

Abstract
Marizomib (NPI-0052) is a naturally derived irreversible proteasome inhibitor that potently induces apoptosis via a caspase-8 and ROS-dependent mechanism in leukemia cells. We aim to understand the relationship between the irreversible inhibition of the proteasome and induction of cell death in leukemia cells by using analogs of marizomib that display reversible and irreversible properties. We highlight the importance of sustained inhibition of at least two proteasome activities as being key permissive events for the induction of the apoptotic process in leukemia cells. These data provide the basis for the development of new approaches to generate more effective anti-proteasome therapies.
Keywords: Proteasome inhibitors, leukemia, apoptosis, caspase-8, oxidative stress
1. Introduction
Small molecule inhibitors have afforded improved understanding of the proteasome, the large multicatalytic complex responsible for degrading the majority of cellular proteins. Marizomib, formerly known as NPI-0052, is a second-generation proteasome inhibitor that is currently in clinical trials for solid and liquid tumors. Studies in multiple myeloma and our work in leukemia have demonstrated that this irreversible inhibitor of the proteasome effectively blocks the enzymatic activities associated with the proteasome: the chymotrypsin-like (CT-L), caspase-like (C-L) and trypsin-like (T-L) activities. These activities are located in beta subunits (β5, β1 and β2, respectively) that contain a catalytic N-terminal threonine (Thr1) responsible for hydrolyzing substrates [1]. In leukemia cells, marizomib displays the distinct profile of blocking the CT-L and C-L activities potently, while T-L activity is inhibited to a lesser extent [2].
In both leukemia and multiple myeloma cells, marizomib potently triggers apoptosis by a caspase-8 dependent mechanism. [2, 3]. Further examination in leukemia cells revealed that oxidative stress contributes to the cytotoxicity of marizomib since depleting intracellular reactive oxygen species (ROS) levels with antioxidants rescued the cells from apoptosis [2]. Our previous analyses comparing the irreversible β-lactone inhibitor, marizomib, and the reversible peptidyl proteasome inhibitor, bortezomib, showed that bortezomib was less potent at targeting proteasomal activities and inducing ROS levels than marizomib in the Jurkat ALL cell line [2, 4]. This prompted us to question whether the reversible versus irreversible nature of these inhibitors was critical in apoptosis induction via caspase-8 and ROS dependent pathways. Since irreversible proteasome inhibitors should cause longer lasting inhibition of the proteasome, we wanted to determine if duration of proteasome inhibition was influential in these biochemical events.
Marizomib’s structure contains a β-lactone ring that is uniquely substituted with a chloroethyl group that plays a role in its irreversible properties. In co-crystallization studies, this group has been shown to occupy the S2 binding pocket (and is thus referred to as a “P2” residue in analogy with peptidyl inhibitors) [5, 6]. Importantly, the chlorine behaves as a leaving group (LG) that is eliminated to render a stable cyclic ether end product following acylation of the catalytic enzyme active site Thr1Oγ by the β-lactone of the inhibitor [6]. In order to test the hypothesis that LG elimination confers irreversible binding properties, analogs comprising P2 substituents with a range of LG potentials have been synthesized, including halogen LGs, non-halogen LGs, and non-LGs. Inhibition/recovery experiments using dialysis of the analogs in complex with purified proteasomes demonstrated that the presence of a LG resulted in an irreversible inhibitor, prolonging the duration of proteasome inhibition, whereas the non-LG compounds recovered proteasome activity over time. Thus, the non-LG compounds were identified as slowly reversible inhibitors [7].
The LG and non-LG analogs of marizomib offer the unique opportunity to study the significance of reversible versus irreversible properties within one structural class of proteasome inhibitor in regards to inhibiting proteasomal enzymatic activities and promoting apoptotic biochemical endpoints. Since proteasome inhibition has been shown to be a viable therapeutic strategy for hematological malignancies such as leukemia, it is important to understand the properties of proteasome inhibitors that limit or contribute to their efficacy. Our work is the first to relate proteasome inhibition by these analogs to key initiating events in the induction of cell death in whole cells and highlights the importance of sustained inhibition of at least two proteasome activities as being critical for induction of the apoptotic program in leukemia cells.
2. Materials and methods
2.1 Cells, tissue culture and reagents
The T-acute lymphocytic leukemia (ALL) leukemia cell line, Jurkat, was purchased from ATCC (American Type Culture Collection; Manassas, VA), and maintained in RPMI medium prepared with 10% fetal bovine serum (FBS; Hyclone, Logan, UT), 100 UI/mL penicillin and 100 μg streptomycin and 2 mM L-glutamine (Cellgro, Mediatech Inc, Herndon, VA). The Molt-4 cell line was provided by Dr. Patrick Zweidler-McKay, M.D., Ph.D. (MD Anderson Cancer Center, Houston, TX) and was grown under the same culture conditions as the Jurkat cell line except that the media contained 20% FBS. Cells were grown at 37°C with 5% CO2. All cell lines were fingerprinted to insure authenticity. Cultured cells are replaced after every two months in culture with early passage cells stocks that were frozen within two weeks after purchasing from ATCC. Marizomib, the LG analogs (NPI-2053, NPI-2070, NPI-2056 and NPI-2151) and non-LG analogs (NPI-2104, NPI-0047, NPI-2078 and NPI-2077) were kindly provided by Nereus Pharmaceuticals (San Diego, CA).
2.2 Proteasome activity assays
As previously described, proteasome activities were analyzed by following the hydrolysis of fluorogenic substrates suc-LLVY-amc, z-LLE-amc (AG Scientific, Inc.,San Diego, CA), and boc-LLR-amc (Bachem, King of Prussia, PA), to measure the chymotrypsin-, caspase-, and trypsin-like activities respectively. A spectrofluorometer (SpectraMax Gemini EM, Molecular Devices, Sunnyvale, CA) with an excitation 380 nm and emission of 460 nm analyzed fluorescence released, shown as relative fluorescence units (RFU). Graphs display RFU values or % proteasome activity when results of analog-treated cells were normalized to control (DMSO-treated) cells, which were considered to display 100% activity.
2.3 Cell viability assays
Jurkat cells were harvested after exposure to marizomib and analogs for 24 h, washed and stained with trypan blue. Cell viability was assessed by trypan blue exclusion using a Beckman Coulter Counter (Brea, CA).
2.4 Assessment of DNA fragmentation and cell size
Cells were treated with analogs for 24 h and stained with propidium iodide (PI) over night at 4°C and analyzed by flow cytometry (FACSCalibur; Becton Dickinson; Franklin Lakes, NJ), as previously described [8]. Apoptosis was determined by measuring the percent of subdiploid population using Cell Quest Software (BD Bioscience). Plots of forward scatter versus side scatter were used to examine cell size, and gating of the control population was used to determine changes in size after treatment with the analogs.
2.5 Hoecsht Staining
Cells were treated with 100 nM marizomib or NPI-2078 for 24 h, following which 10,000 cells were suspended in 150 μL of media and stained with 5 μM Hoechst 33342 stain for 20 min (Invitrogen, Carlsbad, CA). Cells were then transferred to slides using a Wescor Cytopro centrifuge and viewed by fluorescence microscopy.
2.6 ROS measurements
After treatment, cells were stained with either dihydroethidine (HEt) or CM-H2DCF-DA (DCF; Molecular Probes, Eugene, OR) for 30 min in the dark to measure superoxide and hydrogen peroxide levels, respectively. Samples were stained and analyzed by flow cytometry either in the FL-3 or FL-1 channel as previously described [9].
2.7 Western blotting
Protein lysates from 5x106 cells were collected and 50 μg of protein was separated by SDS-PAGE and transferred to nitrocellulose membrane. Membranes were blotted with caspase-8 antibody at a 1:500 dilution (Cell Signaling, Beverly, MA), β5 subunit antibody at a 1:1000 dilution (Enzo Life Sciences, Farmingdale, NY) or PARP antibody at a 1:2000 dilution (kindly provided by Scott Kauffman, M.D., Ph.D., Mayo Clinic, Rochester, MN), followed by mouse or rabbit secondary antibodies used at 1:5000 dilutions. All antibodies were diluted in 5% milk in TBS with 0.005% Tween. Bound antibodies were detected by using enhanced chemiluminescense (ECL western blotting detection system, Amersham Biosciences UK limited, Little Chalfont Buckinghamshire, England). Densitometry values were determined using ImageJ software. Intensity readings were standardized using the loading control actin (Figures 1C and 2D) or by determining a ratio of cleaved (active) versus whole (inactive) caspase 8 (Figure 4A-B).
Figure 1. Non-LG analogs and LG analogs differ in blocking the CT-L activity at equimolar concentrations.

A. Inhibition of chymotrypsin-like activity. Jurkat cells were treated with 100 nM of each analog for 1 h. The fluorescent substrate suc-LLVY-amc was used to measure CT-L activity. Control cells (treated with DMSO) were normalized to represent 100% activity and cells treated with analogs were compared to control. Shown is the percent of chymotrypsin-like activity observed for each sample for three independent experiments. P values were determined by one way ANOVA and the Bonferroni post-hoc test. B. CT-L activity is recovered over time by non-LG analogs and sustained with LG analogs. Enzymatic activity was analyzed after harvesting cells treated with analogs for 1, 6, 12 and 16 h. C. Level of β5 protein is not decreased after treatment with the LG analog marizomib or the non-LG analog NPI-2078. Jurkat cells treated with 100 nM marizomib or 100 nM NPI-2078 for 1, 6, and 12 h were probed for the chymotryptic-like catalytic subunit, β5. Densitometry is the β5 value normalized to actin.
Figure 2. Inhibition of proteasome catalytic activity by the analogs correlates with cytotoxicity.

A. Cell viability is decreased with LG analogs but not with non-LG analogs. Jurkat cells were treated with 100 nM of the analogs for 24 h, and then their viability was assessed by trypan blue exclusion. B. The LG analog marizomib, but not the non-LG analog NPI-2078, leads to a change in Jurkat cell size compared to the control population. Cell size measurements were obtained from the forward scatter/side scatter plot from flow cytometry analysis. C. Marizomib-treated Jurkat cells, but not NPI-0078-treated cells, display shrunken and fragmented nuclei as visualized by Hoechst staining. D. PARP is cleaved in Jurkat cells treated with marizomib, but not in cells treated with NPI-2078. Cells treated with 100 nM of each drug for 16 h were examined for cleavage of PARP, which is a molecular event characteristic of apoptosis. Densitometry values are for the cleaved PARP band, normalized to actin. E. LG analogs induce DNA fragmentation. A portion of the cells plated and treated for (A) were stained with propidium iodide and analyzed by flow cytometry for DNA fragmentation. F. Molt-4 cells undergo more DNA fragmentation when treated with LG analogs than when treated with equimolar doses of non-LG analogs.
Figure 4. Caspase-8 activation by analogs of marizomib.

A. LG analogs but not non-LG analogs activate caspase-8 at equimolar concentrations. Protein lysates from Jurkat cells treated with 100 nM of LG and non-LG analogs for 6 h were analyzed for caspase-8 activation by western blot using an antibody that recognizes both pro-caspase 8 and cleaved, active caspase-8. B. At equipotent concentrations, both LG and non-LG analogs activate caspase-8. Protein extracts from Jurkat cells treated with concentrations that induced 50% of DNA fragmentation (4.5 μM NPI-2078, 1 μM NPI-2104 and 25 nM marizomib) or equivalent concentrations (100 nM of NPI-2078, NPI-2104 and marizomib) for 6 h were probed for caspase-8. Densitometry values for (4A) and (4B) reflect the ratio of cleaved (active) to uncleaved, pro-caspase-8. C. DNA fragmentation induced by the analogs is dependent on caspase-8. Pre-treatment with the caspase-8 inhibitor z-IETD-fmk prevented DNA fragmentation from occurring in Jurkat cells treated for 24 h with 100 nM of the analogs.
2.8 Inhibition of caspase 8
Cells were pre-treated for 30 min with 25 μM z-IETD-fmk, an inhibitor of caspase 8 (Enzo, Life Sciences, Farmingdale, NY). They were then treated with indicated doses of the analogs for 24 h before being stained with propidium iodide as described in section 2.4.
2.9 Statistical analyses
The data shown in this paper represent the mean ± standard deviation (SD) from three independent experiments, unless otherwise stated. Statistical analyses were conducted with GraphPad Prism software. Statistical significance of differences between analogs versus DMSO-treated cells was determined by one way analysis of variance (ANOVA). Time course experiments were evaluated using two way ANOVA. Each ANOVA was followed by post-hoc analysis with Bonferroni's multiple comparison test. The minimal level of significance was P < 0.05.
3. Results
3.1 CT-L activity is not inhibited at equimolar concentrations by non-LG versus LG analogs to the same degree and its inhibition correlates with cytotoxicity
We analyzed LG analogs versus non-LG analogs listed on Table 1 to determine if analogs of marizomib were able to inhibit the proteasome. The CT-L proteasomal activity was measured using the fluorescent substrate suc-LLVY-amc in Jurkat cells treated with marizomib or its analogs after 1 h exposure. This specific time point was chosen based on our previous detailed time course work demonstrating that marizomib induced caspase-8 activation as early as 2 h after proteasome inhibition and maximum cell death is observed at 24 h[2]. Following an 1 h incubation with 100 nM of NPI analogs, we observed that the CT-L activity was blocked more effectively in cells that had been treated with LG analogs, displaying less than 10% CT-L activity, compared to non-LG agents, some of them retaining more than 30% activity post-exposure (Figure 1A). The only exception was the non-LG analog NPI-2104, which achieved similar inhibition to LG analogs (about 90%). Since the CT-L activity was analyzed using equivalent concentrations (100 nM), these results suggest that equimolar concentrations are not equipotent at blocking this enzymatic activity. Based on results from inhibition/recovery experiments measuring proteasome activity after dialysis from purified proteasomes with analogs [7], we next evaluated CT-L proteasome activity over a period of time with analogs in leukemia whole cell lysates. Examination of CT-L activity overtime revealed that LG analogs maintained inhibition of this proteolytic activity from 1 h to 16 h (Figure 1B). In contrast, non-LG analogs suppressed this activity to a lesser extent at 1 h compared to LG analogs with the exception of NPI-2104, which consistently displayed a similar inhibition profile to LG analogs at 1 h (Figures 1A and 1B). However, as expected based on the absence of a LG as the P2 substituent, CT-L activity started recovering with non-LG analogs over the 16 h period, firmly establishing them as slowly reversible inhibitors. To see whether the ability of the agents to inhibit the proteasome was due to diminished proteasome protein content in the cells, β5 protein (the chymotryptic-like subunit of the proteasome) was examined by Western blot between 1 h and 16 h after treatment with marizomib and a representative non-LG analog (NPI-2078). Neither inhibitor decreased levels of β5 protein, suggesting that the differential ability of the inhibitors to decrease chymotrypsin-like activity is not due to decreased levels of the targeted subunit (Figure 1C).
Table 1.
Marizomib (NPI-0052) and P2 analog structures.
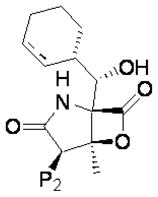 | |||
|---|---|---|---|
| NPI-# | P2 | Structural Class | MWt |
| Marizomib (NPI-0052) | CH2CH2Cl | LG | 313.78 |
| NPI-2056a | CH2CH2Cl | LG | 315.80 |
| NPI-2053 | CH2CH2Br | LG | 358.23 |
| NPI-2070 | CH2CH2I | LG | 405.24 |
| NPI-2151 | CH2CH2OTosyl | LG | 449.53 |
| NPI-2104 | CH2CH2SCN | Non-LG | 336.41 |
| NPI-2078 | CH2CH2N3 | Non-LG | 320.35 |
| NPI-2077 | CH2CH2OH | Non-LG | 295.34 |
| NPI-0047 | CH2CH3 | Non-LG | 279.34 |
Cyclohexenyl is replaced with cyclohexyl in NPI-2056 only.
Next, we wanted to correlate proteasome inhibition with cytotoxicity. Therefore we measured cell viability by trypan blue exclusion in Jurkat cells exposed to marizomib and its analogs at equimolar concentrations. After 24 h exposure, LG analogs but not non-LG analogs decreased cell viability (Figure 2A). Cell size was also analyzed by gating the control population in the plot of forward scatter versus side scatter from flow cytometry of Jurkat cells. As indicated in Figure 2D, cells treated with the LG analog marizomib displayed altered cell size as shown by fewer cells matching the control population size, whereas cells treated with the non-LG analog NPI-2078 did not show a large change in cell size. Jurkat cells treated with these analogs were also Hoechst stained so that nuclear morphology could be examined. Nuclei of cells treated with marizomib show shrinkage and fragmentation of the DNA, whereas cells treated with NPI-2078 do not (Figure 2C). To further implicate apoptosis as the pathway initiated by the LG analogs, the caspase 3/7 mediated cleavage of PARP was examined after 16 h treatment of Jurkat cells with marizomib and the non-LG analog NPI-2078. PARP cleavage occurred in cells treated with marizomib, but not in cells treated with NPI-2078 (Figure 2D). Accordingly, only LG analogs induced DNA fragmentation, a hallmark of apoptosis, as determined by propidium iodide staining followed by flow cytometry analysis, in the leukemia cells compared to non-LG analogs (Figure 2E). These results reflected the CT-L time course observations (Figure 1B). For example, a partial recovery of proteasome activity was detected with NPI-2056 at 16 h and this correlated with the observation that cells treated with this compound displayed more viable cells and less DNA fragmentation compared to cells exposed with the other LG analogs, which sustained CT-L inhibition after 16 h and achieved more DNA fragmentation and less cell viability (Figure 1B, 2A and 2E).
To show that the cell death trend seen in Jurkat cells can be expanded to other cell lines, the ALL cell line Molt-4 was treated with the analogs and DNA fragmentation was examined. Similarly to the Jurkat cells, the LG analogs caused more DNA fragmentation in these cells than an equimolar dose of the non-LG analog NPI-2078.
3.2 Both LG and non-LG analogs similarly block the CT-L at equipotent concentrations
Since our results suggested that equimolar doses do not target the CT-L activity to the same extent, we next wanted to identify doses that would exert similar effects on cell death. Therefore, we determined IC50 values for each of the analogs based on cell viability as measured by trypan blue staining (Figure 3A-B). Graphical representation of this data (Figure 3A) illustrates that the LG analogs consistently have lower IC50 values than non-LG analogs. We also measured DNA fragmentation to evaluate cytotoxicity by select doses nearing the IC50. Dose response experiments (not shown) revealed that 4.5 μM NPI-2078, 1.0 μM NPI-2104 and 25 nM marizomib induced approximately 50% DNA fragmentation (Figure 3C) in the total cell population. Because different endpoints were measured for determination of the doses for IC50 based on trypan blue exclusion (Figure 3A-B) versus doses that cause 50% DNA fragmentation (Figure 3C), the exact dose values are slightly different. However, it should be noted that the trend is comparable between the two methods of determining equipotent doses, with the LG analogs causing a 50% effect in the nanomolar dose range, while non-LG analogs require micromolar doses to achieve a 50% effect.
Figure 3. Doses of analogs that cause equipotent proteasome inhibition are similarly cytotoxic.

A. Higher doses of non-LG analogs are required to cause decreases in viability of Jurkat cells as measured by trypan blue exclusion. B. Non-LG analogs have IC50 values in the micromolar range, compared to the LG analogs with have IC50s in the nanomolar range. IC50 doses were determined from the viability readings in (3A). C. Equipotent concentrations were identified to induce DNA fragmentation. Jurkat cells were treated with increasing concentrations of marizomib, NPI-2078 and NPI-2104 for 24 h. DNA fragmentation was assessed by propidium iodide staining followed by flow cytometry. Shown are effects of concentrations identified to induce DNA fragmentation in 50% of the cell population. D. Analysis of CT-L activity with equipotent concentrations. Jurkat cells were treated with equipotent concentrations identified in (C) of NPI-2078, NPI-2104 and marizomib. Samples were analyzed for CT-L activity after 1 and 12 h of exposure to analogs. Control cells treated with DMSO were defined as exhibiting 100% of CT-L activity. * P < 0.05 control cells compared to treatment (two way ANOVA and post-hoc analysis with Bonferroni’s test). LG: leaving group, Non-LG: non-leaving group.
Next, Jurkat cells were treated with NPI-2078, NPI-2104 or marizomib at equivalent cytotoxic concentrations for 1 h and the CT-L activity was analyzed. Results showed that these concentrations of LG and non-LG analogs blocked the CT-L activity to the same degree (Figure 3D), displaying only 10% of activity compared to control levels (100% activity). However, 10-fold lower concentrations of NPI-2104 (100 nM) also resulted in 90% inhibition (Figure 1A) but this activity was recovered over a 16 h period at this dose (Figure 1B), thus, the higher dose of 1.0 μM NPI-2104 could potentially be sufficient to initially saturate proteasome binding sites and also provide an excess reservoir of ligand in the cell over time. The CT-L activity 12 h interval results provide an explanation by demonstrating that activity is not recovered as robustly with 1 μM NPI-2104, displaying 28.6% activity (Figure 3D), compared to 100 nM NPI-2104 which exhibits 64.5% activity (Figure 1B).
3.3 Equivalent cytotoxic concentrations induce similar caspase-8 activation and ROS generation with LG and non-LG analogs
We next extended our study to examine ROS production and caspase-8 activation, two important events we had previously described in marizomib-induced apoptosis in leukemia cells. Using caspase-8 deficient cells we had previously established that marizomib relies on this initiator caspase to exert its cytotoxic effects in leukemia cells [2]. Therefore, here we analyzed cleavage of pro-caspase-8 by LG and non-LG compounds. After a 6 h incubation with analogs at equimolar concentrations (100 nM), western blot results showed that caspase-8 activation was detected after treatment with LG analogs but not with non-LG analogs, with the exception of NPI-2104, for which caspase-8 was weakly activated (Figure 4A). Next, after previously establishing equipotent concentrations that exhibited similar cytotoxicity profiles by representative analogs (Figure 3C), we assessed caspase-8 activation by western blot with these concentrations. Analysis of caspase-8 cleavage revealed detection of cleaved products, suggesting caspase-8 activation occurred in cells treated with each of the three analogs, NPI-2078, NPI-2104 and marizomib (Figure 4B). However, much higher (1.6 – 2.3 log) concentrations of non-LG analogs were required to achieve levels of caspase-8 activation obtained with low concentrations (25 nM) of marizomib.
Since apoptosis is mediated by caspases, activation of caspase-8 at 6 h by analogs also suggested that the DNA fragmentation observed at 24 h (Figure 2E and 3C) was a result of triggering the apoptotic process. We demonstrated that the DNA fragmentation caused by the LG analogs is caspase-8 dependent by using the caspase-8 inhibitor z-IETD-fmk. Pre-treatment with this caspase-8 inhibitor prevented NPI analog-induced DNA fragmentation (Figure 4C).
Our previous data demonstrate that marizomib induces oxidant-dependent cell death in leukemic cells [2], thus we next wanted to evaluate ROS expression with analogs and determine whether reversible versus irreversible analogs would differ in their production of ROS levels in a leukemia cell line at both equipotent and equimolar concentrations. Results demonstrate that in a time-dependent manner, LG analogs (NPI-2053, NPI-2070, NPI-2056 and NPI-2151) steadily increased both intracellular superoxide (over 7-fold increase at 16 h) and hydrogen peroxide levels compared to cells treated with diluent. The time course also revealed that elevated superoxide levels were persistently increasing over a 24 h period (Figure 5A, left graph), while hydrogen peroxide levels peaked at 12 h and then decreased (Figure 5B, right graph). In these experiments, all of the LG analogs generated ROS to a similar degree as marizomib. In contrast, non-LG analogs (NPI-0047and NPI-2077) expressed similar levels of ROS compared to control treated cells (Figure 5B). NPI-2104, and to a lesser extent NPI-2078, were the only non-LG analogs that displayed some ROS production, showing a “weak” effect since it was significantly less than levels achieved with marizomib. Previously, our lab showed that treatment with the antioxidant N-acetyl cysteine (NAC) diminished accumulation of ROS without affecting the degree of proteasome inhibition achieved by marizomib, indicating that ROS production occurs downstream from proteasome inhibition [2]. Since NPI-2104 was the most potent inhibitor of CT-L activity among non-LG analogs (Figure 1A) and because ROS generation occurs downstream of proteasome inhibition, it was hypothesized that the failure of less potent non-LG analogs to generate ROS might be overcome with higher concentrations of inhibitor. Indeed, monitoring intracellular superoxide levels with representative LG and non-LG analogs at equipotent doses showed that there were a higher percentage of cells displaying superoxide levels with all analogs, compared to control cells (Figure 5C, left graph). In addition, an increase was observed when examining hydrogen peroxide levels in cells treated with the three analogs in comparison to cells treated with diluent (Figure 5C, right graph). All together these results suggested that at equipotent concentrations, both LG and non-LG analogs behaved similarly to marizomib, generating more ROS and caspase-8 activation. Therefore, dose escalation of non-LG analogs was sufficient to compensate for their reversible properties, however, as in the case of caspase-8, the required dose-escalation was dramatic (> 1 log).
Figure 5. Production of intracellular superoxide and hydrogen peroxide levels by LG and non-LG analogs.

A. LG analogs increase superoxide and hydrogen peroxide levels over time. Left graph: Hydroethidium (HEt) staining assessed intracellular superoxide levels in Jurkat cells treated with 100 nM of LG analogs. Graphed are the mean fluorescent values from flow cytometry readings. * P < 0.05 cells treated with analogs for 12 h compared to cells treated with DMSO for 12 h. Right graph: DCF staining determined hydrogen peroxide levels in cells. * P < 0.05 cells treated with analogs for 12 h compared to cells treated with DMSO for 12 h. B. The non-LG analogs, NPI-2104 and NPI-2078, induce weak ROS levels. Cells were treated with 100 nM of non-LG analogs and analyzed for superoxide expression (left graph) or hydrogen peroxide levels (right graph). C. Superoxide and hydrogen peroxide levels were increased with LG and non-LG analogs at equipotent concentrations. After treatment with indicated concentrations of NPI-2078, NPI-2104 and marizomib for over a period of 16 h, cells were analyzed for superoxide or hydrogen peroxide expression. Inset. Representative histograms of cells treated with DMSO (control) or 25 nM marizomib for 16 h. * P < 0.05 compares analog treated cells compared to control cells in HEt and DCF experiments. The level of significance of differences between means was determined by two way ANOVA with Bonferroni post-hoc comparisons.
3.4 Oxidative stress contributes towards cytotoxicity
Previously, we showed that marizomib induces reactive oxygen species production and that this contributes to its cytotoxicity [2]. Therefore, we wanted to look at the contribution of reactive oxygen species generation to the cytotoxicity of the reversible analogs. We examined whether the ROS production by analogs was influencing the lethal effects of these compounds. To implicate oxidative stress, cells were pretreated with the antioxidant NAC followed by subsequent exposure to analogs. Cell viability experiments demonstrated that NAC offered protection against LG analogs, which at 100 nM significantly decreased viability (Figure 6A). No benefits were observed with NAC in cells treated with 100 nM NPI-2104 or NPI-2078, most likely since at this concentration these analogs did not affect viability in Jurkat cells. In addition, NAC attenuated the DNA fragmentation induced by LG analogs (Figure 6B). Together, the protection offered by an antioxidant when assessing viability and DNA fragmentation indicates that the cytotoxicity observed with LG analogs is oxidant dependent.
Figure 6. Oxidant dependent cytotoxicity by LG analogs.

A. An antioxidant protects against LG analogs. Jurkat cells were treated with 24 mM NAC, which did not have toxic effects when added as a single agent to Jurkat cells, prior to exposure to analogs (100 nM concentration). After 24 h treatment with analogs viable cells were identified by trypan blue exclusion. P < 0.05 control (DMSO treated) cells compared to cell treated with LG analogs or comparison between cells exposed to LG analog with or without NAC. B. DNA fragmentation induced by LG analogs is prevented with an antioxidant. A portion of the cells treated in (A) were stained with propidium iodide and DNA fragmentation was assessed by flow cytometry. P < 0.05 comparing cell treated with LG analogs versus DMSO treated cells. P < 0.05 comparing cells treated with LG analogs versus cells exposed to NAC prior to treatment with LG analogs. One way ANOVA followed by Bonferroni post-hoc analyses determined P values.
3.5 Effects of equipotent doses on proteasome enzymatic activities
We then investigated whether the concentrations equipotent at inducing DNA fragmentation, ROS generation and caspase-8 activation were exerting similar effects on blocking all three proteolytic activities of the proteasome. Analyzing CT-L activity with suc-LLVY-amc substrates demonstrated that at 1 h, the three analogs significantly blocked this activity (> 90%), an inhibition that was sustained by marizomib after 12 h of exposure. In contrast, activity was gradually being recovered with NPI-2078 and NPI-2104 (70% inhibition after 12 h; Figure 7). Results with z-LLE-amc, examining the C-L activity, showed a similar effect, with all three compounds potently inhibiting this enzymatic activity. Specifically, 1 h incubation with marizomib resulted in 88% inhibition that was sustained over a period of 12 h (Figure 7). Inhibitors NPI-2078 and NPI-2104 significantly (P < 0.05) blocked this activity by ≥ 95% after 1 h when compared to control cells, but this activity was partially recovered with time (> 65% inhibition after 12 h). NPI-2104 and marizomib partially inhibited the T-L activity (11% and 24%, respectively, at 1h), and most significantly at a later time (> 30% at 12 h). In contrast, NPI-2078 had minimal effects on T-L activity, displaying about a 5% inhibition after 12 h. Overall, our results evaluating all three enzymatic activities of the 20S proteasome with these analogs at equipotent concentrations indicate that marizomib (a LG compound) and NPI-2104 (a non-LG compound) were able to target all three enzymatic activities of the proteasome to different degrees, whereas non-LG analog NPI-2078 blocked only two activities. Regardless of whether the analogs were able to block two or three enzymatic activities or whether they behaved as irreversible and slow-reversible inhibitors, all analogs were able to achieve similar caspase-8 cleavage and ROS production and ultimately cell death, as shown by DNA fragmentation, but only when equipotent concentrations were used. Together, our results examining programmed cell death associated end-points and proteasomal enzymatic activities highlight that perhaps inhibiting only two activities, in this case the chymotrypsin-like and caspase-like activities, is sufficient to induce these apoptosis associated events.
Figure 7. The CT-L and C-L activities are inhibited by LG and non-LG analogs at equipotent concentrations.

Jurkat cells were monitored for proteasomal enzymatic activities using fluorogenic peptides. Cells were treated with indicated concentrations of NPI-2078, NPI-2104 and marizomib for 1 h or 12 h prior to analysis. For CT-L and C-L activity * P < 0.05 compared to control 1 h (two way ANOVA and the Bonferroni post-hoc test).
4. Discussion
The structurally unique proteasome inhibitor marizomib has been shown to bind irreversibly to the proteasome based on its ability to eliminate its chlorine group in the P2 position to form a stable cyclic ether product that is irreversibly bound to the active site of the proteasome. Recent studies have reported the generation of analogs of marizomib that display either irreversible or slowly reversible properties based on the presence or absence of a LG in the P2 position (Table 1). While a previous study demonstrated that the LG analogs are more cytotoxic to multiple myeloma and prostate cancer cells than non-LG analogs, no work has yet been done to examine the effectiveness of these analogs in leukemia [10]. Importantly, possible apoptotic mechanisms that are key to the differential cytotoxicity of these drugs, such as activation of caspases and generation of reactive oxygen species, have not yet been explored. Also of note is the focus of past work on the chymotrypsin-like proteasomal activity, while the contribution of inhibition of the caspase-like and trypsin-like activities to the effects of the analogs has not yet been elucidated.
We took advantage of the availability of these structurally related analogs of marizomib to evaluate inhibition of proteasomal enzymatic activities and specific apoptotic biochemical end points that we had previously shown to be important in marizomib cytotoxicity. Taken together, our results with both equipotent and equimolar concentrations revealed that regardless of the binding nature of the inhibitor, all the analogs can block proteasome activity to some degree in Jurkat cells. Similar to previous results reported in purified 20S proteasomes [7], here we show that in leukemia cells proteasome activities are recovered over time with non-LG inhibitors. We further extend these observations by demonstrating the consequences of this inhibition profile on the concentrations required to trigger apoptotic signaling events.
We examined caspase-8 activation and ROS generation given our previous results that these two events were critical for marizomib to induce apoptosis in leukemia cells [2]. To date, these biochemical end-points have not been studied with analogs of marizomib. At equimolar concentrations, we initially observed that caspase-8 activation and ROS generation were only occurring with LG analogs but not with non-LG analogs, suggesting that perhaps irreversible and reversible properties were influencing these events. However, weak activation of caspase-8 and ROS production were induced by the most potent non-LG analog NPI-2104 (inhibiting CT-L activity by 90% at 1 h; Figure 1A), suggesting that the differential behavior might be overcome via dose escalation. A closer examination of these end-points using concentrations equipotent at inducing DNA fragmentation revealed that with much higher concentrations, non-LG analogs were able to achieve similar ROS production and caspase-8 activation as LG-containing marizomib. Interestingly, the ROS experiments demonstrate that LG analogs, at both equipotent and equimolar concentrations, and equipotent non-LG analogs consistently increased superoxide levels over time while hydrogen peroxide levels were initially elevated but after 12 h began to decrease. This information may lead to future studies examining the role of protective antioxidant systems in these cells. Previous work by another group found that heme oxygenase-1 (HO-1) attenuated hemin-induced oxidative stress and cell death in astrocytes. Interestingly, the proteasome inhibitor MG-132 protected against the oxidative injury caused by hemin via upregulation of HO-1 [11]. In fact, several studies implicate MG-132 and lactacystin as responsible for increasing expression of HO-1 mRNA and protein in astrocytes and macrophages [11–13]. Approaches to address the possible involvement of HO-1 and other antioxidant systems, such as catalase, peroxiredoxin or glutathione mediated systems in leukemia cells will be undertaken in future studies. Furthermore, our NAC experiments clearly indicate that the decreased viability and DNA fragmentation induced by LG analogs is being influenced by oxidant dependent systems, highlighting the importance of ROS not only in marizomib induced apoptosis but also with its analogs.
The activation of caspase-8 and ROS by non-LG analogs suggested that dose escalation was sufficient to compensate for their reversible properties. Although proteasomal activities were slowly recovered over time in the case of non-LG analogs, the equipotent concentrations provided marked levels of inhibition, even after 12 h (70% inhibition of CT-L and C-L activities). Since 90% initial inhibition in the case of non-LG analog NPI-2104 was insufficient to activate caspase-8 and generate ROS, we hypothesize that increased concentrations of inhibitor help to sustain inhibition over time, allowing the downstream events to proceed. Thus, the high concentrations of non-LG inhibitors effectively compensate for their reversible nature. Interestingly, we observed a difference at targeting proteasome activities between non-LG analogs NPI-2078 and NPI-2104. NPI-2104, similar to marizomib, was able to target all three enzymatic activities. In contrast, the non-LG analog NPI-2078 was not able to block the trypsin-like activity. However, it is important to note that the degree of inhibition of the T-L activity with any of the analogs was less than that achieved against CT-L- and C-L activities which supported earlier studies by Ruiz et al and Miller et al in chronic lymphocytic leukemia (CLL), acute myelogenous leukemia (AML) and ALL patient derived leukemia cells [2, 14]. Based on our findings, targeting of at least two proteasomal activities are key permissive events that promote these apoptotic signals. However, targeting of all three proteolytic sites can result in apoptotic signaling at lower levels of proteasome inhibition [3].
Although reversible inhibitors can also target the proteasome and induce apoptotic events, it must be cautioned that much higher concentrations are required to achieve the same effect with a non-LG reversible inhibitor. This may be attributed to a kinetic effect, such that higher concentrations of slowly reversible non-LG analogs are required to initially saturate the proteasome binding sites and to provide a reservoir of inhibitor to replenish the ligand population that has been eliminated from the binding site over time due to the reversible nature of binding. This idea is similar to that proposed by Dick et al in which omuralide, a non-LG β-lactone, reacted with glutathione in cultured cells to form an adduct to serve as a reservoir for later release of the active β-lactone [15]. Unlike competitive inhibitors, the non-LG inhibitors in this class may become inactivated during the course of removal from the active site and/or by hydrolysis after elimination due to irreversible opening of the β-lactone ring. Hence, fresh ligand is required to sustain inhibition. Thus, high levels of non-LG analogs not only help saturate binding sites 'initially' but may sustain inhibition over time, as evidenced by the marked (60–70%) inhibition that remains 12 h after exposure to high concentrations of non-LG analogs (Figure 7 and 3D) but not observed with the lower concentration of non-LG analogs, with inhibition levels ranging from 0–36% inhibition at 12 h (Figure 1B). Therefore the slower and/or sustained delivery of non-LG analogs may achieve similar anti-tumor effects as the potent, irreversible and fast-acting inhibitor marizomib. The potential to regulate proteasome inhibition duration and selective subunit activity with non-LG analogs provides for exciting new approaches to develop more specific anti-proteasome therapies. Since combination chemotherapy is the mainstay of acute leukemia treatment, our results suggest that use of the non-LG analogs in proteasome inhibitor based regimens where a short and reversible repression could potentiate the effect of other agents is a viable option. Improved understanding of the mechanism of these inhibitors will enable us to fine tune the clinical application of proteasome inhibitors, honing these therapeutics into ever more effective agents.
Highlights.
We use irreversible and reversible analogs of the proteasome inhibitor, marizomib.
Irreversible leaving group (LG) analogs of marizomib are more potent inducers of apoptosis in leukemia cells.
Inhibition of two proteasomal activities and the duration of inhibition are key factors in apoptosis induction.
Acknowledgments
This work was supported, in part, by the National Institutes of Health with grant R01 CA115811 to J.C. and fellowship F31CA123645-03 to C.P.M. The Cancer Prevention and Research Institute of Texas Research Training Award RP101502 provided additional support for C.A.M. We thank Rao Manam for providing some of the analogs used in this study. We also acknowledge Raelene Endersby for her assistance with formatting figures and Mary Irwin for her help with use of Prism software.
Footnotes
Conflict of interest. V.R.M., B.C.P., and M.A.P. are employees of Nereus Pharmaceuticals, Inc., the company that manufactures marizomib and its analogs. The remaining authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev. 2007;107(3):687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- 2.Miller CP, Ban K, Dujka ME, McConkey DJ, Munsell M, Palladino M, Chandra J. NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood. 2007;110(1):267–277. doi: 10.1182/blood-2006-03-013128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, Mitsiades C, Mitsiades N, Yasui H, Letai A, Ovaa H, Berkers C, Nicholson B, Chao TH, Neuteboom ST, Richardson P, Palladino MA, Anderson KC. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005;8(5):407–419. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 4.Miller CP, Rudra S, Keating MJ, Wierda WG, Palladino M, Chandra J. Caspase-8 dependent histone acetylation by a novel proteasome inhibitor, NPI-0052: a mechanism for synergy in leukemia cells. Blood. 2009 doi: 10.1182/blood-2008-08-174797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew Chem Int Ed Engl. 2003;42(3):355–357. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- 6.Groll M, Huber R, Potts BC. Crystal Structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in Complex with the 20S Proteasome Reveal Important Consequences of beta-Lactone Ring Opening and a Mechanism for Irreversible Binding. J Am Chem Soc. 2006;128(15):5136–5141. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]
- 7.Manam RR, McArthur KA, Chao TH, Weiss J, Ali JA, Palombella VJ, Groll M, Lloyd GK, Palladino MA, Neuteboom ST, Macherla VR, Potts BC. Leaving groups prolong the duration of 20S proteasome inhibition and enhance the potency of salinosporamides. J Med Chem. 2008;51(21):6711–6724. doi: 10.1021/jm800548b. [DOI] [PubMed] [Google Scholar]
- 8.Chandra J, Hackbarth J, Le S, Loegering D, Bone N, Bruzek LM, Narayanan VL, Adjei AA, Kay NE, Tefferi A, Karp JE, Sausville EA, Kaufmann SH. Involvement of reactive oxygen species in adaphostin-induced cytotoxicity in human leukemia cells. Blood. 2003;102(13):4512–4519. doi: 10.1182/blood-2003-02-0562. [DOI] [PubMed] [Google Scholar]
- 9.Chandra J, Tracy J, Loegering D, Flatten K, Verstovsek S, Beran M, Gorre M, Estrov Z, Donato N, Talpaz M, Sawyers C, Bhalla K, Karp J, Sausville E, Kaufmann SH. Adaphostin-induced oxidative stress overcomes BCR/ABL mutation-dependent and -independent imatinib resistance. Blood. 2006;107(6):2501–2506. doi: 10.1182/blood-2005-07-2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obaidat A, Weiss J, Wahlgren B, Manam RR, Macherla VR, McArthur K, Chao TH, Palladino MA, Lloyd GK, Potts BC, Enna SJ, Neuteboom ST, Hagenbuch B. Proteasome Regulator Marizomib (NPI-0052) Exhibits Prolonged Inhibition, Attenuated Efflux, and Greater Cytotoxicity than Its Reversible Analogs. J Pharmacol Exp Ther. 337(2):479–486. doi: 10.1124/jpet.110.177824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Regan RF. Increasing expression of heme oxygenase-1 by proteasome inhibition protects astrocytes from heme-mediated oxidative injury. Curr Neurovasc Res. 2005;2(3):189–196. doi: 10.2174/1567202054368344. [DOI] [PubMed] [Google Scholar]
- 12.Wu WT, Chi KH, Ho FM, Tsao WC, Lin WW. Proteasome inhibitors up-regulate haem oxygenase-1 gene expression: requirement of p38 MAPK (mitogen-activated protein kinase) activation but not of NF-kappaB (nuclear factor kappaB) inhibition. Biochem J. 2004;379(Pt 3):587–593. doi: 10.1042/BJ20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamamoto N, Izumi Y, Matsuo T, Wakita S, Kume T, Takada-Takatori Y, Sawada H, Akaike A. Elevation of heme oxygenase-1 by proteasome inhibition affords dopaminergic neuroprotection. J Neurosci Res. 2010;88(9):1934–1942. doi: 10.1002/jnr.22363. [DOI] [PubMed] [Google Scholar]
- 14.Ruiz S, Krupnik Y, Keating M, Chandra J, Palladino M, McConkey D. The proteasome inhibitor NPI-0052 is a more effective inducer of apoptosis than bortezomib in lymphocytes from patients with chronic lymphocytic leukemia. Mol Cancer Ther. 2006;5(7):1836–1843. doi: 10.1158/1535-7163.MCT-06-0066. [DOI] [PubMed] [Google Scholar]
- 15.Dick LR, Cruikshank AA, Destree AT, Grenier L, McCormack TA, Melandri FD, Nunes SL, Palombella VJ, Parent LA, Plamondon L, Stein RL. Mechanistic studies on the inactivation of the proteasome by lactacystin in cultured cells. J Biol Chem. 1997;272(1):182–188. doi: 10.1074/jbc.272.1.182. [DOI] [PubMed] [Google Scholar]