils in the presence of wild type neutrophils that could generate inflammatory arthritis. Congenically marked mixed bone marrow chimeras were generated from various ratios of wild type (CD45.1) and sykf/f MRP8-cre+ (CD45.2) bone marrow. The mixed chimeric mice were then treated with K/BxN serum. The disease course in mixed bone marrow chimeras depended on the ratio of wild type to Syk-deficient neutrophils, as assessed by peripheral blood examination. Chimeras with a neutrophil compartment comprised of 75% or greater Syk-deficient neutrophils developed less severe arthritis, with decreased maximum clinical scores (Fig. 5A). Animals with a lower percentage of Syk-deficient cells were not significantly different than wild type (data not shown). Syk-deficient neutrophils were easily found in the synovial fluid of inflamed joints based on their congenic marker (CD45.2) and lack of Syk expression by intracellular staining (Fig. 5B). Importantly, the ratio of wild type to Syk-deficient cells in the joint was a linear reflection of the extent of chimerism in the peripheral blood over multiple chimeric mice (Fig. 5C). These results indicate that Syk-deficient neutrophils are able to migrate normally into the inflamed joint. Hence, the lack of disease in sykf/f MRP8-cre mice must reflect an impaired ability of the neutrophils to become activated rather than a migratory defect. Indeed, Syk-deficient neutrophils show a profound block in respiratory burst when stimulated with immune complexes in vitro (Supplemental Fig. 3) (21).
Figure 5.
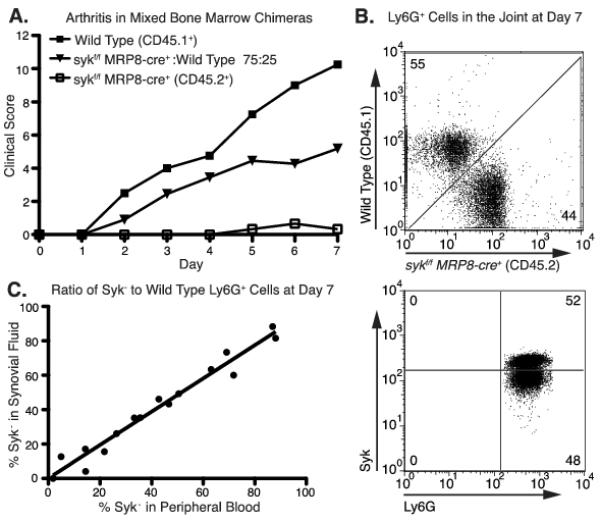
Syk-deficient neutrophils migrate equivalently to the inflamed joint. A, Mixed bone marrow chimeras were generated from sykf-f MRP8-cre+ (CD45.2+) and congenically marked wild type mice (C56BL/6; CD45.1+) in a ratios varying from 75% sykf/+MRP8-cre+ with 25% wild type, to 25% sykf/+MRP8-cre+. Control chimeras were also made with 100% wild type or sykf/f MRP8-cre+ bone marrow. Eight weeks following bone marrow transfer, chimeras were injected with K/BxN serum and clinical score was recorded as described in Material and Methods. Data shown are for control chimeras and the 75% sykf/fMRP8-cre+ mix only (n=5 mice per group). B, Synovial fluid neutrophils were isolated at day 7 following serum transfer from a mixed chimeric mouse (containing roughly 50% sykf/−MRP8-cre+ and 50% wild type cells) then stained for CD45.1, CD45.2 (top panel) and Syk protein (bottom panel). Peripheral blood neutrophils from a syk−/− chimeric mouse were used to define Syk negative cells (data not shown). C, Peripheral blood and synovial neutrophils from mixed chimeric mice were stained for Ly5.1 versus Ly5.2 to determine the percentage of sykf/−MRP8-cre+ versus wild type cells in each compartment as shown in B. The percentage of sykf/−MRP8-cre+ (designated % Syk-) in peripheral blood versus synovial fluid is shown for each individual mouse (R2 = 0.96).
Syk deficiency affects specific effector functions in the inflamed joint
To determine the particular functional defects of Syk-deficient neutrophils in the K/BxN model, we induced arthritis in mixed chimeras to compare expression activation markers, apoptosis, and cytokine production by the two neutrophil types present in the same inflammatory joint environment. Syk-deficient neutrophils in the inflamed joint showed equivalent upregulation of CD11b and shedding of CD62L (Fig. 6A, B) compared to wild type cells in the same joint, indicating that Syk-deficiency did not reduce exocytosis of secretory granules (3, 42). Further, activation in the peripheral blood is unaffected by Syk deficiency, as wild type and Syk-deficient peripheral blood neutrophils from K/BxN serum treated chimeras equivalently shed CD62L compared to wild type neutrophils from untreated mice. However, a greater proportion of Syk-deficient neutrophils in the joint were undergoing cell death than wild type cells, as defined by being both Annexin V and propidium iodide (PI) positive (Fig. 6C). No difference was seen in the proportion of neutrophils in earlier stages of apoptosis, defined as Annexin V positive but PI negative (2, 43).
Figure 6.
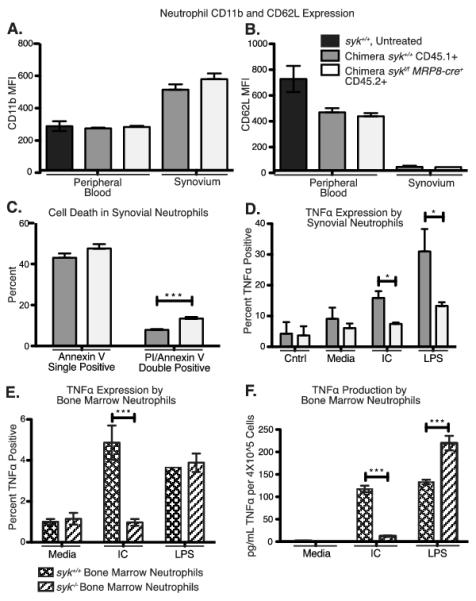
Altered cytokine production, but not activation marker expression, by Syk-deficient neutrophils in the arthritic joint. A, B, Ly6G+ Neutrophils were isolated from the peripheral blood and synovial fluid of arthritic mixed bone marrow chimeras on day 7 following K/BxN serum transfer and stained for CD11b and CD62L (L-selectin). Mean fluorescence intensity of CD11b and CD62L for Syk-deficient (CD45.2+, white bars) or wild type (CD45.1+ gray bars) neutrophils are shown compared to peripheral blood neutrophils from a control B6 mouse not treated with serum (black bars). C, Day 7 synovial fluid neutrophils from mixed chimeric mice were stained with Ly6G, annexin V and propidium iodide (PI) and the percentage of positive cells for each marker are shown. A-C, n=7. D, Day 7 synovial fluid neutrophils were pooled from 8 mixed chimeric mice and stained for intracellular TNFα as described in Materials and Methods following 6 hours of incubation with media or media plus immune complexes (indicated as IC) or media plus 10 ng/ml LPS. Wild type versus sykf/f MRP8-cre+ cells were distinguished by CD45.1 versus CD45.2 staining. E, F, Naïve bone marrow neutrophils were isolated from either wild type or syk−/− fetal liver chimeric mice and stimulated in vitro with the indicated agonists for 6 hours, then E, stained for intracellular TNFα or F, culture supernatant was collected for TNFα ELISA as described in Materials and Methods (n = 3). Statistics analyzed by two-way ANOVA. IC = insoluble immune complex. *=p<0.05, **=p<0.01, *** = p<0.001
Though early markers of neutrophil activation were intact, there was evidence that Syk-deficient neutrophils in the inflamed joint were less primed for late stage effector functions. The Syk-deficient neutrophils were less competent to induce TNFα production, as determined by intracellular staining following stimulation, compared to wild type cells (Fig. 6D). As expected, immune complex stimulation elicited TNFα production in wild type, but not Syk-deficient cells. Similarly, bone marrow derived Syk-deficient neutrophils failed to produce TNFα when stimulated with immune complexes (Fig. 6E, F). Surprisingly, Syk-deficient neutrophils from the inflamed joint did not respond to LPS as robustly as the wild type cells. In contrast, the percentage of naïve Syk-deficient neutrophils from the bone marrow that induced TNFα expression was equivalent to wild type bone marrow neutrophils (Fig. 6E), and on a per cell basis, they produced more TNFα in total, consistent with the increased TLR responses reported in Syk-deficient macrophages (Fig. 6F) (44). This indicates that the reduced TNFα production in Syk-deficient synovial neutrophils is not due to a general defect in responsiveness, but instead suggests that these cells are poorly primed in the inflamed joint. Since Syk is required for FcγR and integrin but not G-protein coupled receptor (GPCR) signaling (45), which would be a major inducer of cell migration, these data suggest that neutrophils are responding directly to immune complexes to mediate disease in this arthritis model.
Mast cells and basophils are not required for antibody-mediated arthritis
Previous research suggested that mast cells play a central role in the K/BxN disease process, since the c-kitW/Wv mast cell-deficient strain of mice are protected from disease (46). A general model for the progression of arthritis in K/BxN serum transfer has been recognition of joint immune complexes by resident mast cells followed by production of chemokines that lead to neutrophil recruitment and disease (8, 9, 16, 47). Given that we observed disease protection in mice with a neutrophil-specific mutation, we sought to re-evaluate the role mast cells. Indeed, we found that the c-kitSh/Sh strain of mast cell-deficient mice responded normally to K/BxN serum challenge (Fig. 7A). In contrast, the mast cell-deficient c-kitW/Wv strain showed only minimal response K/BxN serum challenge (Fig. 7B) as previously reported (46). We confirmed that both strains of mice lacked mast cells in the peritoneum (Fig. 7C), and joint (Fig. 7D). Mast cells were also readily detectable in the joints of Sykf/f MRP8-cre+ mice following K/BxN serum transfer (Fig. 7E), showing that this population was grossly unaffected by MRP8-cre-induced Syk deletion. We conclude that the lack of arthritis in c-kitW/Wv mice reflects the relative neutropenia and poor neutrophil recruitment found in these animals (48).
Figure 7.
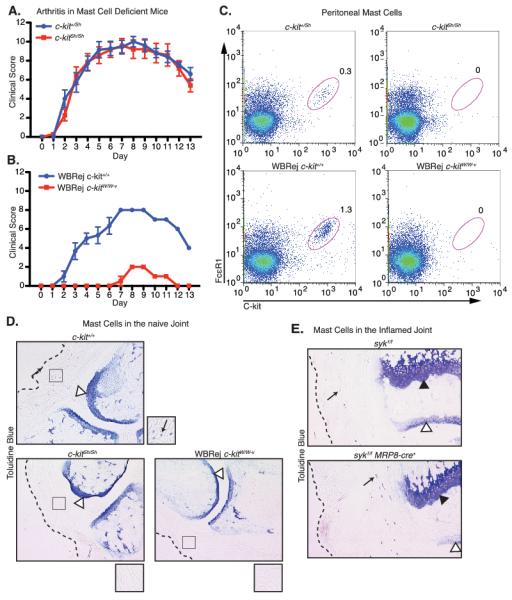
Mast cell deficiency does not affect the development or persistence of antibody-mediated arthritis. A, B, K/BxN serum was administered to c-kit+/Sh versus c-kitSh/Sh mice or wild-type versus c-kitW/W-v mice and disease was followed as described in Materials and Methods. C, Flow cytometric staining of resting peritoneal cells, stained for the mast cell markers C-kit and FcεR1 from c-kitSh/Sh, c-kitW/W-v and control c-kit+/+ or c-kit+/Sh mice. D, E, Toluidine blue staining as described in the Materials and Methods on D, forepaws from serum untreated c-kitSh/Sh, c-kitW/W-v and control mice, and E, hindpaws from sykf/f and sykf/f MRP8-cre+ mice 7 days after serum transfer. Magnified views of the regions highlighted by the squares are shown to the lower right of each panel. Dotted lines approximate the epidermis, arrows indicate mast cells, white arrowheads indicate cartilage, and black arrowheads indicate the epiphysial plate. Data shown are averaged from 3-5 mice per cohort.
Basophils have been recently shown to play a large role in immune complex-mediated inflammation (49). As some MRP8-cre expression, marked by GFP, was seen in splenic basophils (Supplemental Fig. 1), we sought to determine the effect of basophils on K/BxN serum-induced arthritis. For this purpose, we induced arthritis in basophil deficient mice, generated using a basophil-specific Cre under the Mcpt8 promoter (Basoph8+) crossed to mice containing the Rosa-flxDTα allele (Dtα+) (33). Basophil-deficient mice develop a course of arthritis identical to control littermates (Fig. 8A). Expression of Dtα in DX5+ basophils, which can be tracked by the ires-YFP construct within the Basoph8 allele, leads to apoptosis and depletion (Fig. 8B) (33). Combined with the results from the sykf/f MRP8-cre+ mice, we conclude that basophils are not required for the development of K/BxN serum-induced arthritis.
Figure 8.
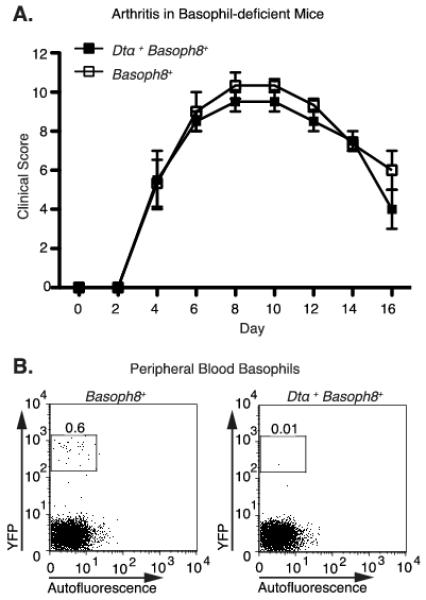
Basophil deficiency does not affect the development or persistence of antibody-mediated arthritis. A, Clinical score was recorded following K/BxN serum injection in mice expressing a basophil-specific YFP-ires-cre (Basoph8+) either with or without the floxed Rosa-flxDTα (Dtα+) gene. B, Representative dot plots from Cre-expressing mice. Peripheral blood basophils were identified as CD4− autofluorescentlo YFP+ by flow cytometry.
DISCUSSION
We have utilized the sykf/f MRP8-cre+ mice, which lack Syk in neutrophils, to examine the role of these cells in the innate immune-mediated effector stage of inflammatory arthritis. Since Syk kinase is required for FcγR-induced signaling, the neutrophils in these mice fail to respond to immune complexes. Loss of Syk signaling in neutrophils is sufficient to protect mice from K/BxN serum induced arthritis. Clinical swelling is greatly reduced or absent in the sykf/f MRP8-cre+ mice; the synovium of these animals is undisturbed, with no evidence of bone and cartilage damage. Sykf/f MRP8-cre+ mice also demonstrate decreased antibody accumulation along the cartilage and lower serum cytokine levels. Further, Syk signaling pathways in neutrophils are required at several stages of immune response, including early induction of vascular permeability. However, using mixed chimeras, it is clear that Syk is not required for neutrophil migration into the joint if inflammation is already established. Neither mast cells nor basophils are required for arthritis development following K/BxN serum challenge. These observations help re-define the pathogenesis of inflammatory arthritis in this model and emphasize the neutrophil dependence of this disease.
These conclusions are based on the neutrophil specificity of the MRP8-cre gene. Although monocytes/macrophages may express MRP8 (50), we found no significant deletion of Syk or upregulation of Cre expression in these cell types. Therefore, while we cannot completely rule out a contribution from syk deletion in monocytes/macrophages, it would be minor compared to neutrophil deletion.
The original model of disease progression in K/BxN arthritis postulates that tissue resident mast cells are required for the initial recognition of anti-GPI immune complexes, promoting vascular permeability and antibody deposition, and elaborating proinflammatory mediators leading to neutrophil recruitment, activation and tissue injury (8, 9, 16, 47). Indeed, mast cell-deficient c-kitW/Wv mice are protected from K/BxN serum induced arthritis, and disease can be restored by engraftment of wild type mast cells. However, we and others have found that the mast cell-deficient strain c-kitSh/Sh is susceptible to K/BxN serum induced arthritis and the similar anti-collagen mAb/LPS injection model (12, 48). The difference between the c-kitSh/Sh and c-kitW/Wv strains likely reflects the hematopoietic defect in the W/Wv mouse, which includes neutropenia (48). Together, these results show that mast cell Fc/Syk signaling is not required for the disease process as previously thought. In contrast, recent studies suggest a critical role for neutrophil FcγRs. Arthritis progresses normally in human-FcγRIIA transgenic mice that express FcγRs only on neutrophils (20), and in FcγRn/I/IIA/IIB/III/-deficient mice that express FcγRIV only on macrophages and neutrophils (12). In both these models FcγR expression is absent in all other cell lineages, and it is not known whether forcing Fc receptor expression in other cell types would have a compensatory effect on disease development. In contrast, FcγR signaling is intact in all cells except neutrophils in sykf/f MRP8-cre mice. In concert with the above studies, one can generally conclude that neutrophil recognition of immune complexes is the major driver of K/BxN serum induced arthritis.
By generating mixed bone marrow chimeras, we were able to directly compare the behavior of Syk-deficient and wild type neutrophils during inflammation. Combined with data from Sykf/f MRP8-cre+ mice, our results show that Syk deficiency has effects on specific pathways and mechanisms. Despite their expected inability to respond to immune complexes in vitro, in an in vivo inflammatory setting Syk-deficient neutrophils mobilize secretory granules normally, possibly in response to factors produced by wild type neutrophils (2), and migrate normally. Syk deficiency does impede the ability of neutrophils to promote vascular permeability and produce cytokines at the inflammatory site. Syk-deficient neutrophils also show enhanced/accelerated cell death.
Systemic immune complexes induce both acute (5-10 minutes after administration) and long term (hours to days) changes to the vascular endothelium that increase permeability and extravasation (19, 41). Arthritis can occur in the absence of acute vascular leak in the joint, though disease is slightly attenuated (41, 51). Notably, Fc receptor expression by neutrophils can rescue arthritis on an FcRγ−/− background, but does not rescue the vascular leak defect observable at 45 minutes post serum injection (20). This observation appears to be at odds with our finding that deletion of Syk in neutrophils causes a decrease in antibody deposition in the joint. However, our results in the cutaneous Arthus reaction show that edema in response to immune complexes is reduced in sykf/f MRP8-cre+ mice after several hours. We suggest that very early acute vascular changes occur through a separate mechanism than the more long-term process of edema, where the latter at least partially depends on IgG recognition by neutrophils.
Syk is also responsible for signaling through other neutrophil receptors that utilize ITAM-dependent signaling pathways, such as integrins, some C-type lectin receptors and cytokine receptors (52). Reduced integrin signaling may contribute to impaired neutrophil migration, as Syk-deficient neutrophils do show reduced recruitment in the cremaster muscle model following superfusion with chemoattractant peptides (53). However, we have found no defect in Syk-deficient neutrophil recruitment in thioglycollate induced sterile peritonitis (54), hemorrhagic vasculitis in the skin (55) or as shown here in the K/BxN model of arthritis. We conclude that the loss of integrin signaling in Syk-deficient neutrophils has variable effects on cellular migration dependent on the inflammation model. Whether Syk deficiency would alter cellular recruitment in other tissue sites remains to be tested.
Since sykf/f MRP8-cre+ mice are protected from arthritis despite normal migration in Syk-deficient neutrophils, the block in inflammation likely occurs upstream of substantial neutrophil recruitment. Similar findings were reported with mice lacking BLT1, the LTB4 receptor, in which reconstitution with wild type neutrophils induced arthritis and the recruitment of BLT1−/− neutrophils to the joint (19). We propose that Syk-dependent signaling in neutrophils is required for the elaboration of chemokines and cytokines, such as TNFα and LTB4, which induce further recruitment of additional monocytes and neutrophils, leading to more cytokine production and tissue damage, in the fashion of a self-amplifying loop. Activation of tissue resident cells alone is not sufficient to induce significant neutrophil recruitment in sykf/f MRP8-cre+ mice, even though Syk-deficient neutrophils could otherwise migrate into the joint if a sufficient inflammatory signal was present. Undoubtedly, the majority of neutrophil activation occurs in response to tissue-deposited immune complexes. However, the decreased antibody deposition in joints of sykf/f MRP8-cre+ mice suggests that at least part of the neutrophil activation occurs in the peripheral blood.
The role of Fc receptors in human autoimmune disease is complex, since these molecules mediate both activating and inhibitory signaling. Hypomorphic alleles of the human FcγRIIA, RIIIA, and RIIIB are associated with increased disease severity and nephritis in patients with SLE (56, 57). These hypoactive FcγRs could result in decreased immune complex clearance, paradoxically leading to accumulation of IgGs in tissues that would mediate chronic immune cell activation (58). Syk is therefore at attractive therapeutic target, as it is required for signaling through all FcγRs. Indeed, Syk inhibitors are efficacious in multiple animal models of autoimmune arthritis and SLE; phase II clinical trials with rheumatoid arthritis patients show promise (59). Our data suggests that part of the efficacy of Syk inhibitors could stem from the inhibition of immune-complex induced activating signals in innate immune cells (7, 20, 26).
Overall, these results suggest that signaling through Syk in neutrophils is the major mediator of arthritis in the K/BxN model, while immune complex recognition by other cells, in particular mast cells and basophils, plays a less important role in disease development. In combination with similar findings in immune complex nephritis (7), this suggests that neutrophils are the dominant pathogenic cell in most immune complex-mediated diseases. Obviously, this hypothesis will require further testing, but it does significantly alter the pathogenic models of immune complex inflammation, particularly diminishing the role of mast cells. This would have direct implications in the development of cell-targeted therapeutics for treatment of immune complex disease.
Supplementary Material
ACKNOWLEDGMENTS
We thank Clare Abram and Lynn Kamen for suggestions and comments. Additionally we thank the A. Luster (Harvard) lab for their IgG staining protocol, as well as the S. Rosen (UCSF) lab for help with the crytostat tissue sectioning. We also thank the A. Weiss (UCSF) lab for use of the 5F5.2 anti-mouse Syk hybridoma.
This work was supported by the National Institutes of Health (AI65495 and AI68150 to C.A.L.) and the NSF Graduate Research Fellowship Program (to E.R.E.).
Abbreviations used in this paper
- GPI
glucose-6 phosphate isomerase
- i.d.
intradermally
- LTB4; ITAM
immunoreceptor tyrosine-based activation motifs; leukotriene B4
- MRP8
myeloid-related protein 8, also known as S100A8
- LysM
lysozyme M gene
- NTS
nephrotoxic serum
- polyI:C
polyinosinic:polycytidylic acid
- RA
rheumatoid arthritis
- Syk
spleen tyrosine kinase
Footnotes
The authors have no conflicting financial interests.
REFERENCES
- 1.Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33:657–670. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 2.Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010;10:427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- 3.Van Ziffle JA, Lowell CA. Neutrophil-specific deletion of Syk kinase results in reduced host defense to bacterial infection. Blood. 2009;114:4871–4882. doi: 10.1182/blood-2009-05-220806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cassatella MA, Locati M, Mantovani A. Never underestimate the power of a neutrophil. Immunity. 2009;31:698–700. doi: 10.1016/j.immuni.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Cascao R, Rosario HS, Souto-Carneiro MM, Fonseca JE. Neutrophils in rheumatoid arthritis: More than simple final effectors. Autoimmun. Rev. 2010;9:531–535. doi: 10.1016/j.autrev.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 6.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 7.Tsuboi N, Asano K, Lauterbach M, Mayadas TN. Human neutrophil Fcγ receptors initiate and play specialized nonredundant roles in antibody-mediated inflammatory diseases. Immunity. 2008;28:833–846. doi: 10.1016/j.immuni.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ditzel HJ. The K/BxN mouse: a model of human inflammatory arthritis. Trends. Mol. Med. 2004;10:40–45. doi: 10.1016/j.molmed.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Asquith DL, Miller AM, McInnes IB, Liew FY. Animal models of rheumatoid arthritis. Eur. J. Immunol. 2009;39:2040–2044. doi: 10.1002/eji.200939578. [DOI] [PubMed] [Google Scholar]
- 10.Matsumoto I, Maccioni M, Lee DM, Maurice M, Simmons B, Brenner M, Mathis D, Benoist C. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat. Immunol. 2002;3:360–365. doi: 10.1038/ni772. [DOI] [PubMed] [Google Scholar]
- 11.Solomon S, Rajasekaran N, Jeisy-Walder E, Snapper SB, Illges H. A crucial role for macrophages in the pathology of K/B x N serum-induced arthritis. Eur. J. Immunol. 2005;35:3064–3073. doi: 10.1002/eji.200526167. [DOI] [PubMed] [Google Scholar]
- 12.Mancardi DA, Jonsson F, Iannascoli B, Khun H, Van Rooijen N, Huerre M, Daeron M, Bruhns P. The murine high-affinity IgG receptor FcγRIV is sufficient for autoantibody-induced arthritis. J. Immunol. 2011;186:1899–1903. doi: 10.4049/jimmunol.1003642. [DOI] [PubMed] [Google Scholar]
- 13.Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF, Lee DM. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J. Exp. Med. 2006;203:837–842. doi: 10.1084/jem.20052371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji H, Pettit A, Ohmura K, Ortiz-Lopez A, Duchatelle V, Degott C, Gravallese E, Mathis D, Benoist C. Critical roles for interleukin 1 and tumor necrosis factor α in antibody-induced arthritis. J. Exp. Med. 2002;196:77–85. doi: 10.1084/jem.20020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kneilling M, Hultner L, Pichler BJ, Mailhammer R, Morawietz L, Solomon S, Eichner M, Sabatino J, Biedermann T, Krenn V, Weber WA, Illges H, Haubner R, Rocken M. Targeted mast cell silencing protects against joint destruction and angiogenesis in experimental arthritis in mice. Arthritis. Rheum. 2007;56:1806–1816. doi: 10.1002/art.22602. [DOI] [PubMed] [Google Scholar]
- 16.Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. 2002;297:1689–1692. doi: 10.1126/science.1073176. [DOI] [PubMed] [Google Scholar]
- 17.Shin K, Nigrovic PA, Crish J, Boilard E, McNeil HP, Larabee KS, Adachi R, Gurish MF, Gobezie R, Stevens RL, Lee DM. Mast cells contribute to autoimmune inflammatory arthritis via their tryptase/heparin complexes. J. Immunol. 2009;182:647–656. doi: 10.4049/jimmunol.182.1.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J. Immunol. 2001;167:1601–1608. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 19.Kim ND, Chou RC, Seung E, Tager AM, Luster AD. A unique requirement for the leukotriene B4 receptor BLT1 for neutrophil recruitment in inflammatory arthritis. J. Exp. Med. 2006;203:829–835. doi: 10.1084/jem.20052349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsuboi N, Ernandez T, Li X, Nishi H, Cullere X, Mekala D, Hazen M, Kohl J, Lee DM, Mayadas TN. Human neutrophil FcγRIIA regulation by C5aR promotes inflammatory arthritis in mice. Arthritis. Rheum. 2011;63:467–478. doi: 10.1002/art.30141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berton G, Mocsai A, Lowell CA. Src and Syk kinases: key regulators of phagocytic cell activation. Trends Immunol. 2005;26:208–214. doi: 10.1016/j.it.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Underhill DM, Goodridge HS. The many faces of ITAMs. Trends Immunol. 2007;28:66–73. doi: 10.1016/j.it.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Wong BR, Grossbard EB, Payan DG, Masuda ES. Targeting Syk as a treatment for allergic and autoimmune disorders. Expert Opin. Investig. Drugs. 2004;13:743–762. doi: 10.1517/13543784.13.7.743. [DOI] [PubMed] [Google Scholar]
- 24.Abtahian F, Guerriero A, Sebzda E, Lu MM, Zhou R, Mocsai A, Myers EE, Huang B, Jackson DG, Ferrari VA, Tybulewicz V, Lowell CA, Lepore JJ, Koretzky GA, Kahn ML. Regulation of blood and lymphatic vascular separation by signaling proteins SLP-76 and Syk. Science. 2003;299:247–251. doi: 10.1126/science.1079477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bohmer R, Neuhaus B, Buhren S, Zhang D, Stehling M, Bock B, Kiefer F. Regulation of developmental lymphangiogenesis by Syk(+) leukocytes. Dev. Cell. 2010;18:437–449. doi: 10.1016/j.devcel.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Jakus Z, Simon E, Balazs B, Mocsai A. Genetic deficiency of Syk protects mice from autoantibody-induced arthritis. Arthritis. Rheum. 2010;62:1899–1910. doi: 10.1002/art.27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saijo K, Schmedt C, Su IH, Karasuyama H, Lowell CA, Reth M, Adachi T, Patke A, Santana A, Tarakhovsky A. Essential role of Src-family protein tyrosine kinases in NF-κB activation during B cell development. Nat. Immunol. 2003;4:274–279. doi: 10.1038/ni893. [DOI] [PubMed] [Google Scholar]
- 28.Passegue E, Wagner EF, Weissman IL. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell. 2004;119:431–443. doi: 10.1016/j.cell.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Kiel MJ, Acar M, Radice GL, Morrison SJ. Hematopoietic stem cells do not depend on N-cadherin to regulate their maintenance. Cell. Stem. Cell. 2009;4:170–179. doi: 10.1016/j.stem.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 31.Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, Voehringer D. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J. Exp. Med. 2009;206:549–559. doi: 10.1084/jem.20082394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 2005;167:835–848. doi: 10.1016/S0002-9440(10)62055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sullivan BM, Liang HE, Bando JK, Wu D, Cheng LE, McKerrow JK, Allen CD, Locksley RM. Genetic analysis of basophil function in vivo. Nat. Immunol. 2011;12:527–535. doi: 10.1038/ni.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scapini P, Hu Y, Chu CL, Migone TS, Defranco AL, Cassatella MA, Lowell CA. Myeloid cells, BAFF, and IFN-gamma establish an inflammatory loop that exacerbates autoimmunity in Lyn-deficient mice. J. Exp. Med. 2010;207:1757–1773. doi: 10.1084/jem.20100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimizu K, Bae SJ, Hara T, Iwata Y, Yamaoka T, Komura K, Muroi E, Takenaka M, Ogawa F, Sato S. Involvement of gaseous low molecular monoxides in the cutaneous reverse passive Arthus reaction: cytoprotective action of carbon monoxide. Clin. Exp. Immunol. 2008;153:245–257. doi: 10.1111/j.1365-2249.2008.03688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cassatella MA, Pereira-da-Silva G, Tinazzi I, Facchetti F, Scapini P, Calzetti F, Tamassia N, Wei P, Nardelli B, Roschke V, Vecchi A, Mantovani A, Bambara LM, Edwards SW, Carletto A. Soluble TNF-like cytokine (TL1A) production by immune complexes stimulated monocytes in rheumatoid arthritis. J. Immunol. 2007;178:7325–7333. doi: 10.4049/jimmunol.178.11.7325. [DOI] [PubMed] [Google Scholar]
- 37.Cailhier JF, Partolina M, Vuthoori S, Wu S, Ko K, Watson S, Savill J, Hughes J, Lang RA. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J. Immunol. 2005;174:2336–2342. doi: 10.4049/jimmunol.174.4.2336. [DOI] [PubMed] [Google Scholar]
- 38.Luche H, Weber O, Rao T. Nageswara, Blum C, Fehling HJ. Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. Eur. J. Immunol. 2007;37:43–53. doi: 10.1002/eji.200636745. [DOI] [PubMed] [Google Scholar]
- 39.Colonna L, Catalano G, Chew C, D’Agati V, Thomas JW, Wong FS, Schmitz J, Masuda ES, Reizis B, Tarakhovsky A, Clynes R. Therapeutic targeting of Syk in autoimmune diabetes. J. Immunol. 2010;185:1532–1543. doi: 10.4049/jimmunol.1000983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu HJ, Sawaya H, Binstadt B, Brickelmaier M, Blasius A, Gorelik L, Mahmood U, Weissleder R, Carulli J, Benoist C, Mathis D. Inflammatory arthritis can be reined in by CpG-induced DC-NK cell cross talk. J. Exp. Med. 2007;204:1911–1922. doi: 10.1084/jem.20070285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lister KJ, James WG, Hickey MJ. Immune complexes mediate rapid alterations in microvascular permeability: roles for neutrophils, complement, and platelets. Microcirculation. 2007;14:709–722. doi: 10.1080/10739680701404879. [DOI] [PubMed] [Google Scholar]
- 42.Borregaard N, Sorensen OE, Theilgaard-Monch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol. 2007;28:340–345. doi: 10.1016/j.it.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 43.Scatizzi JC, Hutcheson J, Pope RM, Firestein GS, Koch AE, Mavers M, Smason A, Agrawal H, Haines GK, 3rd, Chandel NS, Hotchkiss RS, Perlman H. Bim-Bcl-2 homology 3 mimetic therapy is effective at suppressing inflammatory arthritis through the activation of myeloid cell apoptosis. Arthritis. Rheum. 2010;62:441–451. doi: 10.1002/art.27198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hamerman JA, Tchao NK, Lowell CA, Lanier LL. Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat. Immunol. 2005;6:579–586. doi: 10.1038/ni1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mocsai A, Zhang H, Jakus Z, Kitaura J, Kawakami T, Lowell CA. G-protein-coupled receptor signaling in Syk-deficient neutrophils and mast cells. Blood. 2003;101:4155–4163. doi: 10.1182/blood-2002-07-2346. [DOI] [PubMed] [Google Scholar]
- 46.Nigrovic PA, Binstadt BA, Monach PA, Johnsen A, Gurish M, Iwakura Y, Benoist C, Mathis D, Lee DM. Mast cells contribute to initiation of autoantibody-mediated arthritis via IL-1. Proc. Natl. Acad. Sci. USA. 2007;104:2325–2330. doi: 10.1073/pnas.0610852103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benoist C, Mathis D. Mast cells in autoimmune disease. Nature. 2002;420:875–878. doi: 10.1038/nature01324. [DOI] [PubMed] [Google Scholar]
- 48.Zhou JS, Xing W, Friend DS, Austen KF, Katz HR. Mast cell deficiency in Kit(W-sh) mice does not impair antibody-mediated arthritis. J. Exp. Med. 2007;204:2797–2802. doi: 10.1084/jem.20071391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karasuyama H, Mukai K, Obata K, Tsujimura Y, Wada T. Nonredundant roles of basophils in immunity. Annu Rev Immunol. 2011;29:45–69. doi: 10.1146/annurev-immunol-031210-101257. [DOI] [PubMed] [Google Scholar]
- 50.Xu K, Geczy CL. IFNγ and TNF regulate macrophage expression of the chemotactic S100 protein S100A8. J. Immunol. 2000;164:4916–4923. doi: 10.4049/jimmunol.164.9.4916. [DOI] [PubMed] [Google Scholar]
- 51.Binstadt BA, Patel PR, Alencar H, Nigrovic PA, Lee DM, Mahmood U, Weissleder R, Mathis D, Benoist C. Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat. Immunol. 2006;7:284–292. doi: 10.1038/ni1306. [DOI] [PubMed] [Google Scholar]
- 52.Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat. Rev. Immunol. 2010;10:387–402. doi: 10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schymeinsky J, Sindrilaru A, Frommhold D, Sperandio M, Gerstl R, Then C, Mocsai A, Scharffetter-Kochanek K, Walzog B. The Vav binding site of the non-receptor tyrosine kinase Syk at Tyr 348 is critical for β2 integrin (CD11/CD18)-mediated neutrophil migration. Blood. 2006;108:3919–3927. doi: 10.1182/blood-2005-12-030387. [DOI] [PubMed] [Google Scholar]
- 54.Mocsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity. 2002;16:547–558. doi: 10.1016/s1074-7613(02)00303-5. [DOI] [PubMed] [Google Scholar]
- 55.Hirahashi J, Mekala D, Van Ziffle J, Xiao L, Saffaripour S, Wagner DD, Shapiro SD, Lowell C, Mayadas TN. Mac-1 signaling via Src-family and Syk kinases results in elastase-dependent thrombohemorrhagic vasculopathy. Immunity. 2006;25:271–283. doi: 10.1016/j.immuni.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 56.Edberg JC, Langefeld CD, Wu J, Moser KL, Kaufman KM, Kelly J, Bansal V, Brown WM, Salmon JE, Rich SS, Harley JB, Kimberly RP. Genetic linkage and association of Fcγ receptor IIIA (CD16A) on chromosome 1q23 with human systemic lupus erythematosus. Arthritis. Rheum. 2002;46:2132–2140. doi: 10.1002/art.10438. [DOI] [PubMed] [Google Scholar]
- 57.Kelley JM, Edberg JC, Kimberly RP. Pathways: Strategies for susceptibility genes in SLE. Autoimmun. Rev. 2010;9:473–476. doi: 10.1016/j.autrev.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, Mangion J, Roberton-Lowe C, Marshall AJ, Petretto E, Hodges MD, Bhangal G, Patel SG, Sheehan-Rooney K, Duda M, Cook PR, Evans DJ, Domin J, Flint J, Boyle JJ, Pusey CD, Cook HT. Copy number polymorphism in Fcγr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 59.Genovese MC, Kavanaugh A, Weinblatt ME, Peterfy C, Dicarlo J, White ML, M OB, Grossbard EB, Magilavy DB. An oral syk kinase inhibitor in the treatment of rheumatoid arthritis: A 3 month randomized placebo controlled phase 2 study in patients with active RA who had failed biologic agents. Arthritis. Rheum. 2011;63:337–345. doi: 10.1002/art.30114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.