

Abstract
Background & Aims
Inflammatory bowel disease increases the risks for colon cancer and colitis-associated cancer (CAC). Epithelial cell-derived matrix metalloproteinase (MMP)9 mediates inflammation during acute colitis and the cleavage and activation of the transcription factor Notch1, which prevents differentiation of progenitor cells into goblet cells. However, MMP9, also protects against the development of CAC and acts as a tumor suppressor. We investigated the mechanisms by which MMP9 protects against CAC in mice.
Methods
C57/B6 wild-type mice were given a single dose of azoxymethane and 2 cycles of dextran sulfate sodium (DSS). Mice were also given the γ-secretase inhibitor DAPT or DMSO (control) during each DSS cycle; they were sacrificed on day 56. We analyzed embryonic fibroblasts isolated from wild-type and MMP9−/− mice and HCT116 cells that were stably transfected with MMP9.
Results
Wild-type mice were more susceptible to CAC following inhibition of Notch1 by DAPT, demonstrated by increased numbers of tumors and level of dysplasia, compared with controls. Inhibition of Notch1 signaling significantly reduced protein levels of active Notch1, p53, p21WAF1/Cip1, Bax-1, active caspase-3, as well as apoptosis, compared with controls. Similar results were observed in transgenic HCT116 cells and embryonic fibroblasts from MMP9−/− mice, upon γ-radiation–induced damage of DNA.
Conclusion
MMP9 mediates Notch1 signaling via p53 to regulate apoptosis, cell-cycle arrest, and inflammation. By these mechanisms, it might prevent CAC.
Keywords: colorectal cancer, mouse model, extracellular matrix, IBD, tumor development
Introduction
Matrix metalloproteinases (MMPs) are extra cellular matrix degrading Zn2+ dependent endopeptidases that play an important role in ulceration and tissue remodeling 1. MMPs share common functional domains and activation mechanisms 2, 3, 4. In comparison to other MMPs, MMP9 is distinguished by i) its absence from normal epithelia, ii) its acute upregulation in response to inflammatory stimuli (LPS, dsDNA, fMLP, adhesion molecules, chemokines and cytokines) and iii) in being the terminal member of the MMP family activated by other MMPs including MMP1, 2, 3, 7 and TIMP2. Further, of the MMPs, MMP9 has been shown to be consistently elevated in inflammatory bowel disease (IBD) and is the predominant MMP that is upregulated in several animal models of colitis as well as in patients with IBD 1, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15. Both MMP2 and MMP9 activity have been detected in lower crypt epithelial cells surrounding degraded matrix as well as underlying ulcerations 8, 9, 16. We and others have demonstrated that genetic ablation of MMP9 protected mice from chemical (DSS- and TNBS-) as well as bacteria-(Salmonella)-induced colitis 13, 15, 17, 18. Our studies also demonstrated that epithelial, but not immune cell derived, MMP9 mediates inflammation 13. In addition to its role in regulating inflammation, MMP9 also plays an important role in epithelial cell differentiation by mediating the preferential differentiation of progenitor cells to enterocytes 19. Specifically, MMP9 mediates proteolytic cleavage/activation of the transcription factor Notch1, which modulates the differentiation of progenitor cells into goblet cells.
IBD, which includes ulcerative colitis (UC) and Crohn’s Disease (CD), are chronic, incurable inflammatory diseases 20, 21. UC and CD are characterized by continuous or discontinuous mucosal inflammation, respectively, with inflammatory cell infiltration, epithelial cell destruction, connective tissue defects and ulceration of the mucosa of the major portion of intestine and/-or colon. Colitis-associated cancer (CAC) is an important and deadly complication of IBD. The lifetime risk of developing CAC in patients with IBD is 15–40% and accounts for approximately 15% of mortality in IBD patients. Surprisingly, the pathogenesis of CAC is poorly understood.
Given its role in intestinal inflammation and differentiation, we examined the role of MMP9 in the development of CAC. We have demonstrated that epithelial cell-derived MMP9, in contrast to its role in mediating tissue damage during acute colitis, plays a protective role in the development of CAC 22. Thus, the overall goal of this study is to comprehend the underlying mechanism of the tumor suppressive function of MMP9 in CAC.
Materials and Methods
Animal models
See supplementary section.
Induction of colitis associated cancer and inhibition of Notch1 by pharmacological inhibitor in animals
See supplementary section.
Protein extraction and Western blot analysis
See supplementary section.
Histological assessment of colitis
Swiss rolls of mouse colons were fixed in formalin. Sections were stained with hematoxylin and eosin. Microscopic sections were analyzed and histological scoring was performed as described by Cooper et. al. 23, 24.
Endoscopic assessment of colitis
See supplementary section.
Terminal deoxynucleotidyl transferase–mediated dUTP nick end (TUNEL) labeling staining
As described previously 22, paraffin sections of colon were deparaffinized and stained for apoptotic nuclei (TUNEL).
Isolation of mRNA and quantitative PCR
Total RNA was isolated from mucosal stripping of mouse colons using the RNeasy Kit (Qiagen, Valencia, CA) as per manufacturer’s instruction. Total RNA was then reverse transcribed using the RevertAid first strand cDNA synthesis kit (Fermentas, Glen-Burnie, MD). As described previously 25, quantitative real-time PCR was performed for TNF-α, IF-γ, IL-1β and IL-6 specific primers 25 using Fermentas Maxima SYBR Green qPCR master mix (Fermentas).
Isolation of MEFs and cell culture
See supplementary section.
γ-irradiation
Both MEFs and HCT116 cells are apoptotic resistant, so to enhance the apoptotis γ-irradiation of cultured cells was performed using a 137Cs γ-irradiator at 0.8Gy/min for 15 min, for a total of 12Gy 26, 27. Cells were harvested 24h after γ-irradiation for subsequent assays.
Inhibition of Notch1 signaling by pharmacological inhibitor DAPT in MEFs
MEFs isolated from WT mice as described above were cultured in 6-well plates. 60% confluent cell cultures were exposed to γ-irradiation of the above stated dose and treated with 30 μM (a dose that does not affect cell viability, yet inhibits Notch1) of DAPT or DMSO (vehicle-treated MEFs) per ml of culture medium. Cell lysates were collected after 24 hours of γ-irradiation.
Statistical analysis
The data are presented as means ± SE. Groups were compared by Student’s t-test. P values <0.05 was considered statistically significant.
Results
Inhibition of Notch1 signaling results in increased susceptibility to CAC
WT and MMP9−/− mice treated with azoxymethane (AOM) and two cycles of dextran sodium sulfate (DSS) were given γ-secretase inhibitor DAPT (Difluorophenacetyl-L-alanyl-S-phenylglycine t-butyl ester) (10 μmol/kg) intraperitoneally, an inhibitor of Notch1, daily for five consecutive days during the DSS cycle. Control mice received vehicle/solvent alone (vehicle-treated mice) during each DSS cycle. There was a slight (<5%), but significant decrease in the body weight of WT mice treated with DAPT after two cycles of 3% DSS, compared to vehicle-treated mice (Figure 1A), however, there was no significant difference in body weight at the end of 56 days as shown in Figure 1A. Figure 1B shows the colonoscopic view (left panel) and gross images (right panel) indicating increased number of polyps in DAPT-treated mice compared to vehicle-treated mice. Figure 1C is the graphical representation of the number of polyps among WT and MMP9−/− mice and indicates that DAPT had no effect on polyp number in MMP9−/− mice. This suggests that inhibition of Notch1 signaling in the absence of MMP9 was not important for CAC sensitivity. On the other hand, in WT mice there was a significantly higher number of polyps among DAPT-treated mice (10.14±1.22) compared to vehicle-treated mice (2.5±0.63), indicating more tumor multiplicity among DAPT-treated mice. Figure 1D is the graphical representation of polyp size in the range of <1–2mm diameter and ≥2–4mm diameter indicating that there were a significantly higher number of polyps in both the size range of <1–2mm diameter (4.0±1.21 vs. 1.88±0.84) as well as ≥2–4 mm diameter (7.14±1.26 vs 1.0±0.38) among DAPT-treated WT mice compared to vehicle-treated WT mice in CAC. This result signifies an increased tumor burden among DAPT-treated WT mice compared to vehicle-treated WT mice. Histological examination showed increased dysplasia among DAPT-treated mice compared to vehicle-treated mice (Figure 1E). The histological inflammatory score (based on three parameters: inflammatory cells in lamina propria, crypt damage and evaluation of ulcer 23, 24), was significantly higher in DAPT-treated mice (6.3±0.5) compared to vehicle-treated mice (4.5±0.3) (Figure 1F). These results together indicate that inhibition of Notch1 signaling exhibited increased susceptibility to CAC with increased inflammation, tumor multiplicity and tumor burden.
Figure 1. Inhibition of Notch1 signaling results in increased susceptibility to CAC.
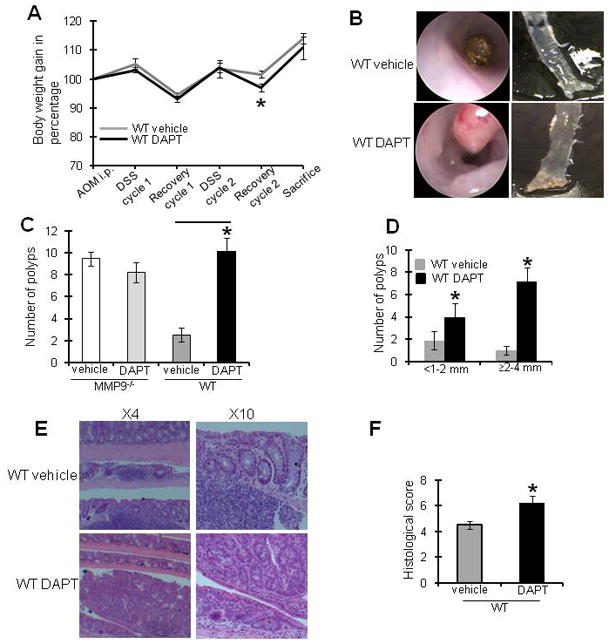
CAC was induced in WT and MMP-9−/− mice as described in the Methods section. Notch1 signaling was inhibited by 5 consecutive i.p. injections of DAPT or vehicle during each DSS cycle. Mice were weighed once a week and sacrificed after 56 days. A) graphical presentation of change in body weight of mice treated with Notch1 inhibitor and placebo. B) left panel shows representative colonoscopy images and right panel shows representative gross anatomy of the colon of mice treated with Notch1 inhibitor and vehicle. C) polyp count and D) polyp size. E) H&E staining of Swiss rolls showing dysplasia. F) histological score of mice treated with Notch1 inhibitor and vehicle. Each bar represents mean ± S.E. Data is representative of three experiments (n=20/group). *p< 0.05.
Inhibition of Notch signaling is associated with altered mRNA levels of cytokines
To examine the effect of inhibition of Notch1 signaling on inflammation in CAC, cytokines expressions were measured by quantitative PCR assays. RNA were isolated from mucosal stripping of WT mice treated with water or AOM and two cycles of DSS, then injected with DAPT or vehicle alone during each cycle of DSS and sacrificed after 56 days. Figure 2A and 2B show that DAPT treatment resulted in a significant increase in TNF-α and IFN-γ mRNA levels (4.6±0.8 and 51.7±17.5 fold increase respectively, compared to WT mice treated with water). There was also a significant increase in mRNA levels of cytokines TNF-α and IFN-γ in DAPT-treated mice compared to vehicle-treated mice (Figure 2A and 2B). Interestingly, inhibition of Notch1 signaling in DAPT-treated mice resulted in 6.9±2.7 and 1.8±0.4 fold increase in mRNA levels of IL-1β and IL-6 respectively compared to WT mice treated with water (Figure 2C and 2D). Though, these mRNA levels were significantly lower compared to vehicle-treated mice as represented in Figure 2C and 2D respectively. These results show that inflammatory cytokines TNF-α and IF-γ are upregulated, while IL-6 and IL-1β were downregulated by Notch1 inhibition in mice treated with AOM and 2 cycles of DSS, despite an increase in inflammatory parameters in the DAPT-treated group of mice.
Figure 2. Inhibition of Notch1 signaling alters mRNA levels of cytokines.
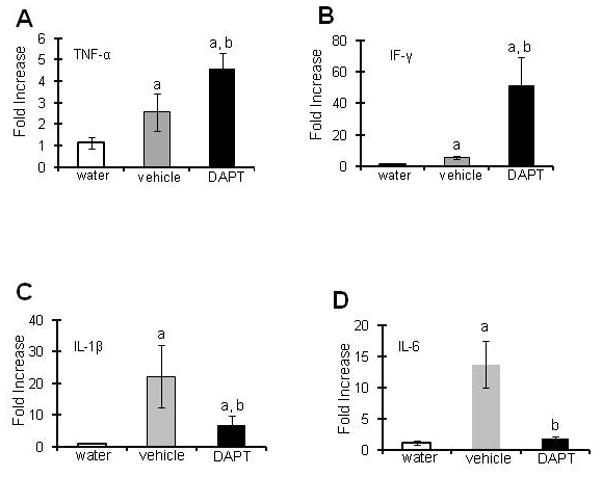
As described in the Methods section, Notch1 signaling was inhibited by 5 consecutive i.p. injections of DAPT or vehicle alone at each DSS cycle during CAC induction and mice were sacrificed after 56 days. RNA was extracted from colonic mucosal stripping and mRNA levels were quantified using real time PCR A) TNF-α mRNA, B) IF-γ mRNA, C) IL-1β mRNA and D) IL-6 mRNA. Each bar represents mean ± S.E., n=6, ap<0.05 vs water, bp<0.05 vs vehicle.
Inhibition of Notch1 is associated with altered levels of p53, p21WAF1/Cip1 and Bax-1
Given the profound effect of MMP9 knock down 22 or Notch1 inhibition on CAC, we examined the levels of p53, a tumor suppressor known to play a important role in CAC and other cancers, and its downstream targets Bax-1 and p21WAF1/Cip1. p53 regulates cell survival by inducing cell cycle arrest or apoptosis. As expected, there was a significant decrease of NICD (Notch intracellular domain)- active form of Notch1 in mice treated with DAPT (89.0±20.0 percent inhibition) compared to vehicle-treated mice (Figure 3B). Inhibition of Notch1 signaling resulted in decreased expression of p53 (45.0 ±20.0 percent inhibition), compared to vehicle-treated mice (Figure 3C). Pro-apoptotic factor Bax-1 was also inhibited in mice treated with DAPT (65.0±7.0 percent inhibition) compared to vehicle-treated mice (Figure 3D). There was no significant difference in MMP9 protein expression in DAPT-treated mice compared to the vehicle-treated mice (Figure 3A). Interestingly, inhibition of Notch1 signaling also showed a decrease in the protein expression of p21WAF1/Cip1 (40.0±15.0 percent inhibition), a well known inhibitor of cyclin dependent kinases (CDKs). These results together suggest that inhibition of Notch1 signaling is associated with decreased levels of p53, p21WAF1/Cip1 and Bax-1 levels.
Figure 3. Inhibition of Notch1 is associated with altered levels of p53, Bax-1 and p21WAF1/Cip1.

As described in the Methods section, Notch1 signaling was inhibited by 5 consecutive i.p. injections of DAPT or vehicle alone at each DSS cycle during CAC induction and mice were sacrificed after 56 days. Western blots of proteins (30μg/lane) from the mucosal stripping of the colons were probed with A) anti-MMP9, B) anti-NICD, C) anti-p53, D) anti-Bax-1 and E) anti-p21WAF1/Cip1. Western blots are quantified by scanning densitometry. Values are representative of three experiments, each bar represents mean ± S.E., *p<0.05.
Inhibition of Notch signaling is associated with decreased apoptosis during CAC
Swiss rolls of colons of mice treated with AOM and two cycles of DSS to induce CAC and injected with DAPT or vehicle alone were used to perform TUNEL staining. Figure 4A left panel shows the TUNEL staining as depicted by fluorescent green nuclei staining among the crypts of colonic epithelium of DAPT-treated and vehicle-treated mice with CAC, middle panel shows the fluorescent blue DAPI nuclear staining and the right panel shows the merged image of TUNEL and DAPI indicating no significant level of apoptosis among DAPT-treated mice compared to vehicle-treated mice with CAC. Figure 4B shows the graph representing the number of nuclei positive for TUNEL staining per crypt. WT mice treated with DAPT showed dramatically decreased apoptosis (2.9±0.5) compared to the vehicle-treated mice (14.4±1.1) (Figure 4B). These data indicate that inhibition of Notch1 signaling is associated with decreased apoptosis. Figure 4C shows that inhibition of Notch1 signaling resulted in decreased expression of active caspase-3 (90.0±10.0 percent inhibition) compared to vehicle-treated mice. Together, the foregoing data demonstrate the inhibition of Notch1 signaling by DAPT in CAC is associated with increased tumor burden, altered cytokine expression, dramatic decrease in p53 and p21WAF1/Cip1 levels and apoptosis.
Figure 4. Inhibition of Notch signaling is associated with decreased apoptosis during CAC.
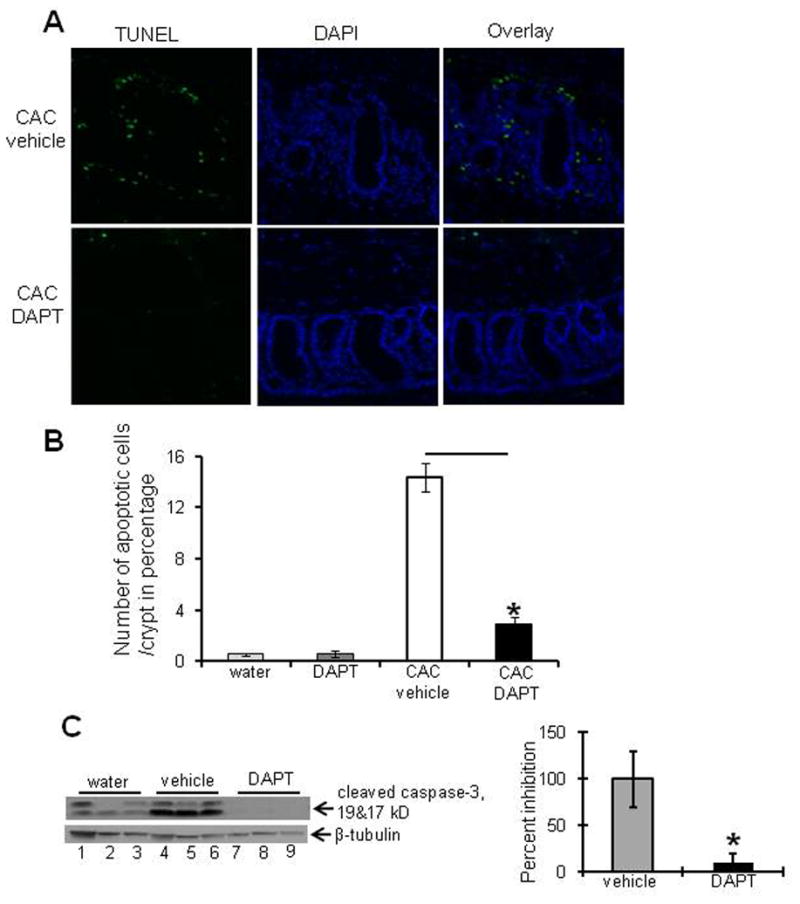
CAC was induced in WT mice as described in the Methods section. Notch1 signaling was inhibited by 5 consecutive i.p. injections of DAPT or vehicle alone during each DSS cycle. A) colons of mice were processed for immunofluorescence using TUNEL and DAPI staining (X20 magnification). B) bar graph presentation of the percentage of apoptotic cells/crypt (12 crypts/mice) among mice treated with Notch1 inhibitor and placebo. C) Western blot of proteins (30μg/lane) from the mucosal stripping of the colons probed with anti-caspase-3. Western blot was quantified by scanning densitometry. Values are representative of three experiments, each bar represents mean ± S.E., *p<0.05.
MMP9 is required for p53 activation
MEFs (mouse embryonic fibroblast cells) isolated from MMP9−/− mice compared to MEFs isolated from WT mice. As described in the Methods section, MEFs isolated from WT and MMP9−/− mice were exposed to γ-radiation to enhance p53 levels and/or apoptosis. Cell lysates for western blot were obtained after 24 hours of irradiation. Figure 5A(i) shows γ-radiated MMP9−/− MEFs had significantly less p53 protein levels (40.0±14.0 percent inhibition) compared to γ-radiated WT MEFs (Figure 5A). Interestingly, γ-radiated MMP9−/− MEFs (lanes 4–6) also exhibited a significant decrease in protein levels of NICD (32.0±5.0 percent inhibition) as well as a significant decrease in protein levels of pro-apoptotic factor Bax-1 (29.0±18.0 percent inhibition) compared to γ-radiated WT MEFs (Figure 5A(ii) and 5A(iii) respectively). γ-radiated MMP9−/− MEFs also exhibited significant decrease in protein levels of p21WAF1/Cip1 (32.0±9.0 percent inhibition), compared to γ-radiated WT MEFs (Figure 5A(iv)).
Figure 5.
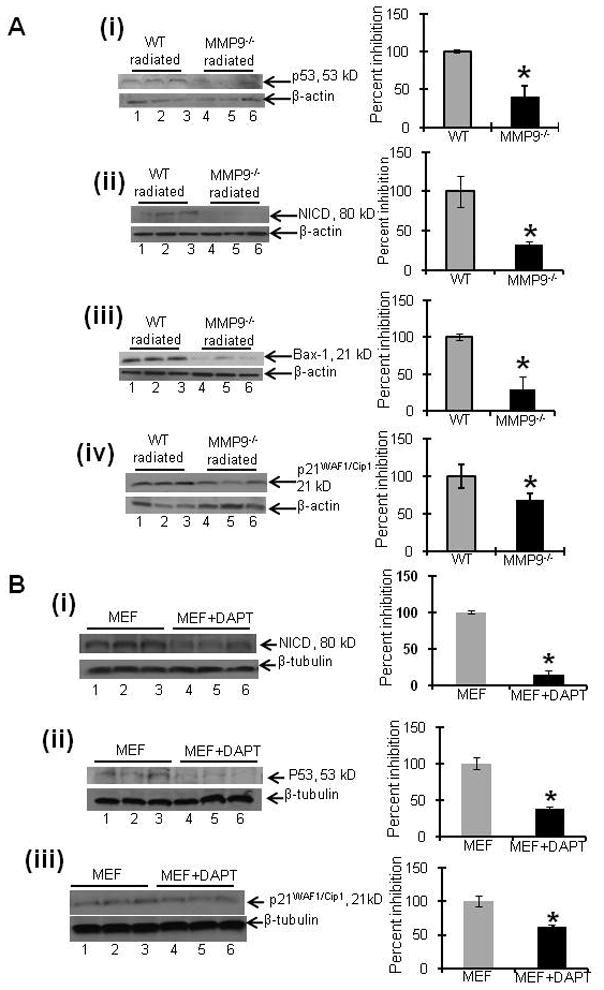
A) MEFs exposed to γ-radiation induce DNA damage and indicate that MMP9 activates p53 expression. MEFs obtained from WT and MMP9−/− mice embryos were harvested on six well plates. Cultured cells were γ-irradiated (12Gy) and cell lysates were collected after 24 hrs. Western blots of proteins (15μg/lane) probed with i) anti-p53, ii) anti-NICD, iii) anti-Bax-1 and iv) anti-p21WAF1/Cip1. Western blots were quantified by scanning densitometry. Values are representative of three experiments, each bar represents mean ± S.E., *p<0.05. B) Inhibition of Notch1 signaling among MEFs exposed to γ-radiation exhibited decreased NICD, p53 and p21WAF1/Cip1 expression. MEFs obtained from WT mice embryos were harvested on six well plates. Cultured cells were γ-irradiated (12Gy) and then treated with DAPT (Notch1 inhibitor) or vehicle and cell lysates were collected after 24 hrs. Western blot of proteins (15μg/lane) probed with i) anti-NICD, ii) anti-p53 and iii) anti-p21WAF1/Cip1. Western blots are quantified by scanning densitometry. Values are representative of three experiments, each bar represents mean ± S.E., *p<0.05.
To understand the underlying role of Notch1 activated by MMP9 in regulating p53 expression, we exposed WT MEFs to γ-radiation in the presence of DAPT (as described in methods section).
There was a decreased expression of active Notch1 i.e. NICD (85.0±5.6 percent inhibition) among DAPT-treated MEFs compared to vehicle-treated MEFs as represented by Figure 5B(i). Interestingly, inhibition of Notch1 signaling by DAPT in γ-irradiated MEFs showed a decrease in protein levels of p53 (62.0±3.5 percent inhibition) compared to vehicle-treated MEFs as represented by Figure 5B(ii). There was also a significant decrease in protein levels of p21WAF1/Cip1 (38±3.7 percent inhibition) among DAPT-treated MEFs compared to vehicle-treated MEFs as shown in Figure 5C(iii).
To support our hypothesis that MMP9 mediated Notch1 activation is necessary for p53 activation. We performed transient transfection on MMP9−/− MEFs with active Notch1 i.e. NICD. Supplementary Figure 1A shows the transfection efficiency of NICD as indicated by increased expression of NICD (2.1±0.2 fold) compared to vector. Supplementary Figure 1B indicates that MMP9−/− MEFs transiently transfected with NICD resulted in increased expression of p53 (3.2±0.5) compared to vector. These results together show that MMP9 mediated Notch1 activation is necessary for p53 activation and its downstream targets.
MMP9 overexpression regulates NICD, p53, Bax-1 and p21WAF1/Cip1 levels
Our next goal was to determine the effect of excess MMP9 on p53 expression and its downstream targets that regulate cell survival. We used the HCT116 cell line (stably transfected with a pEGFP plasmid with or without the MMP9 gene), as described in the Methods section, as our in vitro model to study the effect of MMP9 on the expression of NICD, p53, Bax-1 and p21WAF1/Cip1. As HCT116 cells are apoptosis resistant, we used γ-radiation to induce apoptosis as described in Methods section, γ-irradiated HCT116 cells stably transfected with MMP9 showed increased expression of MMP9 (4.5±0.5 fold) at 120kD (Figure 6A). MMP9 overexpression in HCT116 cells after γ-irradiation produced increased expression of NICD, p53, Bax-1 and p21WAF1/Cip1 (2.8±0.3, 2.4±0.6, 1.85±.04 and 2.35±0.09 fold compared to vector) (Figure 6B-6E respectively).
Figure 6. MMP9 overexpression results in altered expression of NICD, p53, Bax-1 and p21WAF1/Cip1.
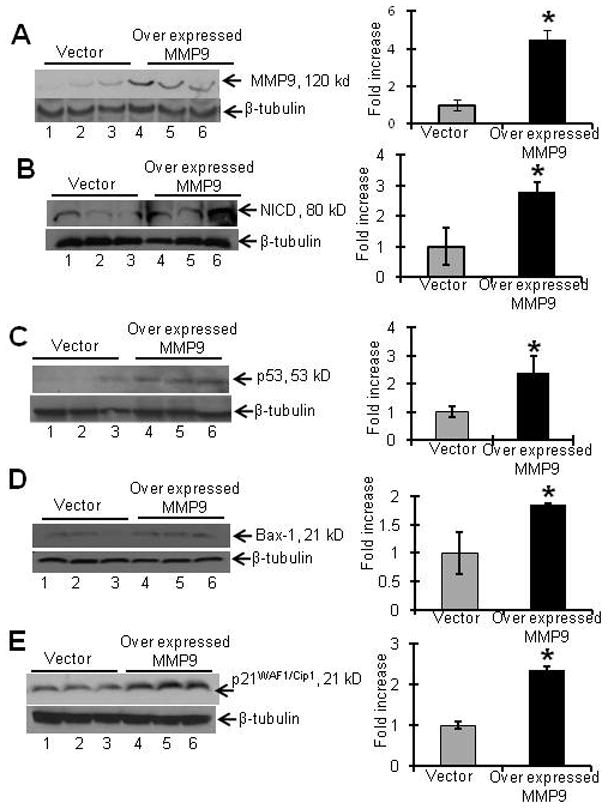
Stably transfected HCT116 cells overexpressing MMP9 were harvested from six well plates. Cultured cells were γ-irradiated (12Gy) and collected after 24 hrs. Western blot of proteins (15μg/lane) probed with A) anti-MMP9, B) anti-NICD, C) anti-p53 D) anti-Bax-1 and E) p21WAF1/Cip1. Western blots were quantified by scanning densitometry. Values are representative of three experiments, each bar represents mean ± S.E., *p<0.05.
Thus our in vitro data corroborate with in vivo results indicating that MMP9 activation in CAC induces Notch1 signaling, which in turn regulates cell survival through p53 activation (Figure 7).
Figure 7. Schematic representation of mechanistic pathway showing that MMP9 acts as a tumor suppressor in CAC.
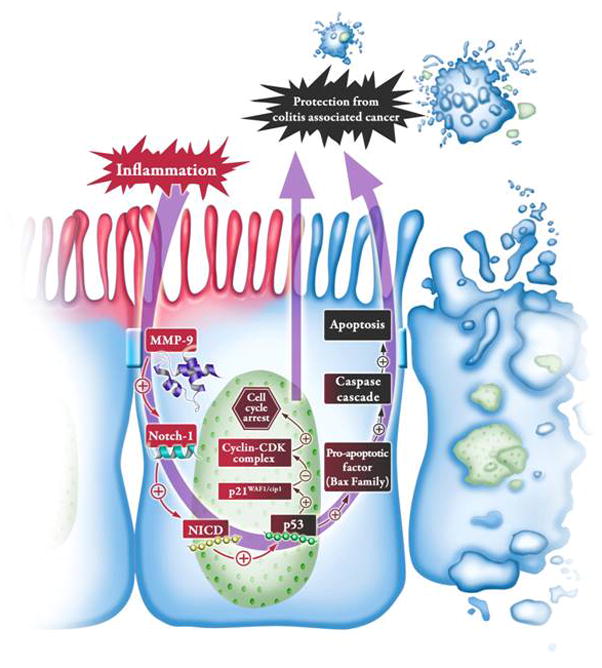
During inflammation, MMP9 expression is elevated and we have shown previously that MMP9 activates Notch1. Active Notch1 (NICD) then translocates to nucleus and activates p53. Activation of p53 activates p21WAF1/Cip1 which promotes cell cycle arrest and thereby promoting the protection from CAC and/or activates caspase cascade leading to an increase in apoptosis and thereby resulting in protection from CAC. Thus MMP-9 acts as a tumor suppressor during CAC, likely through p53 activation via activation of Notch1.
Discussion
Inflammation is no longer just the reaction of cells to external injury but recently its role in association with a wide variety of diseases including cancer has now been well accepted. CAC is associated with IBD, is difficult to treat, and has high mortality 28. Immune-mediated mechanisms have been documented to link IBD and CAC 29, 30, though the molecular mechanisms by which inflammation promotes CAC is still unexplored. We have previously reported that MMP9−/− mice showed increased susceptibility to CAC as indicated by polyp number, polyp size and mortality rate 22. Based on our observation that MMP9 mediates cleavage of Notch1 19, we hypothesized that MMP9, via activation of Notch1 signaling, acts as a tumor suppressor in CAC. In the present study, we used a pharmacological inhibitor, DAPT (γ-secretase inhibitor), to inhibit Notch1 activation to test our hypothesis.
We observed that inhibition of Notch1 signaling by DAPT showed increased susceptibility to CAC as indicated by tumor multiplicity, tumor burden and dysplasia compared to vehicle-treated mice. We observed that there were increased mRNA levels of pro-inflammatory cytokines TNF-α and IF-γ, which are well known to promote colitis associated tumor development. In contrast, mRNA levels of IL-6 and IL-1β cytokines were low. This observation is in accordance with Grivennikov et al 31, 32, suggesting a protective role of IL-6 in CAC. In this manuscript we observed that upregulation of MMP9 in an inflammatory environment activates Notch1 signaling, p53 expression, p21WAF1/Cip1 and increased apoptosis. This was in accordance with the data generated using MEFs isolated from WT and MMP9−/− mice as well as by an in vitro model using stably transfected HCT116 cells overexpressing MMP9. Further, MEFs isolated from WT mice treated with DAPT showed significant decrease in p53 expression signifying the necessity of MMP9 mediated Notch1 activation for p53 expression and its downstream targets.
Notch1 has various important roles during embryogenesis and tissue homeostasis, however, its role in chronic inflammation or CAC is unclear. Chronic inflammation is critical for tumor progression through the proliferation and survival of tumor cells by regulating mediators or effectors 33, 34. So far, cross talk of Notch1 with other signaling pathways like hedgehog, BMP, WNT, NF-κB etc has been reported in the literature during chronic inflammation 35. Studies have shown minimal side effects with short-term blockade of either Notch1 or its ligand Delta-like 4, but long-term side effects were not investigated. On the other hand, Notch1 signaling in cancer biology is well illustrated and its contrasting role is surprising as it can act as tumor promoter in some circumstances, while it can also act as a tumor suppressor in others. Therefore, the two sides of Notch1, one that promotes and the other that suppresses tumorigenesis, is cellular context dependent and also depends on its crosstalk with other signal transduction pathways 36, 37, 38, 39. Studies in mice have shown that Notch1 signaling in sporadic colon cancer is oncogenic and is required for adenoma formation in response to elevated Wnt signaling that occurs in the APCMin mouse model of colon cancer 40, 41, 42, 43. In contrast to colon cancer, Notch1 signaling has been demonstrated to function as a tumor suppressor in skin cancer 44 and cervical cancer 45, 46. Recently, Liu et al have shown that that chronic Notch1 inhibition leads to vascular tumors in the liver and decreased survival 47. Our data is consistent with a tumor protective role for Notch1 in contrast to its role in sporadic colon cancer.
p53 is a well established tumor suppressor that plays an important role in the development of CAC as well as sporadic colon cancers. In CAC, p53 is one of the first oncogenes activated, in contrast to sporadic colon cancer where p53 is activated late during carcinogenesis 48, 49. p53−/− mice are highly susceptible to CAC 50. During carcinogenesis, unlike Notch1 activation, which can exert entirely opposite effects, p53 functions consistently as a negative regulator of cell proliferation by inducing cell cycle arrest or as an inducer of apoptosis via caspase-3 activation. p21WAf1/Cip1 acts as a blocker of two CDKs- CDK2 and CDC which are active in late G1, S, G2 and M phases and arrests cell cycle 51. On the other hand, during genotoxic damage, activated p53 induces pro-apoptotic factors like Noxa and PUMA which in turn induces another set of pro-apoptotic factors such as Bax and Bak and activates the caspase cascade leading to increased apoptosis 52. The factors involved in the decision of p53 to induce cell cycle arrest over apoptosis or vice versa are still unclear.
Several studies have shown the control of p53 expression by Notch1 signaling. According to these studies, Notch1 signaling can either suppress or increase p53 activity in a context dependent manner that is closely related to tumor promotion or suppression. Studies conducted with human T-ALL (T cell culture acute lymphoblastic leukemia), breast cancer and keratinocyte cell lines reported that Notch1 activation resulted in an increased cell survival though the activation of the PI3K-Akt pathway, which leads to increased MDM2 activity and consequent p53 degradation 53–56. In the case of primary melanomas, breast cancer and gliomas 53, 57, 58, 59, Notch1 activates genes with oncogenic potentials like MYC and CCND1 (which encodes cyclin D1), which in turn results in decreased apoptosis through downregulation of p53 53. In contrast, Notch1 can positively affect p53 expression leading to a growth inhibitor or pro-apoptotic function, e.g., in mouse and human keratinocyte, cervical carcinoma and Ewing’s sarcoma 53, 60, 61, 39, 62. It has been reported that Notch activation modulates activation of p53 via upregulation of HEY1 and HES1 (canonical Notch targets) 53, 63, which may play a role in inhibiting MDM2 transcription, and suppression of Ras and Myc mediated tumorigenesis in a p53-dependent manner 64.
Inhibition of Notch1 signaling has been implicated in a variety of tissue specific cancers 65. Targeting the inhibition of Notch1 signaling is important as it may cause the clinical reevaluation of anti-Notch1 cancer therapies. This will be a big step in elucidating the underlying paradox of Notch1 therapeutics. Our present study therefore implies that, in colonic epithelium, MMP9 mediates CAC by acting as a tumor suppressor through the activation of Notch1 signaling via p53 activation, resulting in increased apoptosis and/or cell cycle arrest. Interestingly our study demonstrates that, in vivo inhibition of Notch1 signaling resulted in a significant decrease of p53 protein expression, which was also supported by MEF data. Our MEF data shows that inhibition of Notch1 signaling by DAPT resulted in decreased expression of p53, signifying that MMP9 mediated Notch1 activation is necessary for p53 expression in colonic epithelium. These results together indicate the mechanistic pathway (Figure 7) by which MMP9 acts as a tumor suppressor via activation of Notch1, resulting in increased expression of p53 and, thereby, increased apoptosis or decreased cell growth because of cell cycle arrest due to increased p21WAF1/Cip1 expression. Our study advances the understanding of the role and mechanism by which MMP9 modulates CAC and opens avenues for novel therapeutic targets for CAC.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health of Diabetes and Digestive and Kidney Diseases (R24-DK-064399 center grant, RO1-DK064711 & RO1-DK076825 to S.V.S.; RO1-DK-071594 to D.M.; RO1-DK061417 to A.T.G.); and a career development grant from the Crohn’s and Colitis Foundation of America to P.G.
This work was supported by the National Institute of Health of Diabetes and Digestive and Kidney Diseases (R24-DK-064399 center grant, RO1-DK076825 to S.V.S.; RO1-DK071594 to D.M.; RO1-DK061417 to A.T.G.); and a career development grant from the Crohn’s and Colitis Foundation of America to P.G. We are thankful to Dr. R Kopan, Prof. Department of Molecular Biology and Pharmacology and the Department of Medicine (Division of Dermatology), Washington University (St. Louis, MO) for generously gifting the NICD construct. We appreciate the contribution of Dr. D. Sarma, Prof. Emeritus, Department of Pathology, University of Toronto for helpful discussions. We are thankful to Dr. Tracy S. Obertone, Emory University in proof reading the manuscript.
Footnotes
| List of Authors | Contribution |
| Pallavi Garg | study concept; acquisition analysis and interpretation of data; drafting and critical revision of the manuscript |
| Sabrina Jeppsson | acquisition of data; drafting of the manuscript |
| Guillaume Dalmasso | technical support |
| Amr M. Ghaleb | technical support |
| Beth B. McConnell | critical revision of the manuscript |
| Vincent W. Yang | critical revision of the manuscript |
| Andrew T. Gewirtz | critical revision of the manuscript |
| Didier Merlin | interpretation of data; critical revision of the manuscript |
| Shanthi V. Sitaraman | study concept; analysis and interpretation of data; critical revision of the manuscript |
No conflict of interest exists
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Naito Y, Yoshikawa T. Role of matrix metalloproteinases in inflammatory bowel disease. Mol Aspects Med. 2005;26:379–90. doi: 10.1016/j.mam.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 2.Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: a tail of a frog that became a prince. Nat Rev Mol Cell Biol. 2002;3:207–14. doi: 10.1038/nrm763. [DOI] [PubMed] [Google Scholar]
- 3.Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16:558–64. doi: 10.1016/j.ceb.2004.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baugh MD, Evans GS, Hollander AP, et al. Expression of matrix metalloproteases in inflammatory bowel disease. Ann N Y Acad Sci. 1998;859:249–53. doi: 10.1111/j.1749-6632.1998.tb11139.x. [DOI] [PubMed] [Google Scholar]
- 6.Baugh MD, Perry MJ, Hollander AP, et al. Matrix metalloproteinase levels are elevated in inflammatory bowel disease. Gastroenterology. 1999;117:814–22. doi: 10.1016/s0016-5085(99)70339-2. [DOI] [PubMed] [Google Scholar]
- 7.Pender SL, Tickle SP, Docherty AJ, et al. A major role for matrix metalloproteinases in T cell injury in the gut. J Immunol. 1997;158:1582–90. [PubMed] [Google Scholar]
- 8.Tarlton JF, Whiting CV, Tunmore D, et al. The role of up-regulated serine proteases and matrix metalloproteinases in the pathogenesis of a murine model of colitis. Am J Pathol. 2000;157:1927–35. doi: 10.1016/S0002-9440(10)64831-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seifert WF, Wobbes T, Hendriks T. Divergent patterns of matrix metalloproteinase activity during wound healing in ileum and colon of rats. Gut. 1996;39:114–9. doi: 10.1136/gut.39.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Medina C, Videla S, Radomski A, et al. Increased activity and expression of matrix metalloproteinase-9 in a rat model of distal colitis. Am J Physiol Gastrointest Liver Physiol. 2003;284:G116–22. doi: 10.1152/ajpheart.00036.2002. [DOI] [PubMed] [Google Scholar]
- 11.McKaig BC, McWilliams D, Watson SA, et al. Expression and regulation of tissue inhibitor of metalloproteinase-1 and matrix metalloproteinases by intestinal myofibroblasts in inflammatory bowel disease. Am J Pathol. 2003;162:1355–60. doi: 10.1016/S0002-9440(10)63931-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salmela MT, MacDonald TT, Black D, et al. Upregulation of matrix metalloproteinases in a model of T cell mediated tissue injury in the gut: analysis by gene array and in situ hybridisation. Gut. 2002;51:540–7. doi: 10.1136/gut.51.4.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castaneda FE, Walia B, Vijay-Kumar M, et al. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology. 2005;129:1991–2008. doi: 10.1053/j.gastro.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 14.Kossakowska AE, Medlicott SA, Edwards DR, et al. Elevated plasma gelatinase A (MMP-2) activity is associated with quiescent Crohn’s Disease. Ann N Y Acad Sci. 1999;878:578–80. doi: 10.1111/j.1749-6632.1999.tb07732.x. [DOI] [PubMed] [Google Scholar]
- 15.Garg P, Vijay-Kumar M, Wang L, et al. Matrix metalloproteinase-9-mediated tissue injury overrides the protective effect of matrix metalloproteinase-2 during colitis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G175–84. doi: 10.1152/ajpgi.90454.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garg PRM, Ravi A, Bockbrader K, Epstein S, Vijay-kumar M, Gewirtz AT, Merlin D, Sitaraman SV. Selective ablation of matrix metalloproteinase-2 exacerbates experimental colitis: contrasting role of gelatinases in the pathogenesis of colitis. J Immunol. 2006;177:4103–4112. doi: 10.4049/jimmunol.177.6.4103. [DOI] [PubMed] [Google Scholar]
- 17.Medina C, Santana A, Paz MC, et al. Matrix metalloproteinase-9 modulates intestinal injury in rats with transmural colitis. J Leukoc Biol. 2006;79:954–62. doi: 10.1189/jlb.1005544. [DOI] [PubMed] [Google Scholar]
- 18.Santana A, Medina C, Paz-Cabrera MC, et al. Attenuation of dextran sodium sulphate induced colitis in matrix metalloproteinase-9 deficient mice. World J Gastroenterol. 2006;12:6464–72. doi: 10.3748/wjg.v12.i40.6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garg P, Ravi A, Patel NR, et al. Matrix metalloproteinase-9 regulates MUC-2 expression through its effect on goblet cell differentiation. Gastroenterology. 2007;132:1877–89. doi: 10.1053/j.gastro.2007.02.048. [DOI] [PubMed] [Google Scholar]
- 20.Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–17. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 21.Russo MW, Wei JT, Thiny MT, et al. Digestive and liver diseases statistics, 2004. Gastroenterology. 2004;126:1448–53. doi: 10.1053/j.gastro.2004.01.025. [DOI] [PubMed] [Google Scholar]
- 22.Garg P, Sarma D, Jeppsson S, et al. Matrix metalloproteinase-9 functions as a tumor suppressor in colitis-associated cancer. Cancer Res. 2010;70:792–801. doi: 10.1158/0008-5472.CAN-09-3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooper HS, Murthy SN, Shah RS, et al. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–49. [PubMed] [Google Scholar]
- 24.Cooper HS, Murthy S, Kido K, et al. Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis-associated neoplasia in the human: a study of histopathology, B-catenin and p53 expression and the role of inflammation. Carcinogenesis. 2000;21:757–68. doi: 10.1093/carcin/21.4.757. [DOI] [PubMed] [Google Scholar]
- 25.Dalmasso G, Charrier-Hisamuddin L, Nguyen HT, et al. PepT1-mediated tripeptide KPV uptake reduces intestinal inflammation. Gastroenterology. 2008;134:166–78. doi: 10.1053/j.gastro.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoon HS, Ghaleb AM, Nandan MO, et al. Kruppel-like factor 4 prevents centrosome amplification following gamma-irradiation-induced DNA damage. Oncogene. 2005;24:4017–25. doi: 10.1038/sj.onc.1208576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghaleb AM, Katz JP, Kaestner KH, et al. Kruppel-like factor 4 exhibits antiapoptotic activity following gamma-radiation-induced DNA damage. Oncogene. 2007;26:2365–73. doi: 10.1038/sj.onc.1210022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feagins LA, Souza RF, Spechler SJ. Carcinogenesis in IBD: potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol. 2009;6:297–305. doi: 10.1038/nrgastro.2009.44. [DOI] [PubMed] [Google Scholar]
- 29.Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 30.Terzic J, Grivennikov S, Karin E, et al. Inflammation and colon cancer. Gastroenterology. 2010;138:2101–2114. e5. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 31.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rose-John S, Mitsuyama K, Matsumoto S, et al. Interleukin-6 trans-signaling and colonic cancer associated with inflammatory bowel disease. Curr Pharm Des. 2009;15:2095–103. doi: 10.2174/138161209788489140. [DOI] [PubMed] [Google Scholar]
- 33.Mantovani A, Allavena P, Sica A, et al. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 34.Katoh M. Transcriptional regulation of WNT2B based on the balance of Hedgehog, Notch, BMP and WNT signals. Int J Oncol. 2009;34:1411–5. [PubMed] [Google Scholar]
- 35.Katoh M. Notch signaling in gastrointestinal tract (review) Int J Oncol. 2007;30:247–51. [PubMed] [Google Scholar]
- 36.Weng AP, Nam Y, Wolfe MS, et al. Growth suppression of pre-T acute lymphoblastic leukemia cells by inhibition of notch signaling. Mol Cell Biol. 2003;23:655–64. doi: 10.1128/MCB.23.2.655-664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maillard I, Pear WS. Notch and cancer: best to avoid the ups and downs. Cancer Cell. 2003;3:203–5. doi: 10.1016/s1535-6108(03)00052-7. [DOI] [PubMed] [Google Scholar]
- 38.Wang Z, Zhang Y, Li Y, et al. Down-regulation of Notch-1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol Cancer Ther. 2006;5:483–93. doi: 10.1158/1535-7163.MCT-05-0299. [DOI] [PubMed] [Google Scholar]
- 39.Nicolas M, Wolfer A, Raj K, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33:416–21. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 40.Korinek V, Barker N, Morin PJ, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–7. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 41.Nakamura T, Tsuchiya K, Watanabe M. Crosstalk between Wnt and Notch signaling in intestinal epithelial cell fate decision. J Gastroenterol. 2007;42:705–10. doi: 10.1007/s00535-007-2087-z. [DOI] [PubMed] [Google Scholar]
- 42.Fre S, Huyghe M, Mourikis P, et al. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005;435:964–8. doi: 10.1038/nature03589. [DOI] [PubMed] [Google Scholar]
- 43.Reedijk M, Odorcic S, Zhang H, et al. Activation of Notch signaling in human colon adenocarcinoma. Int J Oncol. 2008;33:1223–9. doi: 10.3892/ijo_00000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Devgan V, Mammucari C, Millar SE, et al. p21WAF1/Cip1 is a negative transcriptional regulator of Wnt4 expression downstream of Notch1 activation. Genes Dev. 2005;19:1485–95. doi: 10.1101/gad.341405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Talora C, Sgroi DC, Crum CP, et al. Specific down-modulation of Notch1 signaling in cervical cancer cells is required for sustained HPV-E6/E7 expression and late steps of malignant transformation. Genes Dev. 2002;16:2252–63. doi: 10.1101/gad.988902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Talora C, Cialfi S, Segatto O, et al. Constitutively active Notch1 induces growth arrest of HPV-positive cervical cancer cells via separate signaling pathways. Exp Cell Res. 2005;305:343–54. doi: 10.1016/j.yexcr.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 47.Liu Z, Turkoz A, Jackson EN, et al. Notch1 loss of heterozygosity causes vascular tumors and lethal hemorrhage in mice. J Clin Invest. 2011;121:800–8. doi: 10.1172/JCI43114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang WC, Coudry RA, Clapper ML, et al. Loss of p53 enhances the induction of colitis-associated neoplasia by dextran sulfate sodium. Carcinogenesis. 2007;28:2375–81. doi: 10.1093/carcin/bgm134. [DOI] [PubMed] [Google Scholar]
- 49.Fujii S, Fujimori T, Kawamata H, et al. Development of colonic neoplasia in p53 deficient mice with experimental colitis induced by dextran sulphate sodium. Gut. 2004;53:710–6. doi: 10.1136/gut.2003.028779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Donehower LA. The p53-deficient mouse: a model for basic and applied cancer studies. Semin Cancer Biol. 1996;7:269–78. doi: 10.1006/scbi.1996.0035. [DOI] [PubMed] [Google Scholar]
- 51.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 52.Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 53.Dotto GP. Crosstalk of Notch with p53 and p63 in cancer growth control. Nat Rev Cancer. 2009;9:587–95. doi: 10.1038/nrc2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mungamuri SK, Yang X, Thor AD, et al. Survival signaling by Notch1: mammalian target of rapamycin (mTOR)-dependent inhibition of p53. Cancer Res. 2006;66:4715–24. doi: 10.1158/0008-5472.CAN-05-3830. [DOI] [PubMed] [Google Scholar]
- 55.Nair P, Somasundaram K, Krishna S. Activated Notch1 inhibits p53-induced apoptosis and sustains transformation by human papillomavirus type 16 E6 and E7 oncogenes through a PI3K-PKB/Akt-dependent pathway. J Virol. 2003;77:7106–12. doi: 10.1128/JVI.77.12.7106-7112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Palomero T, Dominguez M, Ferrando AA. The role of the PTEN/AKT Pathway in NOTCH1-induced leukemia. Cell Cycle. 2008;7:965–70. doi: 10.4161/cc.7.8.5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Balint K, Xiao M, Pinnix CC, et al. Activation of Notch1 signaling is required for beta-catenin-mediated human primary melanoma progression. J Clin Invest. 2005;115:3166–76. doi: 10.1172/JCI25001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Politi K, Feirt N, Kitajewski J. Notch in mammary gland development and breast cancer. Semin Cancer Biol. 2004;14:341–7. doi: 10.1016/j.semcancer.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 59.Purow BW, Haque RM, Noel MW, et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005;65:2353–63. doi: 10.1158/0008-5472.CAN-04-1890. [DOI] [PubMed] [Google Scholar]
- 60.Estrach S, Cordes R, Hozumi K, et al. Role of the Notch ligand Delta1 in embryonic and adult mouse epidermis. J Invest Dermatol. 2008;128:825–32. doi: 10.1038/sj.jid.5701113. [DOI] [PubMed] [Google Scholar]
- 61.Lefort K, Mandinova A, Ostano P, et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007;21:562–77. doi: 10.1101/gad.1484707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang L, Qin H, Chen B, et al. Overexpressed active Notch1 induces cell growth arrest of HeLa cervical carcinoma cells. Int J Gynecol Cancer. 2007;17:1283–92. doi: 10.1111/j.1525-1438.2007.00927.x. [DOI] [PubMed] [Google Scholar]
- 63.Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194:237–55. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- 64.Huang Q, Raya A, DeJesus P, et al. Identification of p53 regulators by genome-wide functional analysis. Proc Natl Acad Sci U S A. 2004;101:3456–61. doi: 10.1073/pnas.0308562100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ryeom SW. The cautionary tale of side effects of chronic Notch1 inhibition. J Clin Invest. 2011;121:508–9. doi: 10.1172/JCI45976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.