

Abstract
Background & Aims
Patients with inflammatory bowel disease who are in remission and those that developed inflammatory bowel syndrome after enteric infection continue to have symptoms of diarrhea or constipation in the absence of overt inflammation, indicating motility dysfunction. We investigated whether oxidative stress during inflammation impairs integrity of the promoter of Cacna1c, which encodes the pore-forming α1C subunit of Cav1.2b calcium channels.
Methods
We used long-extension PCR (LX-PCR) to evaluate DNA integrity in tissues from distal colons of rats; trinitrobenzene sulfonic acid (TNBS) was used to induce inflammation.
Results
H2O2 increased in the muscularis externa 1 to 7 days after inflammation was induced with TNBS. The oxidative stress significantly impaired DNA integrity in 2 specific segments of the Cacna1c promoter: −506 to −260 and −2,193 to −1,542. The impairment peaked at day 3 and recovered partially by day 7 after induction of inflammation; expression of the products of Cacna1c followed a similar time course. Oxidative stress suppressed the expression of Nrf2, an important regulator of anti-oxidant proteins. Intra-peritoneal administration of sulforaphane significantly reversed the suppression of Nrf2, oxidative damage in the promoter of Cacna1c, and suppression of Cacna1c on day 7 of inflammation. The inflammation subsided completely by 56 days after inflammation was induced; however, impairment of DNA integrity, expression of Nrf2 and Cacna1c, and smooth muscle reactivity to acetylcholine remained suppressed at this timepoint.
Conclusion
Oxidative stress during inflammation impairs the integrity of the promoter of Cacna1c; impairment persists partially after inflammation has subsided. Reduced transcription of Cacna1c contributes to smooth muscle dysfunction in the absence of inflammation.
Keywords: IBD, IBS, gastrointestinal inflammation, DNA repair, damage, mutation
Oxidative stress induced by aerobic metabolism continually causes about 100,000 to 1,000,000 DNA lesions in cells of an organism per day1. DNA repair enzymes detect and fix these lesions (primarily base excision) to maintain homeostasis2. In addition, pathogenic DNA damage occurs when reactive oxygen and reactive nitrogen species generated due to inflammation, UV radiation, pollutants, dietary components, overwhelm the endogenous repair mechanisms and impair genomic integrity. The susceptibility to these lesions and the capability of repair mechanisms to fix them are cell-type and DNA sequence-specific3, 4. The accumulation of DNA lesions is deleterious to cellular functions and organism’s survivability. Depending upon the location of the lesions in the genome, the loss of DNA integrity might result in the production of mutant/inactive proteins, causing senescence, apoptosis, unregulated cell division, or aberrant transcription of genes5. The failure of cells to maintain DNA integrity is a major contributory factor to diseases, such as cancer, Alzheimer’s disease and cystic fibrosis6, 7.
Numerous studies show that persistent or recurring inflammation in the gut- H. pylori infection, ulcerative colitis and reflux esophagitis- leads to cancer in the rapidly dividing epithelial cells8, 9. However, we know very little about the effects of inflammation on DNA integrity in terminally differentiated cells, such as smooth muscle cells in the gut. There is evidence of long-term impairment of their function after inflammation has subsided in at least two prominent conditions. 1) A sizable percent of IBD patients during remission have the classic symptoms of irritable bowel syndrome (IBS) (diarrhea/constipation and intermittent abdominal cramping)10–14. 2) Severe enteric infection, individually or in a community setting accompanied, with comorbid psychological conditions, results in IBS-like symptoms in a subset of the affected subjects (post-infectious IBS; PI-IBS)15–20. In both conditions, the classic inflammatory response is absent when the patients have these symptoms.
Some reports indicate, however, that the colonic mucosal biopsies in these patients show a low-grade inflammation comprised of an increase in T lymphocyte count in the lamina propria and/or epithelium16, 21 and increase in IL-1β mRNA in rectal mucosa22. These studies proposed that the low-grade mucosal inflammation is the cause of IBS-like symptoms – abnormal bowel function. However, there is no known mechanism by which inflammation in mucosal cells could alter the function of smooth muscle and enteric neuronal cells located at the other end of the colon wall. Significant inflammatory infiltrate in the microenvironment of the cells is required to alter their function.
Clinical and experimental studies show that oxidative stress is transmural in IBD23, 24. In this study, we tested a novel hypothesis that oxidative stress during active inflammation in the muscularis externa impairs the integrity of DNA in colonic smooth muscle cells. The impairment of DNA integrity persists after resolution of oxidative stress/inflammation, which causes persistent smooth muscle dysfunction. We tested this hypothesis by inducing inflammation in the rat colon with TNBS and examining the expression of Cacna1c, which encodes the pore-forming α1C-subunit of Cav1.2b (L-type) calcium channels in smooth muscle cells. Previous studies found that alterations in the expression of these channels play a significant role in smooth muscle dysfunction24–26. We found that oxidative stress generated by the inflammatory response causes DNA damage, which is not fully repaired for at least 56 days after the insult, resulting in smooth muscle dysfunction. The DNA damage persists in the absence of any overt inflammation/oxidative stress at that time.
Methods
Induction of Colonic Inflammation
Six to eight week old male adult rats were fasted for 24 hour before the induction of inflammation. During this period, regular water was replaced with Colyte® to cleanse the colon. Colonic inflammation was induced by intraluminal administration of TNBS (68 mg/kg in 250μl), dissolved in 40% ethanol. Age matched control rats received 0.9% saline. The rats were euthanized 1, 3, 7, 28, and 56 days after induction of inflammation.
Long extension PCR (LX-PCR), rolling LX-PCR and real-time PCR
Tables 1 and 2 in supplement materials show all primers. The primers for LX-PCR were designed to amplify the promoter of Cacna1c from −3,053 to +301 and promoter of β-actin gene from −2,480 to +288. Primers for rolling LX-PCR were designed to amplify 6 partially overlapping segments in Cacna1c promoter. The LX-PCR and rolling LX-PCR were performed with 15 ng genomic DNA, 400 nM primers, 300 μM dNTPs, and 5 units of LongAmp DNA polymerase (New England BioLabs, Ipswich, MA) in total volume of 50 ul. The PCR amplification was carried out in 30 cycles as follows; denaturation for 15 seconds at 94°C, 30 seconds at 58°C, and 2 minutes at 68°C. After completion of the cycles, the reaction mixtures were incubated for 7 minutes at 72°C. The PCR products were separated by electrophoresis on 0.8% agarose gels with ethidium bromode and visualized on a UV-transluminator.
Amplification of 235 bp long mitochondrial genomic DNA served as loading control
The level of mRNA for each gene was measured with SYBR-Green– based real-time PCR. The cDNAs were diluted 1:10 with RNase/DNase free water to avoid saturation effect of template during PCR amplification. Each cDNA sample (7 ul of diluted cDNA) was amplified by using SYBR-Green PCR Master Mix according to the manufacturer’s instructions. PCR amplification was performed with Step-One Plus Real-time PCR system (Applied Biosystems, Austin, TX). The level of the housekeeping gene Rpl32 (L32) in each sample was used as an internal control.
Immunofluorescence staining
Immunofluorescence staining was performed using 4 micron thick sections of formalin-fixed parrafin embedded tissues. After deparaffinization by sequential incubation in xylene and 100%, 95% and 85% ethanol, the samples were cooked with antigen retrieval buffer in a steamer for antigen retrieval. The samples were blocked with 10% donkey serum for 1 hour at room temperature. For co-immunofluorescence staining, the sections were incubated with anti-γH2AX antibody and Cy3™-conjugated smooth muscle α-actin overnight at 4°C in the dark, following incubation with Alexa488 anti-rabbit IgG for 1 h at room temperature in the dark. The sections were examined under fluorescence microscope.
MPO, H2O2 and DNA Abasic Site Quantification
For MPO assay, 100 mg of muscularis externae were homogenized in 20 mM phosphate buffer (pH 7.4) and centrifuged at 4°C for 10 minutes. The pellet was sonicated in 50 mM phosphate buffer (pH 6) containing 0.5% hexadecyl trimethyl ammonium bromide (HTAB) and centrifuged at 4°C for 5 minutes. The supernatant was used for MPO assay by incubation of 100 μl supernatant with 16 mM tetramethyl benzidine (TMB) in 50% ethanol, 0.3 mM H2O2 and 8 mM sodium phosphate buffer (pH 5.4) for 3 minutes. The level of MPO was measured by reading the absorbance at 655 nm in plate reader. Abasic site quantification in genomic DNA was assayed with DNA Damage Quantification Kit -AP site Counting- (Dojindo Molecular Tech, Gainthersburg, ND) by following manufacturer’s instructions. The level of hydrogen peroxide was measured with BIOXYTECH H2O2-560 Quantitative Hydrogen Peroxide Assay Kit (Cell biolabs Inc, Portland, OR) by following manufacturer’s instructions.
Results
Inflammation impairs DNA integrity in the promoter region of Cacna1c
TNBS insult significantly increased the expression of GADD45G, a marker for DNA damage, on days 1, 3 and 7 of inflammation (Fig. 1A). LX-PCR showed time-dependent impairment of integrity of the promoter region (−3053/+301) of Cacna1c. The DNA damage (strand breaks) reached the maximum on day-3 of inflammation – and recovered partially by day-7 (Fig.1B). The integrity of the full Cacna1c promoter on day 56 post-inflammation was not different from that of age-matched controls. We used age-matched controls to avoid the possibility of any age-related changes in DNA integrity. Inflammation did not impair integrity of the housekeeping β-actin gene (Actb) (Fig.1C). In addition, TNBS insult did not cause DNA damage in the promoters of genes encoding CPI-17 and MLCK genes on day 1, 3 or 7 post-TNBS insult, suggesting specific damage to the Cacna1c (Fig. S2, supplement). We validated the LX-PCR method by using it to determine integrity of the mitochondrial genomic DNA in response to treatment of muscularis externae strips with 200 μM H2O2 for 30 minutes27. H2O2 decreased the DNA integrity of mitochondrial DNA to 43 ± 0.5 % of control, p<0.05 (Fig. 1D).
Fig. 1.

TNBS inflammation impairs integrity of DNA in the promoter of Cacna1c and suppresses the expression of α1C subunit. (A) TNBS inflammation enhanced the mRNA expression of GDD45G, an indicator of DNA damage, from day 1 to day 7. (B) LX-PCR showed DNA integrity of the entire Cacna1c promoter decreased until day 3 and then started to recover. The DNA integrity of the whole promoter was completely restored by day 56 post-inflammation vs. age-matched controls (C) Inflammation did not affect integrity of the housekeeping β-actin gene. (D) We established the validity of LX-PCR by detecting impairment of mitochondrial DNA integrity due to treatment of muscularis externae tissue with 200 μM H2O2 for 30 minutes. (E) and (F) The mRNA and protein expressions of the Cacna1c bottomed out on Day-3 post-inflammation and recovered partially on day 7 post-inflammation. The gene products remained suppressed on day 56 post-inflammation, compared with age-matched controls. Mit sm = small amplicon of mitochondrial DNA, Mit lg = large amplicon of mitochondrial DNA. N = 4 to 6, *p< 0.05 vs. controls, #p<0.05 vs. age-matched control.
We then investigated whether inflammation impaired integrity of the entire promoter randomly or targeted specific segments, by dividing the ~3 kB (−3,053/+301) Cacna1c promoter into six slightly overlapping segments (Fig. 2A). We could not achieve uniform overlaps due to the presence of significant GC-rich sequences in segments 4 and 5. LX-PCR of individual segments on day-3 of inflammation showed damage of only segments three (−2,193/−1,542) and five (−506/−42) (Figs. 2B to 2D: data not shown for segments 2, 4, and 6). Segment six (−260/+301), which overlaps with segment 5 (−506/−42), showed no drop in the efficiency of PCR amplification. Therefore, we narrowed down the susceptible portion of DNA segment five to −506/−260 bps. Segment 3 had no overlap with its adjacent segments.
Fig. 2.
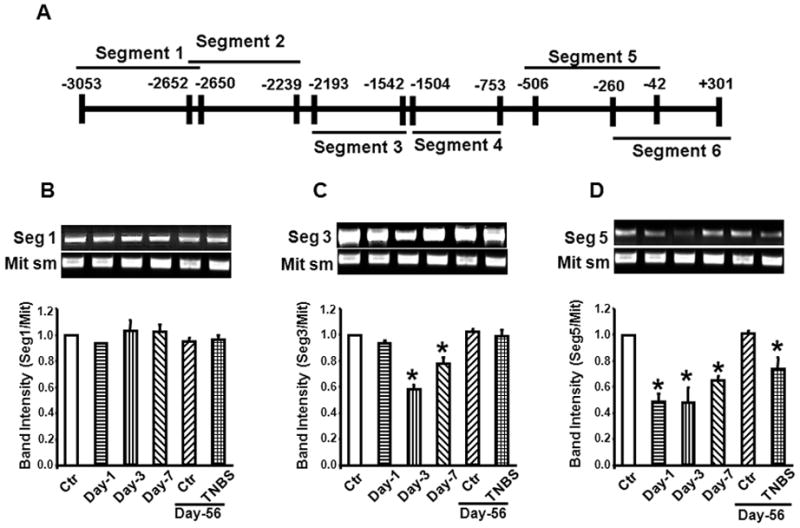
TNBS inflammation induces strand breaks in specific sequences of the Cacna1c promoter. (A) The loci of six sequences in Cacna1c promoter chosen for LXC-PCR. (B) Inflammation did not affect integrity of DNA in segments 1, 2, 4 and 6 at any time during 56 days after induction of inflammation. (Data not shown for segments 2, 4 and 6). (C) and (D) Inflammation impaired integrities of segments 3 and 5 that peaked on day-3 and recovered partially by day 7 post-inflammation. The DNA strand breaks in segment 3 was completely repaired by day 56, while segment 5 was partially repaired. Mit sm= small amplicon of mitochondrial DNA. N = 4 to 10, * p<0.05 vs. controls.
Loss of integrity of DNA in segment 5 was more rapid than in segment 3 (Figs. 2C and 2D). On the other hand, restoration of DNA integrity in segment 5 was slower than in segment 3, suggesting greater vulnerability of segment 5 than segment 3. Specifically, segment 3 DNA was fully repaired by day-56 post-inflammation, while the integrity of segment 5 was significantly less than in age matched controls (Figs. 2C and 2D).
The impairment of mRNA and protein expression of the Cacna1c followed similar time-course as the promoter (Figs. 1E and 1F). We noted that protein and mRNA levels of Cacna1c in the older control rats on day-56 post-inflammation were significantly greater than in younger control rats at the time of induction of inflammation. However, the protein and mRNA expressions of Cacna1c remained significantly suppressed on day-56 post-inflammation, compared with age-matched controls. In accord with this, we found that contractile response to ACh on day-56 was significantly suppressed, compared with age-matched controls (Fig. 3). In addition, the contractile response to 60 mM KCl, which depolarizes the colonic smooth muscle membrane to induce calcium influx through the Cav1.2b channels, was also smaller than that in age-matched controls.
Fig. 3.

The contractile response of circular smooth muscle strips to ACh (Fig. 3A) and to membrane depolarization by KCl (Fig. 3B) remains suppressed 56 day post-inflammation. *p<0.05 vs. controls N = 4, * p<0.05 vs. controls.
DNA base excision and strand break repair
The global level of apurinic/apyrimidinic (AP) sites, an indicator of base excision repair intermediate and DNA strand breaks was determined in genomic DNA extracted from the muscularis externa of the distal colon of TNBS rats on day-3 of inflammation and was found to be significantly greater than that in control rats (Fig. 4A). Immunofluorescence and Western blotting revealed significantly greater phosphorylated γ-H2AX level, an indicator of genomic double -strand breaks, in smooth muscle cells (Figs. 4B and 4C). We investigated whether there were temporal differences in the expression of genes involved in base excision and double strand break repair mechanisms (Supplement Fig. 1). The expression of genes for both repair mechanisms increased similarly from day 1 to day 7 of inflammation.
Fig. 4.
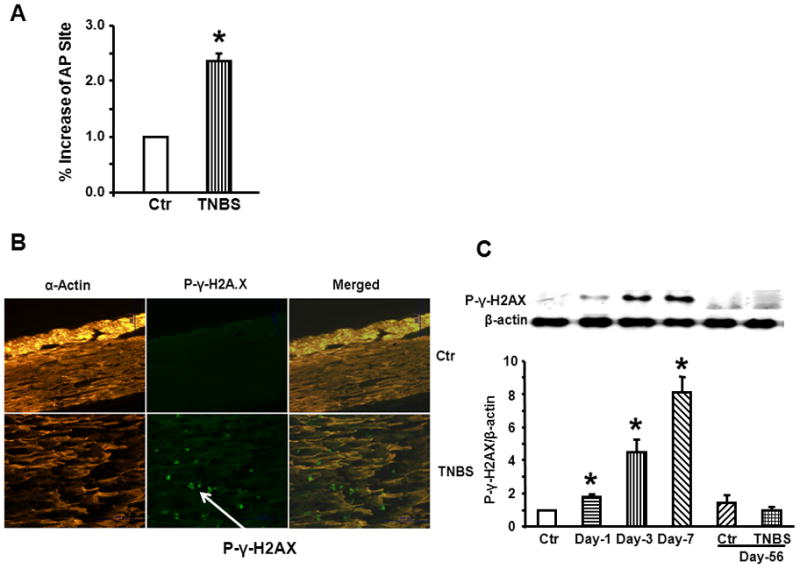
TNBS inflammation significantly increased the markers of DNA damage on day 3 of inflammation. (A) The global level of apurinic/apyrimidinic (AP) sites, an indicator of base excision in genomic DNA, increased significantly on day-3 of inflammation in the muscularis externae tissues. (B) and (C) Immunofluorescence and western blotting revealed significantly greater phosphorylated γ-H2AX, indicator of double strand break, in smooth muscle cells. N = 4 to 6, *p<0.05 vs. controls.
Oxidative stress and impairment of DNA integrity
Hydrogen peroxide and myeloperoxidase activities in the muscularis externa peaked on day-1 of inflammation and declined thereafter. However, both activities remained significantly greater than in naïve controls on day-7 of inflammation. These activities did not differ between animals subjected to TNBS inflammation and their age-matched controls on day-56 post inflammation (Figs. 5A and 5B).
Fig. 5.

TNBS inflammation increased the markers of oxidative stress in the muscularis externae tissues, (A) MPO, (B) H2O2 from days 1 to 7 of inflammation. However, these markers returned to base level by day 56 post-inflammation. (C) By contrast, inflammation suppressed the regulator of antioxidant proteins, which remain suppressed at least until day 56 post-inflammation. N = 4 to 6, *p<0.05 vs. controls, #p<0.05 vs. age-matched controls
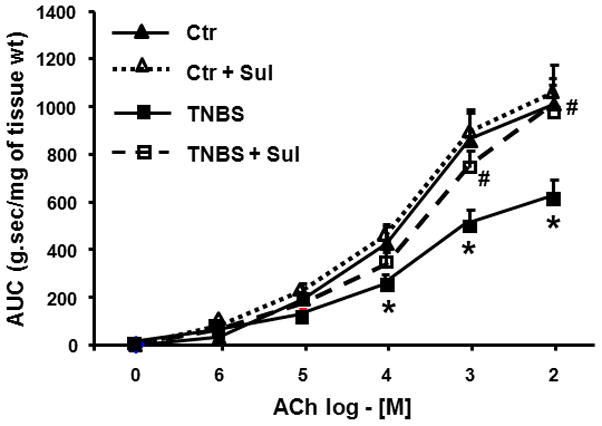
The nuclear factor-erythroid-2-related factor 2 (Nrf2) plays a critical role in the manifestation of oxidative stress in inflammation28. We found that TNBS inflammation significantly suppresses the expression of Nrf2 from day-3 and it does not recover fully at least until day-56 post-inflammation (Fig. 5C). We investigated whether oxidative stress caused impairment of DNA in the promoter sequence of Cacna1c by administering (i.p.) 5 mg/kg sulforaphane [1-isothiocyanato-(4R,S)-(methylsulfinyl)butane], an activator and inducer of Nrf2, daily for 3 days. In one series of experiments, sulforaphane treatment started 3 days prior to the TNBS insult and tissues were collected on day-3 post inflammation. In the second series, the daily administration of sulforaphane started on day 3 after the insult and tissues were collected on day-7 post-inflammation. In both cases, sulforaphane treatment significantly reversed the suppression of Nrf2, the instability of DNA in the promoter of Cacna1c and the suppression of Cacna1c on daty-7 of inflammation (Fig 6; data shown only for treatment starting 3 days prior to inflammatory insult). Prophylactic treatment with sulforaphane prevented the suppression of contractility by TNBS inflammation on day-7 post-TNBS insult (Fig. 7).
Fig. 6.

Administration of sulforaphane in naïve rats had no effect on - (A) the expression of Nrf2, (B) integrity of segment 5 in Cacna1c promoter, (C) and (D) mRNA and protein expressions of Cacna1c. Sulforaphane treatment starting on three days prior to inflammatory insult reverses - (A) the suppression of Nrf2, (B) DNA damage in segment 5 and (C) and (D) mRNA and protein expressions of expressions of Cacna1c on day-3 post-inflammation. Mit sm = small amplicon of mitochondrial DNA, Sul =sulforaphane. N = 4 to 6, *p<0.05 vs. controls.
Fig. 7.
Daily sulforaphane treatment starting 3-days prior to TNBS insult and continuing until day-7 post-inflammation prevented the suppression of smooth muscle reactivity to ACh. Sulforaphane treatment in naïve rats had no effect on reactivity of smooth muscle to ACh. Sul=sulforaphane N=4 rats in each group, * p<0.05 vs. Ctr (controls), #p<0.05 vs. TNBS inflammation.
Discussion
Inflammatory bowel disease (IBD) is recurring inflammation of the gut. It is a complex disease with multiple potential factors contributing to the spontaneous relapse of inflammation. Studies on human tissues and in experimental models of inflammation show an intense immune response comprised of oxidative and nitrosative stresses as well as release of myriad cytokines, chemokines and cell adhesion molecules at various levels and time courses. One of the adverse effects of these inflammatory mediators is impairment of the function of non-immune cells in the gut wall – smooth muscle cells, enteric neurons and epithelial cells, which regulate the motility and epithelial transport. Impairment of smooth muscle function significantly contributes to the morbid symptoms of diarrhea and constipation. Studies on human and animal tissues show that the inflammatory mediators inhibit smooth muscle reactivity to acetylcholine by transcriptional suppression of specific proteins of excitation-contraction coupling in smooth muscle cells24, 29. The impaired contractility of smooth muscle cells suppresses the generation of spontaneous rhythmic phasic contractions, which facilitates the rapid propulsion of fecal material in the colon by giant migrating contractions causing frequent bowel movements30, 31. However, the suppression of smooth muscle rhythmic phasic contractions in the absence of giant migrating contractions results in slower transit and constipation-like conditions32.
Our findings show that colonic inflammation induced by TNBS causes damage in the promoter region of Cacna1c starting at Day 1 of inflammation - reaching the maximum on day 3 and recovers partially by day 7. Further analysis with rolling LX-PCR showed that the DNA damage during active inflammation is confined to two segments of the promoter sequence, one located close to the core promoter (−506/−260) and the other upstream to it (−2,193/−1,542). The DNA damage in the upstream segment is repaired completely by 56 days after inflammation. However, the damage in the segment close to the promoter region persists at this time.
Most studies on DNA damage and repair mechanisms to date have focused in the coding regions of genes in proliferating cells because of their potential in initiating and promoting carcinogenesis. Our understanding of DNA damage and repair mechanisms in terminally differentiated cells is in its infancy. The investigation of these mechanisms is outside the scope of this work. However, one study in the literature points to a link between DNA instability in the promoter sequences and gene transcription. This study reported that the addition of OH· to the C-8 position of guanine within the recognition sequences of transcription factors, resulting in the formation of 8-oxydeoxy-guanosine (8-oxodG), might impair the binding of transcription factor to DNA33. The formation of 8-oxodG impaired the binding of AP-1 and Sp1 transcription factors to their cis-elements. However, 8-oxodG modifications in the NF-κB recognition sequence had no effect on its binding to the transcription factor.
DNA methylation of CpG islands can also suppress gene expression. The screening of Cacna1c promoter (−2910/+263 bp) with MethPath software (http://www.urogene.org/methprimer/index1.html) identified 4CpG islands. However, methylation specific PCR (MS-PCR) showed no change in the methylation status of any of the CpG Islands on the Cacna1c promoter (data not shown).
Our findings show that DNA damage and repair processes are sequence-specific. Oxidative stress induced by TNBS inflammation impaired DNA integrity in two specific segments of the promoter. In addition, the repair mechanisms effectively overcame the damage in one segment after inflammation subsided, but not in the other. We know very little about the mechanisms of this sensitivity. However, folate-sensitive (CGG)n sequences show susceptibility to 8-oxodG formation resulting in DNA instability34. Sequence analysis showed that segment five that is sensitive to DNA damage has the largest numbers of CGG sequences as a percent of total nucleotides (4% vs 0.2, 0.2, 0.9, 1.3, and 2.7% in segments 1, 2, 3, 4, and 6). It is noteworthy that the cis-elements of transcription factor Sp1 known as GC boxes [5′-(GT)GGGCGG(G/A)(GT)-3′] are rich in folate-sensitive sequences. This segment contains Sp1 binding motifs. Sp1 plays a critical role in the transcription of several genes35, 36. We speculate that impairment in the Sp1 cis-elements might contribute to the attenuated transcription of Cacna1c gene.
The suppression of mRNA and protein expressions of the pore-forming α1C subunit of Cav1.2b channels37 during and after inflammation followed a time course similar to that of DNA instability, which suggests that it contributes to impaired transcription of Cacna1c. Specifically, the DNA instability of segment 5 persisted for at least 56 days after induction of inflammation. The mRNA and protein expressions of the α1C subunit remained suppressed at this time, compared with those in age-matched controls. There was no evidence of inflammation at this time.
Other studies show that the density of voltage-dependent Ca2+ channels in hippocampal neurons increases with aging38, 39. We found that mRNA and protein expressions of the pore-forming α1C subunit increased significantly in control rats 56 days after inflammation, at which time they were 8 weeks older than at the time of inflammatory insult. However, in the presence of persistent DNA damage after the inflammatory insult the normal increase in the expression of α1C subunit was blunted. We do not know the effect of aging on the expression of Cav1.2b channels in human colonic myocytes. However, it is noteworthy that the IBS-like symptoms during remission in IBD patients and those in PI-IBS patients persist for several years after the onset of disease13, 40. Therefore, it is likely that changes in the age-related expression of Cacna1c as well as DNA instability due to prior inflammatory episodes together account for persistent smooth muscle dysfunction following inflammatory episodes.
Nrf2 is a basic leucine zipper (bZIP) transcription factor that serves as a central regulator of genes encoding antioxidant proteins and electrophile enzymes. In resting cells, Kelch like-ECH-associated protein 1 (Keap1) sequesters Nrf2 in the cytoplasm as an inactive complex41. The interaction of Nrf2 with Keap1 presents Nrf2 for ubiquitination and subsequent proteasomal degradation42, 43. Oxidative or nitrosative stress modifies this complex by several potential mechanisms, including thiol modification of Keap1 and phosphorylation of serine or threonine residues on Nrf2. Both mechanisms result in dissociation of the complex, accumulation of Nrf2 in the cytoplasm and eventual translocation to the nucleus44–49. Nrf2 forms heterodimers with a group of small musculoaponeurotic fibrosarcoma (Maf) proteins that lack transactivation domain28. Heterodimerization of Nrf2 with these proteins enhances the binding to a cis-acting enhancer ARE/EpRE located in the promoters of several genes encoding antioxidant proteins50–53.
We found that oxidative stress due to TNBS inflammation suppresses the expression of Nrf2 in the muscularis externa, which bottoms out on Day 3 and then recovers partially by day 7- the period during which integrity of the DNA comprising the Cacna1c promoter is impaired. Systemic administration of sulforaphane, an inducer and activator of Nrf2, prior to the induction of TNBS inflammation (prophylaxis) or after its induction (curative) significantly reduced the DNA instability as well as its biological effects on Cacna1c. Sulforaphane, present in cruciferous vegetables, exerts its chemopreventive and cytoprotective effects by the induction of phase 2 detoxifying and antioxidant enzymes through the induction of Nrf2 signaling. These findings suggest that DNA damage results due to the suppression of Nrf2.
Clinical findings show that the expression of antioxidant proteins in the inflamed segments of Crohn’s disease and ulcerative colitis patients is impaired54. Our findings suggest that this is likely due to the suppression of Nrf2. A notable finding in our study is that the partial, but significant, suppression of Nrf2 persists after inflammation has subsided. We speculate that persistent suppression of Nrf2 might exaggerate the inflammatory response to a subsequent insult resulting in greater DNA damage and organ dysfunction. It is noteworthy that sulfasalazine/masalazine, containing 5-ASA, used as a maintenance drug in IBD patients are radical scavengers that might restore balance between oxidants and antioxidants in the face of suppression of Nrf2 during remission.
The persistence of impaired motility function during remission in IBD patients (IBD-IBS) or that following an episode of severe inflammation (PI-IBS) remains an enigma. Some reports found a low-grade inflammatory response, comprised of increase in T lymphocytes in the lamia propria and epithelial layer in the mucosal biopsies of the IBD patients in remission12, 55 or in PI-IBS patients16, 21,22, 56, 57. However, the increase in T lymphocytes is not significantly different between patients who develop the symptoms of IBS and those who do not58. In addition, prednisone treatment significantly reduces lamina propria T-lymphocytes in PI-IBS patients, but it has no effect on the symptoms of diarrhea/constipation indicative of smooth muscle dysfunction21. Therefore, the low-grade mucosal inflammation is not the cause of smooth muscle dysfunction resulting in the symptoms of diarrhea/constipation in these patients. Our findings show that a single episode of inflammation causes persistent impairment of smooth muscle dysfunction due to persistent DNA instability in the Cacna1c promoter. In addition, the persistent low-grade inflammatory response after severe inflammation might be due to the persistent suppression on Nrf2 resulting in an imbalance in the oxidant-antioxidant response. The data on Nrf2 expression in the mucosa of IBD patients in remission or in PI-IBS patients are not available.
Human diseases are complex and multi-factorial. Animal models mimic only specific features of a human disease. Our model of TNBS inflammation shows that a single episode of robust colonic inflammation24 impairs the stability of specific segments of DNA comprising the promoter region of the Cacna1c gene. The stability of DNA recovers partially during active inflammation but remains significantly suppressed in long-term after inflammation has subsided. The impairment of DNA stability contributes to suppression of the pore-forming α1C subunit of Cav1.2b channels and hence smooth muscle contractility. This model does not mimic conditions under which the smooth muscle contractility is enhanced, resulting in diarrhea-like conditions59. Human patients are subject to environmental factors, such as chronic stress and medications that are absent in this model, but they might alter the outcomes.
Supplementary Material
Acknowledgments
Supported in part by NIDDK Grants DK 032346 and DK 072414 (SKS)
Study concept and design (SKS); acquisition of data (KC and JC); analysis and interpretation of data KC, SKS, JC and SM); drafting of the manuscript (SKS, KC); critical revision of the manuscript for important intellectual content (SKS, KC, SM) statistical analysis (KC); obtained funding (SKS); technical, or material support (SM)
Footnotes
There are no conflicts of interest to disclose for all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Williams GM, Jeffrey AM. Oxidative DNA damage: endogenous and chemically induced. Regul Toxicol Pharmacol. 2000;32:283–92. doi: 10.1006/rtph.2000.1433. [DOI] [PubMed] [Google Scholar]
- 2.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirakawa K, Midorikawa K, Oikawa S, et al. Carcinogenic semicarbazide induces sequence-specific DNA damage through the generation of reactive oxygen species and the derived organic radicals. Mutat Res. 2003;536:91–101. doi: 10.1016/s1383-5718(03)00030-5. [DOI] [PubMed] [Google Scholar]
- 4.Fernando LP, Kurian PJ, Fidan M, et al. Quantitation of gene-specific DNA damage by competitive PCR. Anal Biochem. 2002;306:212–21. doi: 10.1006/abio.2002.5705. [DOI] [PubMed] [Google Scholar]
- 5.Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–47. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 6.Liang Y, Lin SY, Brunicardi FC, et al. DNA damage response pathways in tumor suppression and cancer treatment. World J Surg. 2009;33:661–6. doi: 10.1007/s00268-008-9840-1. [DOI] [PubMed] [Google Scholar]
- 7.Gros L, Saparbaev MK, Laval J. Enzymology of the repair of free radicals-induced DNA damage. Oncogene. 2002;21:8905–25. doi: 10.1038/sj.onc.1206005. [DOI] [PubMed] [Google Scholar]
- 8.Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 9.Bojarski C. Malignant transformation in inflammatory bowel disease: prevention, surveillance and treatment - new techniques in endoscopy. Dig Dis. 2009;27:571–5. doi: 10.1159/000233300. [DOI] [PubMed] [Google Scholar]
- 10.Isgar B, Harman M, Kaye MD, et al. Symptoms of irritable bowel syndrome in ulcerative colitis in remission. Gut. 1983;24:190–2. doi: 10.1136/gut.24.3.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minderhoud IM, Oldenburg B, Wismeijer JA, et al. IBS-like symptoms in patients with inflammatory bowel disease in remission; relationships with quality of life and coping behavior. Dig Dis Sci. 2004;49:469–74. doi: 10.1023/b:ddas.0000020506.84248.f9. [DOI] [PubMed] [Google Scholar]
- 12.Keohane J, O’Mahony C, O’Mahony L, O’Mahony S, Quigley EM, Shanahan F, et al. Irritable bowel syndrome-type symptoms in patients with inflammatory bowel disease: a real association or reflection of occult inflammation? Am J Gastroenterol. 2010;105:1788, 1789–94. doi: 10.1038/ajg.2010.156. quiz 1795. [DOI] [PubMed] [Google Scholar]
- 13.Simren M, Axelsson J, Gillberg R, et al. Quality of life in inflammatory bowel disease in remission: the impact of IBS-like symptoms and associated psychological factors. Am J Gastroenterol. 2002;97:389–96. doi: 10.1111/j.1572-0241.2002.05475.x. [DOI] [PubMed] [Google Scholar]
- 14.Farrokhyar F, Marshall JK, Easterbrook B, et al. Functional gastrointestinal disorders and mood disorders in patients with inactive inflammatory bowel disease: prevalence and impact on health. Inflamm Bowel Dis. 2006;12:38–46. doi: 10.1097/01.mib.0000195391.49762.89. [DOI] [PubMed] [Google Scholar]
- 15.Dunlop SP, Jenkins D, Spiller RC. Distinctive clinical, psychological, and histological features of postinfective irritable bowel syndrome. Am J Gastroenterol. 2003;98:1578–83. doi: 10.1111/j.1572-0241.2003.07542.x. [DOI] [PubMed] [Google Scholar]
- 16.Spiller RC, Jenkins D, Thornley JP, et al. Increased rectal mucosal enteroendocrine cells, T lymphocytes, and increased gut permeability following acute Campylobacter enteritis and in post-dysenteric irritable bowel syndrome. Gut. 2000;47:804–11. doi: 10.1136/gut.47.6.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKendrick MW, Read NW. Irritable bowel syndrome--post salmonella infection. J Infect. 1994;29:1–3. doi: 10.1016/s0163-4453(94)94871-2. [DOI] [PubMed] [Google Scholar]
- 18.Gwee KA, Leong YL, Graham C, et al. The role of psychological and biological factors in postinfective gut dysfunction. Gut. 1999;44:400–6. doi: 10.1136/gut.44.3.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gwee KA, Graham JC, McKendrick MW, et al. Psychometric scores and persistence of irritable bowel after infectious diarrhoea. Lancet. 1996;347:150–3. doi: 10.1016/s0140-6736(96)90341-4. [DOI] [PubMed] [Google Scholar]
- 20.Neal KR, Barker L, Spiller RC. Prognosis in post-infective irritable bowel syndrome: a six year follow up study. Gut. 2002;51:410–3. doi: 10.1136/gut.51.3.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunlop SP, Jenkins D, Neal KR, et al. Randomized, double-blind, placebo-controlled trial of prednisolone in post-infectious irritable bowel syndrome. Aliment Pharmacol Ther. 2003;18:77–84. doi: 10.1046/j.1365-2036.2003.01640.x. [DOI] [PubMed] [Google Scholar]
- 22.Gwee KA, Collins SM, Read NW, Rajnakova A, Deng Y, Graham JC, McKendrick MW, Moochhala SM, et al. Increased rectal mucosal expression of interleukin 1beta in recently acquired post-infectious irritable bowel syndrome. Gut. 2003;52:523–6. doi: 10.1136/gut.52.4.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grisham MB. Oxidants and free radicals in inflammatory bowel disease. Lancet. 1994;344:859–61. doi: 10.1016/s0140-6736(94)92831-2. [DOI] [PubMed] [Google Scholar]
- 24.Shi XZ, Winston JH, Sarna SK. Differential immune and genetic responses in rat models of Crohn’s colitis and ulcerative colitis. Am J Physiol Gastrointest Liver Physiol. 2011;300:G41–51. doi: 10.1152/ajpgi.00358.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Rusch NJ, Striessnig J, et al. Down-regulation of L-type calcium channels in inflamed circular smooth muscle cells of the canine colon. Gastroenterology. 2001;120:480–9. doi: 10.1053/gast.2001.21167. [DOI] [PubMed] [Google Scholar]
- 26.Shi XZ, Sarna SK. Gene therapy of Cav1. 2 channel with VIP and VIP receptor agonists and antagonists: a novel approach to designing promotility and antimotility agents. Am J Physiol Gastrointest Liver Physiol. 2008;295:G187–G196. doi: 10.1152/ajpgi.00047.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ayala-Torres S, Chen Y, Svoboda T, et al. Analysis of gene-specific DNA damage and repair using quantitative polymerase chain reaction. Methods. 2000;22:135–47. doi: 10.1006/meth.2000.1054. [DOI] [PubMed] [Google Scholar]
- 28.Itoh K, Igarashi K, Hayashi N, et al. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol Cell Biol. 1995;15:4184–93. doi: 10.1128/mcb.15.8.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snape WJ, Jr, Williams R, Hyman PE. Defect in colonic smooth muscle contraction in patients with ulcerative colitis. Am J Physiol. 1991;261:G987–91. doi: 10.1152/ajpgi.1991.261.6.G987. [DOI] [PubMed] [Google Scholar]
- 30.Sethi AK, Sarna SK. Contractile mechanisms of canine colonic propulsion. Am J Physiol. 1995;268:G530–8. doi: 10.1152/ajpgi.1995.268.3.G530. [DOI] [PubMed] [Google Scholar]
- 31.Annese V, Bassotti G, Napolitano G, et al. Gastrointestinal motility disorders in patients with inactive Crohn’s disease. Scand J Gastroenterol. 1997;32:1107–17. doi: 10.3109/00365529709002989. [DOI] [PubMed] [Google Scholar]
- 32.Rao SS, Holdsworth CD, Read NW. Symptoms and stool patterns in patients with ulcerative colitis. Gut. 1988;29:342–5. doi: 10.1136/gut.29.3.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghosh R, Mitchell DL. Effect of oxidative DNA damage in promoter elements on transcription factor binding. Nucleic Acids Res. 1999;27:3213–8. doi: 10.1093/nar/27.15.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Entezam A, Lokanga AR, Le W, et al. Potassium bromate, a potent DNA oxidizing agent, exacerbates germline repeat expansion in a fragile X premutation mouse model. Hum Mutat. 2010;31:611–6. doi: 10.1002/humu.21237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olofsson BA, Kelly CM, Kim J, et al. Phosphorylation of Sp1 in response to DNA damage by ataxia telangiectasia-mutated kinase. Mol Cancer Res. 2007;5:1319–30. doi: 10.1158/1541-7786.MCR-07-0374. [DOI] [PubMed] [Google Scholar]
- 36.Pazdrak K, Shi XZ, Sarna SK. TNFalpha suppresses human colonic circular smooth muscle cell contractility by SP1- and NF-kappaB-mediated induction of ICAM-1. Gastroenterology. 2004;127:1096–109. doi: 10.1053/j.gastro.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 37.Dai B, Saada N, Echetebu C, et al. A new promoter for alpha1C subunit of human L-type cardiac calcium channel Ca(V)1.2. Biochem Biophys Res Commun. 2002;296:429–33. doi: 10.1016/s0006-291x(02)00894-x. [DOI] [PubMed] [Google Scholar]
- 38.Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272:1017–20. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- 39.Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21:9744–56. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marshall JK, Thabane M, Garg AX, et al. Eight year prognosis of postinfectious irritable bowel syndrome following waterborne bacterial dysentery. Gut. 2010;59:605–11. doi: 10.1136/gut.2009.202234. [DOI] [PubMed] [Google Scholar]
- 41.Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cullinan SB, Gordan JD, Jin J, et al. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24:8477–86. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi A, Kang MI, Okawa H, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eggler AL, Liu G, Pezzuto JM, et al. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc Natl Acad Sci U S A. 2005;102:10070–5. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lo SC, Li X, Henzl MT, et al. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006;25:3605–17. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Natl Acad Sci U S A. 2000;97:12475–80. doi: 10.1073/pnas.220418997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009;48:91–104. doi: 10.1002/mc.20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Talalay P, De Long MJ, Prochaska HJ. Identification of a common chemical signal regulating the induction of enzymes that protect against chemical carcinogenesis. Proc Natl Acad Sci U S A. 1988;85:8261–5. doi: 10.1073/pnas.85.21.8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kwak MK, Kensler TW. Targeting NRF2 signaling for cancer chemoprevention. Toxicol Appl Pharmacol. 2010;244:66–76. doi: 10.1016/j.taap.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Motohashi H, O’Connor T, Katsuoka F, et al. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene. 2002;294:1–12. doi: 10.1016/s0378-1119(02)00788-6. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto T, Kyo M, Kamiya T, et al. Predictive base substitution rules that determine the binding and transcriptional specificity of Maf recognition elements. Genes Cells. 2006;11:575–91. doi: 10.1111/j.1365-2443.2006.00965.x. [DOI] [PubMed] [Google Scholar]
- 52.Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266:11632–9. [PubMed] [Google Scholar]
- 53.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–60. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 54.Kruidenier L, Kuiper I, Van Duijn W, et al. Imbalanced secondary mucosal antioxidant response in inflammatory bowel disease. J Pathol. 2003;201:17–27. doi: 10.1002/path.1408. [DOI] [PubMed] [Google Scholar]
- 55.Saverymuttu SH. Clinical remission in Crohn’s disease--assessment using faecal 111In granulocyte excretion. Digestion. 1986;33:74–9. doi: 10.1159/000199277. [DOI] [PubMed] [Google Scholar]
- 56.Gonsalkorale WM, Perrey C, Pravica V, et al. Interleukin 10 genotypes in irritable bowel syndrome: evidence for an inflammatory component? Gut. 2003;52:91–3. doi: 10.1136/gut.52.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barbara G, Stanghellini V, De Giorgio R, et al. Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology. 2004;126:693–702. doi: 10.1053/j.gastro.2003.11.055. [DOI] [PubMed] [Google Scholar]
- 58.Dunlop SP, Jenkins D, Neal KR, et al. Relative importance of enterochromaffin cell hyperplasia, anxiety, and depression in postinfectious IBS. Gastroenterology. 2003;125:1651–9. doi: 10.1053/j.gastro.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 59.Choudhury BK, Shi XZ, Sarna SK. Gene plasticity in colonic circular smooth muscle cells underlies motility dysfunction in a model of postinfective IBS. Am J Physiol Gastrointest Liver Physiol. 2009;296:G632–42. doi: 10.1152/ajpgi.90673.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.