

Abstract
Malignant gliomas contain a population of self-renewing tumorigenic stem-like cells; however, it remains unclear how these glioma stem cells (GSCs) self-renew or generate cellular diversity at the single-cell level. Asymmetric cell division is a proposed mechanism to maintain cancer stem cells, yet the modes of cell division that GSCs utilize remain undetermined. Here, we used single-cell analyses to evaluate the cell division behavior of GSCs. Lineage-tracing analysis revealed that the majority of GSCs were generated through expansive symmetric cell division and not through asymmetric cell division. The majority of differentiated progeny was generated through symmetric pro-commitment divisions under expansion conditions and in the absence of growth factors, occurred mainly through asymmetric cell divisions. Mitotic pair analysis detected asymmetric CD133 segregation and not any other GSC marker in a fraction of mitoses, some of which were associated with Numb asymmetry. Under growth factor withdrawal conditions, the proportion of asymmetric CD133 divisions increased, congruent with the increase in asymmetric cell divisions observed in the lineage-tracing studies. Using single-cell-based observation, we provide definitive evidence that GSCs are capable of different modes of cell division and that the generation of cellular diversity occurs mainly through symmetric cell division, not through asymmetric cell division.
Keywords: cancer stem cell, glioma, asymmetric cell division
Malignant gliomas (World Health Organization grade III–IV tumors) are extremely lethal and have a 5-year survival rate of <3%.1 Despite aggressive clinical treatment – including surgical resection, radiation, and chemotherapy – tumor recurrence is essentially universal. Therapeutic failure is due, in part, to tumor cell heterogeneity, derived from both genetic and non-genetic sources,2 which contributes to therapeutic resistance and tumor progression. Self-renewing, tumorigenic stem-like cancer cells have been identified in several central nervous system tumors including malignant gliomas.3, 4, 5, 6 Glioma stem cells (GSCs) are resistant to radiation7 and chemotherapy,8 underscoring the need to understand their biology. Currently, our knowledge of GSCs is based on comparative studies of populations from bulk tumors enriched or depleted for GSCs. In many studies, populations of enriched cells have been assessed for growth, survival, and tumor formation potential. Such approaches have determined the importance of GSCs in tumorigenesis and established key GSC regulators and molecular targets. However, it remains elusive how GSCs are maintained and generate cellular diversity, key criteria for consideration as a stem cell. Additionally, it is unclear which modes of cell division (symmetric and/or asymmetric) GSCs utilize for tumor maintenance and the generation of differentiated progeny.
Elegant, single-cell-based studies from the Drosophila neuroblast system and developing mammalian brain have demonstrated that the generation of differentiated progeny from stem/progenitor cells is tightly controlled. This process is regulated by cell polarity and the segregation of cellular components indicative of cell fate determination, such as Numb.9 Interestingly, studies in the mammalian brain have shown that CD133 (prominin-1), which marks neural stem and progenitor cells, is asymmetrically distributed during the generation of differentiated progeny in the developing neuroepithelium.10 Imaging of neural stem/progenitor cells using single-cell time-lapse microscopy has also led to a better understanding of lineage specification.11 Several recent studies in the hematopoietic stem cell system have shown the utility of single-cell time-lapse imaging to delineate symmetric and asymmetric divisions in response to extrinsic and intrinsic stimuli12 and cell fate choice instructed by cytokines.13 In this report, we used single-cell-based analytical methods to examine the modes of cell division used to maintain the GSC population.
Results
Glioma cells originated from a single CD133-positive cell develop heterogeneity in vivo and in vitro
To examine single-cell behavior of GSCs, we isolated individual CD133-positive cells from human glioma specimens. Using the extracellular matrix (ECM) protein laminin as a substrate, previously shown to facilitate long-term adherent culture,14 we expanded the cells over several passages. The long-term passage of the culture demonstrated consistent, sustained, logarithmic cell growth, indicating that these culture conditions support GSC cell divisions (Figure 1a). Xenografts resulting from the expanded cultures had histological hallmarks of malignant gliomas, including perivascular invasion (Figures 1b and c), aberrant vasculature (Figure 1d), and areas of necrosis (Figure 1e). These results confirmed that the in vitro expansion of GSCs supported their growth and tumorigenic capacity.
Figure 1.

Clonal GSCs can be expanded in culture and contain heterogeneity. (a) Population doublings of T4302 A3 clonal cells demonstrated exponential growth over time starting from 100 000 initial cells. (b–e) Representative histology for an intracranial tumor generated from 5000 clonal T4302 A3 cells is shown. Tumors (n=4/4) formed with clone T4302 A3. Additional clones assayed had identical outcomes (data not shown). Intracranial tumors had typical properties of GBM; poorly defined tumor margins (b), infiltrating cells (c), aberrant vasculature (d), and regions of necrosis (e). (f) Immunofluorescence micrographs of T4302 A3 GSCs demonstrated that culturing under defined adherent expansion conditions maintained heterogeneous GSC marker expression (CD133, green; Olig2, red; and marker-negative cells, yellow arrowheads). (g) Neural (Map2, red) and glial (GFAP, green) differentiation was also detected in these cultures (white arrowhead). Nuclei were counterstained with Hoechst 33342 (blue). Scale bar indicates 50 μm
To detect heterogeneity within the culture, cells were stained for stem cell and differentiation markers. The majority of the clonally derived cells were positive for the stem cell markers CD133 and Olig2 (Figure 1f). Few cells in the culture, however, were positive for the glial or neuronal differentiation markers GFAP or Map2, respectively (Figure 1g, Supplementary Figure 1). These observations suggest that the culture conditions can maintain GSC propagation with a low incidence of differentiation, which generates non-stem cells. Since epidermal growth factor (EGF) and fibroblast growth factor (FGF) are established critical components of stem cell culture medium,15, 16 we next removed these growth factors to determine effects on GSC propagation. As expected, we observed more frequent differentiation with EGF and FGF withdrawal (Supplementary Figure 1). Together, these data support the use of defined cell culture medium, including EGF and FGF, for maintaining GSCs in the culture.
Time-lapse lineage tracing detected asymmetric cell division
The expansion of GSCs in our defined cell culture medium also infrequently generated differentiated progeny, which represented a small fraction of cells (Figure 1f, Supplementary Figure 1). To evaluate the emergence of these differentiated progenies, and understand the expansion of GSCs at a single-cell resolution, we used time-lapse lineage tracing to monitor GSC cell divisions (Figure 2). For these experiments, CD133-positive cells were enriched by fluorescence-activated cell sorting (FACS) and directly plated onto culture dishes coated with Geltrex basement membrane matrix, a laminin-rich ECM substrate. Cell divisions were recorded with time-lapse microscopy, and cells were subsequently fixed and stained for the differentiation markers GFAP and Map2 (Figure 2a). We identified cells that had divided once and analyzed the fate of the resulting daughter cells (Figure 2a). Differentiation marker-positive cells were scored as differentiated non-GSCs and differentiation marker-negative cells as putative GSCs. Through this analysis, we detected four distinct modes of cell division: (1) symmetric GSC expanding division; (2) symmetric differentiating division; (3) asymmetric self-renewing/differentiating division; and (4) asymmetric survival/death division (Figure 2b). The vast majority of GSCs (>80%) underwent symmetric division, which expanded the stem cell population (Figure 2b). Among the rarer differentiation events, differentiation was threefold more likely to be induced through symmetric differentiating division than asymmetric division. A few divisions that resulted cell death were detected, although this was <1% of all events in the presence of expansive culture conditions. When growth factors were removed from the culture during time-lapse recording, we observed a decrease in symmetrical stem cell expansion division incidence to <70% of events. Growth factor withdrawal caused a concomitant fivefold increase in asymmetric self-renewal/differentiation division frequency. These data demonstrate that GSCs are capable of utilizing different modes of cell division to maintain a stem cell population and GSCs are maintained mainly by symmetric, not by asymmetric cell divisions.
Figure 2.

Growth factor withdrawal increased asymmetric cell division in time-lapse lineage tracing. CD133-positive GSCs were enriched by FACS and were plated onto Geltrex-coated plates in the presence or absence of growth factors. Cell divisions were recorded by time-lapse video microscopy for 42 h. At the end of image capturing, cells were fixed and immunostained for differentiation markers for neural (Map2) and glial (GFAP) lineages. (a) Example phase (left) and fluorescence (right) images of tracking and immunophenotyping results for daughter cells at the end of a 42-h observation period when Map2 was used as a neural marker. Red and green circles with corresponding number represent progeny generated from the same parent cell (cell 17 shown as an example). Pixel intensity values normalized to background levels are displayed for each cell in the fluorescence micrographs. The scale bar indicates 50 μm. (b) Analysis of time-lapse video microscopy detected four types of cell divisions. Single-cell lineage-tracing time-lapse microscopy movies demonstrated that growth factor withdrawal (n=111 cell divisions for EGF and basic FGF conditions, n=29 cell divisions for growth factor withdrawal conditions) decreased the number of symmetric stem/progenitor cell divisions and increased the generation of differentiated progeny and cell death. Proportional comparison of cell division types between conditions indicated a statistically significant difference, P<0.05, using Fisher's exact test. (N, neural marker positive cell (white arrow); S, stem/progenitor cell (yellow arrow))
Asymmetry of CD133 segregation during mitosis
Asymmetric cell divisions are a major route for stem cells to give rise to differentiated cells while simultaneously maintaining a stem cell population. Similarly, asymmetric cell divisions may have an important role in gliomas to maintain a GSC pool as well as increase heterogeneity of tumor cells by generating more differentiated progeny. We therefore sought to confirm the asymmetric cell division of GSCs using a method complementary to time-lapse imaging. Mitotic GSCs were enriched using mitotic shake off following release from thymidine block, and the segregation pattern of molecules at the time of mitosis was monitored. We examined the distribution of a panel of stem cells, GSCs, and differentiation markers and found that only CD133 was capable of equal and unequal distribution during mitosis (Figure 3; data not shown). The symmetric or asymmetric distribution of CD133 was confirmed with three different antibodies recognizing separate glycosylated or peptide epitopes17 (Supplementary Table 1; Figure 4a). The analyzed mitotic cells were genuinely dividing, which was confirmed by α-tubulin staining at cleavage furrows regardless of CD133 distribution (Figure 4b). Staining for additional stem cell markers, including integrin-α6,18 L1CAM,19 and CD1520 in combination with CD133, further confirmed selective asymmetric distribution of CD133 (Figure 5a). However, analysis with the previously defined asymmetric cell division marker Numb detected asymmetric segregation in a fraction of mitotic GSCs (Figure 5b). Distribution of Numb did correlate with CD133 when Numb showed asymmetry during mitosis (Figure 5b), further confirming the ability of CD133 to be asymmetrically distributed. To ensure that the unequal distribution of CD133 was not an artifact of our enrichment method, we examined cultures without synchronization or mitotic shake off. We observed unequal distribution of CD133 in some cells at each phase of mitosis: prophase, metaphase, anaphase, and telophase (Supplementary Figure 2).
Figure 3.

A screen of stem/progenitor markers on mitotic cell pairs reveals that CD133 is unequally distributed in a fraction of GSCs. Confocal micrographs of mitotic cell pairs derived from T4302 clone A3 cell populations grown in expansion conditions stained for the indicated markers of stem/progenitor cells (green). Symmetric distribution between nascent daughter cells was observed for Bmi1, CD15, integrin-α6, L1CAM, Nestin, Olig2, and Sox2. In contrast, CD133 exhibited both symmetric and asymmetric distributions between progenies (asterisk). Nuclei were counterstained with Hoechst 33342 (blue). Scale bar indicates 5 μm
Figure 4.

Equal and unequal distribution of CD133 is observed using various anti-CD133 antibodies. (a) Confocal micrographs of T4302 clone A3 cells revealed that symmetric and asymmetric distributions of CD133 were detected with multiple CD133 antibodies. W6B3C1 or 80B258 mouse monoclonal antibody (green, mCD133) was used together with ab19898 rabbit polyclonal antibody (red, rCD133). (b) Confocal micrographs of double staining of CD133 (ab19898, green, rCD133) with α-tubulin (red) confirmed that mitotic pairs of T4302 A3 clonal cells were undergoing cell division. Arrows indicate the mid-body. Similar data were observed with non-clonal T4121 cells (data not shown). Nuclei were visualized with Hoechst 33342. Scale bar indicates 5 μm
Figure 5.

Equal and unequal distribution of CD133 irrespective of other GSC markers, but Numb. (a) Confocal micrographs of mitotic cell pairs derived from T4302 A3 clonal cells grown in expansion conditions demonstrated constant uniform expression patterns for integrin-α6, L1CAM, and CD15 (red). In contrast, symmetric or asymmetric CD133 distribution was detected using ab19898 rabbit antiserum (green). Identical results were obtained with non-clonal T4121 cells (data not shown). (b) Fluorescent micrographs of mitotic cell pairs derived from T4302 A3 clonal cell populations grown in expansion conditions demonstrated consistent asymmetric distribution of CD133 and Numb. Nuclei were counterstained with Hoechst 33342 (blue). Scale bars indicate 5 μm for a and 15 μm for b
To define the rate of asymmetric cell division, we developed a control analysis by the segregation pattern of α-tubulin, a molecule that should not asymmetrically segregate. From quantification of α-tubulin distribution between the two emerging daughter cells, symmetrical division was defined when the percent deviation of the stained marker between daughter cells was <25% (see Materials and Methods for details). Using this criterion, we found that the percentage of mitoses with asymmetric CD133 distribution was <30% in cell culture conditions, promoting GSC propagation (Figure 6b). However, removal of growth factors increased the frequency of CD133 asymmetric cell division by at least 44% in GSCs derived from two different gliomas analyzed with two different CD133 antibodies (Figure 6b). Thus, GSCs can undergo asymmetric cell division, and their mode of cell division is impacted by culture conditions using two independent methods.
Figure 6.

CD133 asymmetry increased upon growth factor deprivation. (a) Representative phase and fluorescence micrographs of GSCs analyzed during mitosis stained with rabbit antiserum ab19898 against CD133 (green). Similar images were produced with mouse monoclonal antibody W6B3C1 against CD133 (images not shown). Cells were counterstained with Hoechst 33342 (blue) to mark DNA. Analysis occurred by comparing pixel intensities of DNA with CD133 staining in the area containing each emerging daughter cell, as indicated by the red and yellow lines. Examples of symmetrical and asymmetrical distributions of CD133 are shown. Percent deviation in staining between daughter cells is displayed below micrographs. The scale bar indicates 10 μm. (b) Quantification of images as illustrated in a demonstrated that withdrawal of EGF and basic FGF (bFGF) increased the fraction of mitotic pairs with asymmetric CD133 distribution when compared with cultures with EGF and bFGF in both T4121 and T4302 cells. N, number of cell divisions. **P<0.01; ***P<0.001 as assessed using Pearson's χ2-test
Polarized localization of CD133 in interphase GSCs
The relationship between cell polarity and orientation of cell division determines whether a neural stem cell undergoes symmetric or asymmetric cell division during development. In normal tissue, CD133 associates with cholesterol-rich membrane microdomains (lipid rafts)10, 21 and localizes to special subdomains of the plasma membrane of cells.22 Furthermore, CD133 polarization has been related to cellular motility and cell division of hematopoietic stem and progenitor cells.23 We therefore examined whether CD133 localization is polarized in GSCs to contribute to asymmetric distribution during mitosis. Asynchronous, actively proliferating GSC monolayer cultures were fixed and immunostained for CD133. We detected three types of CD133 distribution patterns: (1) homogenous expression throughout the cells; (2) expression limited to a segment(s) of the cell; and (3) expression localized to scattered patches (Figures 7a–c). As the cell membrane has fluidity, these expression patterns may dynamically change. However, one daughter cell receives higher amounts of CD133 if uneven distributions of CD133 take place at the time of mitosis. Indeed, we detected metaphase cells with uneven patchy concentrations of CD133 on the opposite side of the cell cleavage line. However, concentration of CD133 to patches does not necessitate asymmetric distribution of CD133, as the total sum of CD133 can be equivalently expressed in both emerging daughter cells (Figures 7d–f). If localized CD133 distribution in the cell membrane is a mechanism of asymmetric distribution of CD133, the frequency of GSCs with polarized CD133 would increase under conditions that induce asymmetric cell division. We therefore determined the frequency of GSCs with polarized CD133 in the presence and absence of growth factors (Figure 7g). Under GSC growth expansive culture conditions, the percentage of cells expressing polarized CD133 expression was ∼12%. Growth factor deprivation resulted in a 65% increase in the fraction of GSCs with localized CD133 (Figure 7g). This observation suggests that polarization of CD133 may be an underlying mechanism of asymmetric CD133 segregation during mitosis. Taken together, our data demonstrate that using single-cell analysis of GSCs, we can confirm that they are capable of self-renewing and generating cellular diversity. Additionally, GSCs utilize symmetric and not asymmetric divisions for their propagation and generation of differentiated progeny under expansion conditions.
Figure 7.

Polarization of CD133 during interphase and mitosis. Actively proliferating glioma cells on Geltrex-coated coverslips were fixed and stained for CD133 using rabbit antiserum ab19898 (green). Nuclei were counterstained with Hoechst 33342 (blue). (a–c) Polarized localization of CD133 was detected in a fraction of interphase cells, as indicated by yellow arrows. However, many cells contained diffuse CD133 staining, as indicated by white arrows. (d–f) Polarized localization of CD133 was also sometimes detected during mitosis. (d) Each daughter cell would receive similar total amounts of CD133 when CD133 was evenly distributed throughout the dividing cells. (e) When CD133 is polarized during division, daughter cells could receive divergent amounts of CD133 if expressions were asymmetrically distributed. (f) Even when CD133 is polarized during mitosis, daughter cells could receive similar amounts of CD133 if expressions were equally distributed. The scale bars indicate 20 μm for a–c and 10 μm for d–f. (g) The frequency of interphase cells with polarized CD133 distribution increased when growth factors were deprived from culture media. N, number of cells analyzed. **P<0.02 (P=0.0146) by Pearson's χ2 analysis
Discussion
Malignant gliomas are aggressive tumors with poor prognosis due to resistance to therapies. Recent studies indicate that GSCs have critical roles not only in tumor propagation but also in therapeutic resistance. Using single-cell -based time-lapse microscopy and quantitative immunofluorescence, we found that GSCs utilize two cell division modes to generate GSCs. The first method is to generate two GSCs through a symmetric cell division, and the other is to generate one GSC and one non-GSC through an asymmetric cell division. Symmetric stem cell expanding divisions were more frequent in the presence of growth factors, suggesting that growth factor signaling contributes to the expansion of the stem cell population in the tumor. Upon growth factor deprivation, asymmetric cell division increased. This division mode would contribute to increased cellular heterogeneity of the tumor, while still maintaining a GSC pool. Under conditions of growth factor withdrawal, asymmetric cell division would provide the benefit of maintaining the GSC pool by generating a CD133 high daughter cell with stem cell capability. Although generation of this GSC would be at the expense of the paired daughter cell with CD133 low, asymmetric division would avoid producing two daughter cells with intermediate CD133 levels in higher risk of generating two non-stem cells that could result from symmetric division.
Among the many molecules we examined, only CD133 and Numb could be asymmetrically segregated. CD133 is a cell surface marker that enriches GSCs, and CD133 high cells survive better than CD133 low cells (Supplementary Figure 3). The fact that other stem cell markers were not co-segregated with CD133 suggests that there are multiple factors contributing to GSC maintenance. In support of this notion, CD15 serves as a GSC marker in CD133-negative glioma cells.20 In fact, CD15-positive cells, similar to CD133-positive cells, survive better and proliferate faster as compared with their negative counterparts (Supplementary Figure 3), indicating its potential to complement some part of CD133 function. In contrast to CD133, Numb is a fate-determining molecule that promotes the differentiation of neural stem cells through its ability to antagonize notch and hedgehog signaling pathways.24, 25 A correlation between CD133 and Numb was observed during cell divisions with Numb asymmetry. The daughter cells receiving less Numb and CD133 can potentially give rise to CD133-negative GSCs due to attenuated Numb function. Such complex combinations of stem cell markers and Numb inheritance would contribute to multiple states of GSCs as proposed.26
When interphase GSCs were stained for CD133, we found a polarized staining pattern in a fraction of the cells. We also detected localized CD133 in patches evenly or unevenly distributed across cell cleavage plane during mitosis. These data suggest localized CD133 expression and its positioning against the mitotic axis determines asymmetric segregation of this molecule. A similar mechanism has been reported for asymmetrical segregation of molecules in the apical membrane of neural stem cells.10 CD133 is concentrated in various plasma membrane protrusions, such as microvilli, filopodia, or lamellipodia, probably due to its association with cholesterol-rich membrane microdomains.21, 27 Although it remains unclear how local CD133 concentration is formed in GSCs, there are reports on the mechanisms of membrane microdomain clustering and polarization. In non-polarized (non-epithelial) cells, cholesterol-rich membrane microdomains can form patchy patterns upon crosslinking plasma membrane proteins and/or glycolipids residing in the domain.28 Membrane microdomains are polarized through cytoskeletal action in neutrophils upon chemoattractant stimulation.29 A similar mechanism may regulate the local concentration of CD133 in GSCs. As growth factor receptors are also concentrated in membrane microdomains,30, 31 it is possible that growth factor availability could alter clustering of membrane microdomains harboring CD133.
Our single-cell-based approach revealed that there are two cell division modes to generate GSCs. The symmetric expansion mode will increase the GSC pool in the tumor, whereas asymmetric cell division will increase cellular heterogeneity of the tumor while maintaining GSC pool. The biological significance of these two division modes is not yet fully understood, but it is expected that therapies that increase asymmetric division would generate decreased numbers of resistant GSCs and be favorable for patient outcome. We anticipate the single-cell-based analyses presented here will be useful to assess drug effects on the mode of cell division. The information collected could help to develop new therapeutic strategies for targeting gliomas by developing a greater understanding of the regulation of GSC cell fate.
Materials and Methods
Isolation of GSCs and generation of adherent colonies
Cells that functionally fulfilled criteria for GSCs (serial self-renewal, sustained proliferation, tumor propagation, and stem cell marker expression) were prepared and maintained as previously described.4, 7, 14 Briefly, GSCs were isolated from surgically obtained glioma specimens, transiently amplified by implantation of unsorted tumor cells into immunocompromised mice, and derived xenografts were dissociated before culture. All specimens were derived from glioma surgical tissues obtained from patients undergoing resection for a newly diagnosed (T4302) or recurrent (T4121) glioma in accordance with protocols approved by the Duke University Medical Center Institutional Review Board. Written consent to utilize excess tissue for research was obtained from each patient, and tissues were de-identified for all studies. Additional information about the tumors is provided in Supplementary Table 1. For xenograft dissociation, single cells were prepared from the bulk tumor by a Papain dissociation kit (Worthington Biochemical, Lakewood, NJ, USA) as per the manufacturer's protocol and cultured using previously reported culturing methods. For the generation of adherent colonies, single GSCs were sorted into marker-positive fractions based on antibodies against CD133 (CD133/2-allophycocyanin (APC)-conjugated antibody, 293C3, Miltenyi Biotech, Auburn, CA, USA) using FACS. A single marker-positive cell was plated into each well of a 96-well plate (BD Biosciences, La Jolla, CA, USA) containing neurobasal media supplemented with B27 (Invitrogen, Carlsbad, CA, USA), 20 ng/ml EGF (R&D Systems, Minneapolis, MN, USA), 20 ng/ml basic FGF (R&D Systems), and 5 μg/ml laminin (mouse EHS sarcoma laminin, Millipore, Billerica, MA, USA or Sigma, St. Louis, MO, USA) using a FACS Aria II (BD Biosciences) equipped with a 100 μm nozzle with low sheath pressure. Adherent clonal colonies were fed every week and expanded from a 96-well plate to 10 cm tissue culture dishes via 24- and 6-well tissue culture plates (BD Biosciences). Although the colony formation efficiency was lower for free floating tumorspheres, as has been reported by Pollard and co-workers,14 colonies that were generated from either adherent or non-adherent culture conditions gave rise to populations of cells that efficiently formed tumors when transplanted in vivo (data not shown).
Immunofluorescence
For immunofluorescence analysis of adherent cultures, cells were fixed with 2% paraformaldehyde (Sigma) at room temperature for 15 min, washed with three times with phosphate-buffered saline (PBS), and blocked with 10% normal goat serum (Sigma) in PBS. Depending on the analysis marker, 0.1% Triton X-100 (Sigma) was added into the blocking buffer for cell permeabilization to detect intracellular antigens. Cells were blocked for 30 min at room temperature and incubated with appropriate primary antibodies overnight at 4°C. A detailed list of antibodies can be found in Supplementary Table 2. Cells were washed three times with PBS and incubated with the appropriate secondary antibody (1:400, goat Alexa 488- or 568-conjugated antibody (IgG), Invitrogen). Nuclei were counterstained with Hoechst 33342 (5 μg/ml in PBS, Invitrogen) and coverslipped before imaging. Fluorescence micrographs were acquired using a Leica DMI3000B microscope (Leica Microsystems GmbH, Wetzlar, Germany) with a × 20 objective, confocal micrographs were acquired using a Leica SP5 confocal microscope with a × 63 objective, and images were processed and assembled in Photoshop CS (Adobe, San Jose, CA, USA).
Flow cytometry analysis
For flow cytometry analysis, cells were expanded in adherent culture, collected, and evaluated on a Coulter EPICS cytometer (Beckman Coulter, Brea, CA, USA) or a FACS Aria II (BD Biosciences) using PE- or APC-conjugated CD133/1 (AC133, Miltenyi Biotech), CD15-FITC-conjugated (BD Biosciences), and Annexin V-FITC-conjugated (BD Biosciences) antibodies, according to the manufacturers' recommendations. Appropriate compensation and isotype controls were used.
Mitotic cell collection and analysis
To collect mitotic cells, adherent cultures grown on Geltrex (Invitrogen)-coated plates were synchronized with 2 mM thymidine for 12–15 h. At 10–15 h following release from thymidine block by three times washing with PBS and changing media with or without growth factors, the mitotic cells were shaken off the plates by vortexing for 30 s. Detached cells were collected and settled onto poly--lysine-coated coverslips at the bottom of the 6-well plates using centrifugation at 1000 r.p.m. for 5 min at room temperature. A minimum of 30 pairs of mitotic cells were analyzed per GSC marker. For immunofluorescence analysis, cells were fixed, blocked, and stained as described above with or without permeabilization. Fluorescence micrographs for a minimum of 30 fields per GSC marker were acquired using a Leica (DMI3000B) × 20 objective, and images were processed and assembled in Photoshop CS (Adobe). Confocal micrographs (composite Z-stacks built from 100 nm optical sections) were obtained using a × 63 oil immersion objective with a Leica SP-5 microscope. Images were processed and assembled in Photoshop CS (Adobe).
Mitotic image analysis
To quantify the degree of asymmetric distribution of molecules of interest between the two daughter cells, the intensity of fluorescence immunostaining was determined for each emerging daughter cell. Images of late anaphase to telophase were collected by fluorescence microscopy as described above. The dividing daughter cells were defined using the morphology of the phase-contrasted image together with condensed DNA detected by Hoechst 33342 staining. α-Tubulin immunofluorescence staining was also utilized to identify dividing daughter cells. Using ImageJ (NIH, Bethesda, MD, USA), each identified daughter cell in a dividing pair was enclosed with the polygon tool, and the integrated fluorescent signals of the defined daughter cells were determined for DNA and stained markers.
Statistical analysis
In order to quantify the asymmetry, we calculated percent (%) deviation of the distribution of the stained markers as follows:
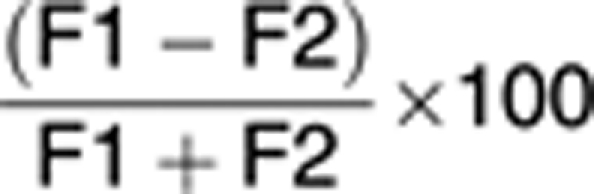 |
where F1 and F2 represent the integrated fluorescent values of a given staining for two dividing daughter cells. As α-tubulin is a critical part of the cell division machinery, both daughter cells should have strong and equal expression. Therefore, we set the asymmetry cell division cutoff to be greater than the 99% confidence interval of α-tubulin (for a bell-shaped distribution, this calculates to the mean difference±2.576 × standard deviation). We calculated the mean distribution of α-tubulin for a minimum of 100 cells to be 6.668% and the standard deviation to be 6.655. For these values, we set the asymmetry cutoff to be greater than a 25% difference between daughter cells, which represents greater than the 99% confidence interval for α-tubulin as detailed above. After determining percent (%) deviation for each dividing pairs, each cell division was categorized as symmetrical or asymmetrical using the 25% difference cutoff value. From four independent shake-off experiments, the variance in percent (%) deviation was determined for each treatment and categorical measures were summarized using frequencies and percentiles. To assess the association between the categorical variables, Pearson's χ2-test or Fisher's exact test was applied to cumulative categorized cell divisions data collected from four independent shake-off experiments. All tests were performed at a significance level of 0.05. SAS 9.2 software (SAS Institute, Cary, NC, USA) was used for all analyses.
Time-lapse microscopy
T4302 A3 adherent clonal cells were grown in 24-well dishes and imaged on a Leica DMIRB Inverted Microscope equipped for time-lapse microscopy with a Roper Scientific CoolSNAP HQ Cooled CCD camera (Roper Scientific, Tucson AZ, USA), temperature controller (37°C) and CO2 (5%) incubation chamber (Leica Microsystems GmbH), PeCon incubator (PeCon GmbH, Erbach, Germany), Prior motorized stage with linearly encoded controller with x/y/z drive for time-lapse imaging of multiple fields and heating insert for 12-well plates (Prior Scientific Inc., Rockland, MA, USA), Uniblitz shutter (Vincent Associates, Rochester, NY, USA), and MetaMorph Software (Molecular Devices, Downingtown, PA, USA). Images of multiple fields per well were collected every 3 min for ∼23 h using a dry × 10, 0.3 NA objective lens, and phase-contrast optics. For CD15 time-lapse microscopy, immunostaining and lineage analysis were performed as described.11 For statistical analysis of CD15 data, the dividing cell pool was analyzed separately from the non-dividing cell pool.
Lineage analysis
Phase-contrast, time-lapse image stacks consisting of 857 frames (3 min time intervals) were imported into Image-Pro Plus (v6.2, Media Cybernetics, Silver Spring, MD, USA). Lineage analysis was performed in a semiautomated manner using customized visual basic Image-Pro Plus macros. Briefly, each frame of a stack was flattened to remove regions of uneven illumination and enhance appearance of cell boundaries. A spectral filter (high pass) followed by a morphological ‘closing' filter (11 × 11 kernel) was then applied to preferentially enhance the appearance of cells that were lifting from the plate surface and rounding. Using morphological (aspect ratio, cell area, and heterogeneity) and intensity-based selection criteria, these rounded cells were subsequently segmented from the original sequence as binary objects. Because cells in the process of dividing/dying may remain rounded in multiple frames before separation/apoptosis, a search algorithm was applied to the binary stack that examined each rounded cell and removed every instance of a similar sized cell for the subsequent 100 frames within a 75 × 75 μm region of interest (ROI) around the rounded cell's centroid. This ensured that each division/death was only counted once. To classify rounding cells as dying, dividing, or neither, a similar algorithm was applied that searched for multiple objects (2 for dividing cells, >2 for apoptotic debris, 1 for neither) within the originating rounded cell's ROI. This particular pass utilized user input to confirm classification. Finally, using a combination of Fourier correlation and user-guided input, progeny following each division were traced from the appearance of rounding to the last frame or frame in which the cell left the field of view or died. Cell-tracing coordinates and corresponding classifications were automatically exported to Excel and pseudo-colored, final frame images of surviving progeny as well as tracing paths sequences were saved for each stack. For the determination of the mode of cell division (asymmetric or symmetric), cells were fixed and stained for Map2, a neuronal differentiation marker, and GFAP, a glial differentiation marker, by indirect immunofluorescence. The pixel intensities of stained cells were measured by ImageJ after enclosing each cell with the polygon tool. A mean value of above 300 was established by visual confirmation as positive staining, and asymmetric divisions were scored when one daughter cell has a mean pixel intensity of above 300 and the other was 300 or below, and at the same time the difference in mean intensities between the daughter cells was at least 25%.
Acknowledgments
We thank Dr. Mike Cook, Dr. Beth Harvat, Cathy Shemo, Sage O'Bryant, and Moneen Morgan for assistance with flow cytometry, and the members of the Rich laboratory for critical review of the manuscript. We also thank Dr. Judy Drazba and Eric Diskin from LRI Imaging Core and Kevin Ryan from W. Nuhsbaum Inc. for critical assistance with time-lapse microscopy, and Linda Vargo for assistance with histology. We thank Xiaobo Liu from the Quantitative Health Sciences Department for statistical analysis assistance. We are also grateful to Drs. Nicholas Gaiano, Shideng Bao, Monica Venere, Mahendra S. Rao, Richard Ransohoff, Thomas Egelhoff, and Yogen Saunthararajah for valuable insight and comments on the manuscript. The Rich laboratory is supported by the McDonnell Foundation and NIH Grants NS054276, CA154130, CA129958, and CA116659. ABH was supported by grants from the National Brain Tumor Society and the NIH (CA151522). SPG was supported by an American Cancer Society Silber Summer Research Fellowship. CEE is supported by a National Research Service Award from the National Institutes of Neurological Disorders and Stroke (NINDS F30 NS063496). JDL is supported by an American Brain Tumor Association Basic Research Fellowship (sponsored by the Joelle Syverson Fund) and a National Research Service Award from the National Cancer Institute (NCI F32 CA142159). MH is supported by a grant from the National Institutes of Neurological Disorders and Stroke (NINDS R21 NS063057). DC is supported by the Deutsche Forschungsgemeinschaft (SFB 655).
Glossary
- GSC
glioma stem cell
- ECM
extracellular matrix
- EGF
epidermal growth factor
- FGF
fibroblast growth factor
- FACS
fluorescence-activated cell sorting
- APC
allophycocyanin
- PBS
phosphate-buffered saline
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on Cell Death and Disease website (http://www.nature.com/cddis)
Edited by A Stephanou
Supplementary Material
References
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–361. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe CQ. Neural stem cells: balancing self-renewal with differentiation. Development. 2008;135:1575–1587. doi: 10.1242/dev.014977. [DOI] [PubMed] [Google Scholar]
- Kosodo Y, Roper K, Haubensak W, Marzesco AM, Corbeil D, Huttner WB. Asymmetric distribution of the apical plasma membrane during neurogenic divisions of mammalian neuroepithelial cells. EMBO J. 2004;23:2314–2324. doi: 10.1038/sj.emboj.7600223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravin R, Hoeppner DJ, Munno DM, Carmel L, Sullivan J, Levitt DL, et al. Potency and fate specification in CNS stem cell populations in vitro. Cell Stem Cell. 2008;3:670–680. doi: 10.1016/j.stem.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Wu M, Kwon HY, Rattis F, Blum J, Zhao C, Ashkenazi R, et al. Imaging hematopoietic precursor division in real time. Cell Stem Cell. 2007;1:541–554. doi: 10.1016/j.stem.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC, Schroeder T. Hematopoietic cytokines can instruct lineage choice. Science. 2009;325:217–218. doi: 10.1126/science.1171461. [DOI] [PubMed] [Google Scholar]
- Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Li Z, Wang H, Eyler CE, Hjelmeland AB, Rich JN. Turning cancer stem cells inside out: an exploration of glioma stem cell signaling pathways. J Biol Chem. 2009;284:16705–16709. doi: 10.1074/jbc.R900013200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbanova J, Missol-Kolka E, Fonseca AV, Lorra C, Janich P, Hollerova H, et al. The stem cell marker CD133 (Prominin-1) is expressed in various human glandular epithelia. J Histochem Cytochem. 2008;56:977–993. doi: 10.1369/jhc.2008.951897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. 2010;6:421–432. doi: 10.1016/j.stem.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, McLendon RE, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008;68:6043–6048. doi: 10.1158/0008-5472.CAN-08-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son MJ, Woolard K, Nam DH, Lee J, Fine HA. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. 2009;4:440–452. doi: 10.1016/j.stem.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper K, Corbeil D, Huttner WB. Retention of prominin in microvilli reveals distinct cholesterol-based lipid micro-domains in the apical plasma membrane. Nat Cell Biol. 2000;2:582–592. doi: 10.1038/35023524. [DOI] [PubMed] [Google Scholar]
- Corbeil D, Marzesco AM, Wilsch-Brauninger M, Huttner WB. The intriguing links between prominin-1 (CD133), cholesterol-based membrane microdomains, remodeling of apical plasma membrane protrusions, extracellular membrane particles, and (neuro)epithelial cell differentiation. FEBS Lett. 2010;584:1659–1664. doi: 10.1016/j.febslet.2010.01.050. [DOI] [PubMed] [Google Scholar]
- Bauer N, Fonseca AV, Florek M, Freund D, Jaszai J, Bornhauser M, et al. New insights into the cell biology of hematopoietic progenitors by studying prominin-1 (CD133) Cells Tissues Organs. 2008;188:127–138. doi: 10.1159/000112847. [DOI] [PubMed] [Google Scholar]
- Di Marcotullio L, Ferretti E, Greco A, De Smaele E, Po A, Sico MA, et al. Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination. Nat Cell Biol. 2006;8:1415–1423. doi: 10.1038/ncb1510. [DOI] [PubMed] [Google Scholar]
- McGill MA, Dho SE, Weinmaster G, McGlade CJ. Numb regulates post-endocytic trafficking and degradation of Notch1. J Biol Chem. 2009;284:26427–26438. doi: 10.1074/jbc.M109.014845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17:362–375. doi: 10.1016/j.ccr.2009.12.049. [DOI] [PubMed] [Google Scholar]
- Weigmann A, Corbeil D, Hellwig A, Huttner WB. Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc Natl Acad Sci USA. 1997;94:12425–12430. doi: 10.1073/pnas.94.23.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder T, Scheiffele P, Verkade P, Simons K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J Cell Biol. 1998;141:929–942. doi: 10.1083/jcb.141.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seveau S, Eddy RJ, Maxfield FR, Pierini LM. Cytoskeleton-dependent membrane domain segregation during neutrophil polarization. Mol Biol Cell. 2001;12:3550–3562. doi: 10.1091/mbc.12.11.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MR, Cinar B, Kim J, Mukhopadhyay NK, Di Vizio D, Adam RM, et al. Transit of hormonal and EGF receptor-dependent signals through cholesterol-rich membranes. Steroids. 2007;72:210–217. doi: 10.1016/j.steroids.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra SK. Dissecting lipid raft facilitated cell signaling pathways in cancer. Biochim Biophys Acta. 2008;1785:182–206. doi: 10.1016/j.bbcan.2007.11.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.