

Abstract
MUT056399 is a highly potent new inhibitor of the FabI enzyme of both Staphylococcus aureus and Escherichia coli. In vitro, MUT056399 was very active against S. aureus strains, including methicillin-susceptible S. aureus (MSSA), methicillin-resistant S. aureus (MRSA), linezolid-resistant, and multidrug-resistant strains, with MIC90s between 0.03 and 0.12 μg/ml. MUT056399 was also active against coagulase-negative staphylococci, with MIC90s between 0.12 and 4 μg/ml. The antibacterial spectrum is consistent with specific FabI inhibition with no activity against bacteria using FabK but activity against FabI-containing Gram-negative bacilli. In vitro, resistant clones of S. aureus were obtained at a low frequency. All of the resistant clones analyzed were found to contain mutations in the fabI gene. In vivo, MUT056399, administered subcutaneously, protected mice from a lethal systemic infection induced by MSSA, MRSA, and vancomycin-intermediate S. aureus strains (50% effective doses ranging from 19.3 mg/kg/day to 49.6 mg/kg/day). In the nonneutropenic murine thigh infection model, the same treatment with MUT056399 reduced the bacterial multiplication of MSSA and MRSA in the thighs of immunocompetent mice. These properties support MUT056399 as a very promising candidate for a novel drug to treat severe staphylococcal infections.
INTRODUCTION
Infections due to antibiotic-resistant pathogens are a serious health problem globally, such that standard antibiotic therapies have become less effective. More specifically, methicillin-resistant Staphylococcus aureus (MRSA) strains are now a major concern in hospital settings worldwide. The emergence of vancomycin-intermediate and -resistant S. aureus (VISA and VRSA), as well as community-acquired MRSA, stresses the need for new antibiotics with new mechanisms of action (3, 4, 17, 23, 25). Fatty acid biosynthesis is the first stage of membrane lipid biogenesis and represents a vital aspect of bacterial physiology (8, 21). In most bacteria, a series of small soluble proteins known as fatty acid synthase (FAS) type II enzymes produce a number of essential lipid-containing components included in the cell membrane. Among the FAS type II enzymes, the NADH-dependent trans-2-enoyl-acyl carrier protein (ACP) reductase FabI has been shown to be essential for the growth of S. aureus and Escherichia coli (20, 22). FabI is a key regulator in controlling the elongation of the acyl chain for saturated fatty acid and unsaturated fatty acid (UFA) synthesis in bacteria (42, 43). On this basis and due to the absence of a eukaryotic orthologue, FabI was identified as a novel and promising candidate drug target (28, 34). Several enoyl-ACP reductases were found in bacterial species (30, 31, 34). For instance, FabK found in streptococci is radically different from FabI at the primary sequence level (29, 36). Consequently, a specific FabI inhibitor is expected to be a narrow-spectrum agent specific for bacterial species dependent on FabI for fatty acid synthesis, such as S. aureus and coagulase-negative staphylococci, as well as some Gram-negative enterobacteria (30).
A rational molecular design strategy has been set up using the available structural data on FabI bound to a very simple molecule such as triclosan (26, 32, 38, 39). This drug discovery program has successfully generated a new series of inhibitors (aryloxy-phenol series) exhibiting strong antistaphylococcal activities, with MUT056399 (also named FAB001) (Fig. 1) being identified as one of the most potent compounds.
Fig. 1.
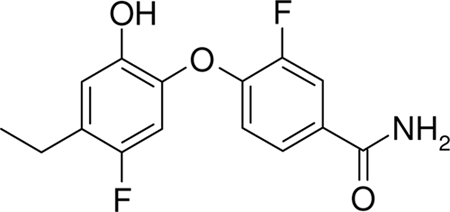
Chemical structure of MUT056399.
MATERIALS AND METHODS
The compound used in this study was MUT056399, synthesized by Mutabilis. The reference compounds vancomycin, linezolid, quinupristin-dalfopristin, levofloxacin, clindamycin, clarithromycin, and triclosan were purchased from commercial sources. Strains from the Mutabilis internal collection were collected from different sources, i.e., the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) and the ATCC (LGC Promochem). Glycopeptide-intermediate S. aureus (GISA) and VISA JUS strains were obtained from the French National Reference Center for S. aureus in Lyon. S. aureus strain ATCC 700699 was used after isolation on vancomycin at 4 μg/ml to maintain the VISA phenotype (MICs of vancomycin ranging from 4 to 8 μg/ml).
The strains and isolates used in susceptibility and spectrum studies came from the Quotient Bioresearch internal collection. The MICs of MUT056399 were determined against 118 S. aureus strains, 165 coagulase-negative staphylococci, 31 other bacterial species listed in Table 1, Candida albicans ATCC 90028, and Candida glabrata NCPF 3309.
Table 1.
MUT056399 in vitro antibacterial activity
| Bacterial strain | No. of isolates | MUT056399 |
Reference antibiotic | MICf range | MIC90f | |
|---|---|---|---|---|---|---|
| MICf range | MIC90f | |||||
| MSSA | 40 | ≤0.03–4 | 0.06 | Linezolid | 2–4 | 2 |
| MRSA (NARSA) | 48 | ≤0.03–4 | 0.12 | Linezolid | 1–≥16 | ≥16 |
| Community-acquired S. aureusa | 30 | ≤0.03–0.12 | ≤0.03 | Linezolid | 1–4 | 2 |
| LinrS. aureusb | 12 | ≤0.03–0.06 | 0.06 | Linezolid | 8–>8 | >8 |
| MRdS. epidermidis | 30 | ≤0.03–16 | 2 | Linezolid | 0.5–1 | 1 |
| MSeS. epidermidis | 30 | ≤0.03–8 | 4 | Linezolid | 0.5–1 | 1 |
| MR S. haemolyticus | 30 | ≤0.03–4 | 0.5 | Linezolid | 1–2 | 1 |
| MS S. haemolyticus | 15 | ≤0.03–8 | 4 | Linezolid | 1–2 | 2 |
| MR S. hominis | 30 | ≤0.03–2 | 0.12 | Linezolid | 0.5–2 | 2 |
| MS S. hominis | 30 | ≤0.03–8 | 2 | Linezolid | 1–2 | 2 |
| Neisseria gonorrhoeae | 10 | 0.06–0.5 | 0.25 | Levofloxacin | ≤0.008–8 | 0.5 |
| Neisseria meningitidis | 10 | 0.06–0.5 | 0.25 | Levofloxacin | ≤0.008 | ≤0.008 |
| Haemophilus influenzae (including resistant strains) | 30 | ≤0.03–1 | 0.5 | Levofloxacin | ≤0.008–0.12 | 0.03 |
| Haemophilus ducreyic | 10 | 4–>64 | Levofloxacin | 0.008–0.5 | ||
| Pseudomonas aeruginosa | 10 | >64 | >64 | Levofloxacin | 0.25–4 | 2 |
| Moraxella catarrhalis | 20 | 1–4 | 2 | Levofloxacin | 0.015–0.06 | 0.03 |
| Acinetobacter baumannii | 20 | 0.5–32 | 16 | Levofloxacin | 0.03–≥16 | 8 |
| Escherichia coli | 20 | 0.5–4 | 1 | Levofloxacin | ≤0.008–≥16 | ≥16 |
| Proteus mirabilis | 10 | 1–2 | 2 | Levofloxacin | 0.03–0.12 | 0.06 |
| Klebsiella spp. | 30 | 1–32 | 16 | Levofloxacin | 0.03–≥16 | ≥16 |
| Enterobacter spp. | 20 | 1–16 | 16 | Levofloxacin | 0.03–≥16 | 4 |
| Morganella morganii | 10 | 16–>64 | ≥64 | Levofloxacin | 0.015–>8 | >8 |
| Serratia marcescens | 10 | >64 | >64 | Levofloxacin | 0.03–1 | 0.12 |
| Citrobacter spp. | 20 | 1–≥128 | ≥128 | Levofloxacin | ≤0.008–2 | 0.25 |
| Helicobacter pylori | 5 | 0.5–2 | Clarithromycin | 0.03–>16 | ||
| Legionella pneumophila | 10 | ≤0.03 | ≤0.03 | Levofloxacin | 0.008–0.015 | 0.015 |
| Chlamydia trachomatis | 10 | 2–4 | 4 | Levofloxacin | 0.12–0.25 | 0.25 |
| Chlamydophila pneumoniae | 5 | 0.5 | Levofloxacin | 0.5–1 | ||
| Mycoplasma pneumoniae | 10 | 64–>64 | 64 | Levofloxacin | 0.5–2 | 1 |
| Mycobacterium fortuitum | 3 | 16 | Levofloxacin | 0.12 | ||
| Bacillus cereus | 3 | ≥64 | Levofloxacin | 0.12–0.25 | ||
| Bacillus subtilis | 3 | 32–>64 | Levofloxacin | 0.06–0.25 | ||
| Enterococcus | 6 | >64 | Linezolid | 0.5–1 | ||
| Streptococcus pneumoniae | 10 | 64–>64 | >64 | Linezolid | 0.5–1 | 1 |
| Streptococcus agalactiae | 3 | >64 | Linezolid | 1–2 | ||
| Beta-hemolytic Streptococcus | 6 | >64 | Linezolid | 1–2 | ||
| Listeria monocytogenes | 10 | 1–64 | 32 | Linezolid | 1–2 | 2 |
| Corynebacterium jeikeium | 10 | >64 | >64 | Linezolid | 0.5–1 | 1 |
| Bacteroides fragilis | 10 | 64>64 | >64 | Clindamycin | 1–4 | 4 |
| Clostridium perfringens | 10 | >64 | >64 | Clindamycin | 0.06–4 | 4 |
| Propionibacterium acnes | 10 | 32–64 | 64 | Clindamycin | 0.12–0.5 | 0.25 |
Defined as having staphylococcal cassette chromosome mec type IV or IVA, being susceptible to gentamicin, and being from outpatients.
The 12 linezolid-resistant (Linr) isolates were part of the 48 NARSA strains.
For 10 isolates, there were two groups consisting of 8 strains with MICs ranging from 4 to 8 μg/ml and 2 strains with MICs of >64 μg/ml. MIC90s were not calculated.
MR, methicillin resistant.
MS, methicillin susceptible.
All MICs are in micrograms per milliliter.
In vitro antibacterial activity.
The MICs were determined according to CLSI methodology against aerobic bacteria and Neisseria gonorrhoeae (10), Listeria monocytogenes, Corynebacterium jeikeium (14), Mycobacterium fortuitum (12), Haemophilus ducreyi (15), anaerobes (11), and yeasts (13). An agar dilution method was used with Wilkins-Chalgren agar enriched with 10% horse blood for Helicobacter pylori, an agar incorporation method with agar supplemented with 1% yeast extract (Oxoid Ltd.) 1.3% Bacteriological Agar No. 1 (Oxoid Ltd.) for Legionella pneumophila, a broth microdilution method in Mycoplasma Broth Base (Oxoid Ltd., Hampshire, United Kingdom) for Mycoplasma pneumoniae. For Chlamydophila pneumoniae and Chlamydia trachomatis, MICs were determined in 96-well flat-bottom microtiter tissue culture plates using a McCoy cell culture line in a supplemented minimal growth medium consisting of Eagle's minimum essential medium (Invitrogen Ltd., Paisley, United Kingdom).
Bactericidal activity was evaluated in time-kill kinetics experiments according to CLSI methods (9) by exposing a mid-log-phase inoculum of 5 × 105 CFU/ml to each compound at 4, 16, and 64 times the MIC in broth medium and by counting the viable bacteria at 0, 4, 6, 8, and 24 h. The criterion for a bactericidal effect was a ≥3-log10 (99.9% killing) decrease in the CFU count at a specified time; a decrease of less than 3 log10 CFU/ml was interpreted as bacteriostatic activity (9). Time-kill kinetics experiments in the presence of 50% human serum were performed using bacteria grown in cation-adjusted Mueller-Hinton broth containing 50% human serum from a commercial source.
Resistance mutants.
The spontaneous resistance frequency was measured by plating 1 × 1010 CFU late-log-phase bacteria onto Mueller-Hinton agar plates with and without MUT056399 at 4 times the MIC and incubating them at 35°C for 48 h. Among the MUT056399-resistant clones obtained, two to four clones were selected per strain. After 5 serial passages on nonselective agar medium, the clones displaying a stable resistance phenotype were subject to fabI gene sequencing. Gene amplification and sequencing were performed using the forward primer 5′-AAATCAAACATTTATCGTTGTAATACGTTT-3′ and the reverse primer 5′-CAAATAATTTTCCATCAGTCCGATT-3′. Sequences of resistant clones were compared to that of the wild-type strain to identify potential mutations.
In vivo antibacterial activity.
For the systemic infection model, groups of six female Swiss mice (6 weeks old) were infected by the intraperitoneal route with MRSA, MSSA, and the GISA JUS strains prepared in 10% mucin. Ten minutes after infection, the mice were treated by the subcutaneous route with MUT056399 in a solution with 20% hydroxypropyl-β-cyclodextrin (HPBCD) and 1% (final concentration) glucose. Each mouse received a single administration of MUT056399 at various dose levels ranging from 6.25 mg/kg to 75 mg/kg. A control group was administered the vehicle alone once, and the positive reference control group was administered vancomycin in 0.9% NaCl. The animals were monitored for 2 days after infection, and deaths were recorded daily. The murine local infection model described by Andes and Craig (2) was also used to assess MUT056399 in vivo activity. In the thigh infection model, groups of five immunocompetent female Swiss mice (6 weeks old) were infected by the intramuscular route with either MRSA or MSSA strains at inoculum levels ranging from 3.1 ×106 to 9.1 ×106 CFU/mouse. One group of mice was used to determine the bacterial count in the thigh just before treatment started, the starting inoculum. At 1.5 h postinfection, the mice were treated with a single subcutaneous administration of MUT056399 in a 20% HPBCD and 1% glucose solution at various dose levels ranging from 25 mg/kg to 75 mg/kg. A positive reference control group was administered linezolid by the subcutaneous route at 50 mg/kg in 0.9% NaCl at 1.5 h postinfection. After 20 h, the thigh muscles were recovered and bacterial counting was performed. The bacteriostatic dose is defined as that which controls bacterial multiplication, resulting in a bacterial burden that is comparable to the starting inoculum.
RESULTS
In vitro antibacterial activity of MUT056399.
MUT056399 is a highly potent inhibitor of the FabI enzyme of both S. aureus and E. coli with 50% inhibitory concentration IC50s of 12 nM and 58 nM, respectively (7). MUT056399 was very active against the 118 S. aureus strains tested, including MSSA and MRSA isolates and linezolid-resistant and multidrug-resistant strains, with MIC90s between ≤0.03 and 0.12 μg/ml (Table 1). The reference compound linezolid (MIC90s, 2 to ≥16 μg/ml) was notably less potent than MUT056399.
MUT056399 also exhibited potent activity against coagulase-negative staphylococci, although more elevated MICs were seen with some of the 165 strains of coagulase-negative staphylococci than with S. aureus. This was previously reported with another FabI inhibitor (33) and could be related to a lower susceptibility of some strain of S. epidermidis to triclosan (38). MUT056399 displayed in vitro antibacterial activity against other bacterial species (Table 1), in particular against FabI-containing Gram-negative bacteria such as E. coli, Haemophilus influenzae, Neisseria gonorrhoeae, Neisseria meningitidis, Helicobacter pylori, and Moraxella catarrhalis. Interestingly, MUT056399 inhibited the replication of several atypical species such as C. trachomatis, Chlamydophila pneumoniae, and L. pneumophila. Finally, MUT056399 was inactive against both C. glabrata and C. albicans, with MICs of >64 μg/ml. The antibacterial spectrum is consistent with specific FabI inhibition with no activity against FabK-containing bacteria (streptococci and enterococci) or bacteria having both FabI and FabK (Pseudomonas aeruginosa and Enterococcus faecalis) or FabL (Bacillus sp.). The higher MICs observed in the susceptible enterobacteria than in S. aureus are probably due to the presence of efflux pumps in these Gram-negative bacteria, as shown for another FabI inhibitor active only in E. coli with its efflux pump deleted (24). MUT056399 showed slow killing kinetics in vitro at 4 times the MIC, with a reduction in viable bacterial counts of about 2 log10 CFU/ml after 8 and 24 h of incubation against most of the S. aureus strains tested, including MSSA, MRSA, and VISA strains (data not shown). With MSSA ATCC 29213, the killing rates were similar from 4 to 64 times the MIC, suggesting time-dependent killing by MUT056399 (Fig. 2A). In the presence of 50% human serum, the MIC of MUT056399 increased by 4- to 16-fold, depending on the strain. This effect could be explained by the strong affinity of MUT056399 for human serum albumin, as determined by high-performance liquid chromatography with 95% binding (18). MUT056399 also demonstrated slow killing kinetics at 4 times the actual MIC against S. aureus ATCC 29213 in the presence of 50% human serum (Fig. 2B), although under these conditions, the killing rate of MUT056399 was lower than those of linezolid and vancomycin.
Fig. 2.

Time-kill curves of MUT056399 activity against MSSA ATCC 29213. The experiments were performed by counting viable bacteria at 4, 6, 8, and 24 h. Mean values represent data from at least two experiments. (A) Filled lozenges, control; open squares, vancomycin at 4 × MIC (4 μg/ml); filled triangles, linezolid at 4 × MIC (16 μg/ml); filled circles, MUT056399 at 4 × MIC (0.25 μg/ml); crosses, MUT056399 at 16 × MIC (1 μg/ml); open circles, MUT056399 at 64 × MIC (4 μg/ml). (B) Values for bacterial cultures that contained 50% (vol/vol) human serum. Filled lozenges, control; open squares, vancomycin at 4 × MIC (4 μg/ml); filled triangles, linezolid at 4 × MIC (16 μg/ml). Under these conditions, the cultures were exposed to MUT056399 at 4 × MIC (4 μg/ml) (filled circles).
In vitro resistance to MUT056399.
The frequency of spontaneous resistance was assessed in eight S. aureus strains and was found to be low, between 2.5 × 10−9 and 7 × 10−9 at 4 times the MIC (7). In these experiments, S. aureus clones with reduced susceptibility to MUT056399 were isolated. They fall into two distinct groups, one with MUT056399 MICs of 0.5 to 4 μg/ml and another with MICs of ≥32 μg/ml (Table 2). Susceptibility to vancomycin was not affected, whereas decreased susceptibility to MUT056399 was associated with decreased susceptibility to triclosan. Sequence analysis of the fabI gene of these clones revealed that four types of single-point mutation occurred in the less susceptible variants. Those nucleotide changes resulted in the amino acid substitution A95V in the FabI protein of all of the clones with MUT056399 MICs of >32 μg/ml, whereas the amino acid substitution F204S, I193F, or Y147H was found in the FabI protein of the clones having MUT056399 MICs of 0.5 to 4 μg/ml (Table 2).
Table 2.
FabI mutations found in S. aureus mutant clones with reduced susceptibility to MUT056399
| S. aureus strain and clone | FabI mutation |
MIC (μg/ml) |
||
|---|---|---|---|---|
| Nucleotide mutation | Amino acid substitution | MUT056399 | Triclosan | |
| MSSA ATCC 29213 | ||||
| Wild type | 0.031 | 0.008 | ||
| 29213-399-2 | T611C | F204S | 1 | 0.5 |
| 29213-399-5 | A577T | I193F | 0.5 | 0.25 |
| MSSA ATCC 12600 | ||||
| Wild type | 0.031 | 0.016 | ||
| 12600-399-1 | T611C | F204S | 2 | 1 |
| 12600-399-16 | C284T | A95V | >32 | 2 |
| VISA ATCC 700699-V4 | ||||
| Wild type | 0.031 | 0.016 | ||
| 700699-399-17 | A577T | I193F | 4 | 1 |
| 700699-399-18 | C284T | A95V | >32 | 2 |
| MRSA Linr NRS 119a | ||||
| Wild type | 0.031 | 0.016 | ||
| 119-399-6 | T611C | F204S | 2 | 1 |
| 119-399-10 | C284T | A95V | >32 | 2 |
| MRSA NRS 384 (USA300) | ||||
| Wild type | 0.031 | 0.012 | ||
| 384-399-5 | T611C | F204S | 2 | 0.5 |
| 384-399-10 | C284T | A95V | >32 | 2 |
| MRSA NRS 385 (USA500) | ||||
| Wild type | 0.031 | 0.008 | ||
| 385-399-1 | T611C | F204S | 1 | 0.5 |
Linr, linezolid resistant.
In vivo antibacterial activity of MUT056399.
In the systemic model of lethal infection of mice, MUT056399 produced a dose-dependent increase in mouse survival at 48 h postinfection with MSSA and MRSA. Mean 50% effective dose (ED50) values ranging from 19.3 to 49.6 mg/kg/day against the different S. aureus strains are reported in Table 3. Under these conditions, the reference compound vancomycin showed ED50s ranging from 1.6 to 9.4 mg/kg/day as previously described (19) and linezolid, the reference compound for the GISA JUS strain, had a mean ED50 of 3.6 mg/kg/day. The data indicate that a single subcutaneous administration of MUT056399 was effective in protecting mice against various S. aureus strains, including MRSA; multidrug-resistant MRSA isolates NRS 382, NRS 384, and NRS 385; and GISA strains. In the thigh infection model, MUT056399 significantly reduced the bacterial loads of three MSSA and MRSA strains in the murine thigh in a dose-dependent manner at a mean static dose of 40 to 45.5 mg/kg/day. The activity of MUT056399 was comparable to that of linezolid in this model using a single dose of 50 mg/kg/day, as shown by the mean reduction of the bacterial burden in the thigh (Table 3).
Table 3.
MUT056399 in vivo activity in the murine systemic and thigh infection modelsa
| S. aureus strain | MIC (μg/ml) |
Mean ED50 (mg/kg/day) in murine systemic infection model |
Murine thigh infection model |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean static MUT056399 dose (mg/kg/day) | Mean bacterial burden reduction (log10)b |
||||||||
| MUT056399 | Vancomycin | Linezolid | MUT056399 | Vancomycin | Linezolid | MUT056399 | Linezolid | ||
| MSSA ATCC 29213 | 0.06 | 1 | 4 | 21.6 | 1.6 | NDc | 40 | 1.5 | 1.4 |
| MRSA ATCC 33591 | 0.06 | 2 | 2 | 31.8 | 6.7 | ND | 45.5 | 1 | 0.7 |
| MRSA NRS 382 (USA100) | 0.06 | 2 | 2 | 19.3 | 6 | ND | 44 | 1 | 1.1 |
| MRSA NRS 384 (USA300) | 0.06 | 1 | 1 | 45.1 | 9.4 | ND | |||
| MRSA NRS 385 (USA500) | 0.06 | 1 | 1 | 28.3 | 5 | ND | |||
| MRSA/GISA JUS | 0.03 | 4 | 4 | 49.6 | ND | 3.6 | |||
In the systemic infection model, the ED50 was the dose that allowed the survival of 50% of each group of treated mice at 48 h postinfection in at least two experiments. In the thigh infection model, bacterial recovery in the thighs of infected animals was performed at 20 h postinfection. All calculations were performed using XL-Stat software. P values were determined with the Dunnett test. Mean values of at least two experiments are shown.
The mean bacterial burden reduction in the thigh was measured after a single administration of MUT056399 or linezolid at 50 mg/kg.
ND, not done.
DISCUSSION
MUT056399 demonstrated very potent in vitro antibacterial activity against a large number of the staphylococci tested, including methicillin-resistant, linezolid-resistant, and multidrug-resistant strains. MUT056399 was designed as a specific FabI inhibitor, and as expected, MUT056399 showed a spectrum specific for bacterial species which are known to be strictly dependent on the FabI enzyme for fatty acid biosynthesis, including Gram-negative bacteria. This spectrum is in contrast to that of other specific FabI inhibitors such as AFN-1252 and CG400549, which have antibacterial activity only against staphylococci (24, 33). The spectrum is also different from that of triclosan, which has a broad spectrum of activity against Gram-positive and Gram-negative bacteria, fungi, and Plasmodium falciparum (20, 27) due to its ability to inhibit related ACPs. The less MUT056399-susceptible mutants of S. aureus isolated in vitro contained FabI with mutations in the same amino acid positions as other mutants resistant to FabI inhibitors previously described (22, 40). These data support the proposed antibacterial mechanism of action of MUT056399 as a specific FabI inhibitor.
In vitro, MUT056399 also showed slow S. aureus killing kinetics in the presence of 50% human serum. This result could indicate that the level of UFA present for 24 h in the culture did not complement the FabI inhibition. Concerns have been raised about compounds targeting fatty acid synthesis due to a report that some Gram-positive bacterial mutants deficient in fatty acid biosynthesis were able to complement auxotrophy for UFA by uptake of UFA from the host (6). The roles of the multiple FAS type II enzymes have been shown to vary among the different bacterial species (1, 31, 41, 42, 43). In particular, it was recently demonstrated that FAS type II inhibitors of the elongation cycle (such as specific FabI inhibitors) cannot be overcome by providing S. aureus with exogenous fatty acids, whereas in Streptococcus pneumoniae they can (35). Furthermore, MUT056399 was found to be very efficacious in vivo by protecting mice against virulent strains of S. aureus, including MSSA, MRSA, and VISA clinical isolates, in two murine infection models. These data, together with other publications of FabI inhibitors (21, 24, 27, 37, 44), do show that FabI is a validated target and that S. aureus is indeed unable to multiply in the presence of these specific inhibitors in animal models of infection (5, 33).
The emergence of MRSA and in vitro resistance to vancomycin, combined with reports of clinical failures with this and other antistaphylococcal agents (4, 16, 23), underscores the need for alternative therapies (17).
In order to address this need, MUT056399 represents a novel antibacterial compound targeting the major Gram-positive pathogen S. aureus and all of its multiresistant variants, such as MRSA, VISA, and VRSA. The key advantage of this compound is its antibacterial activity against a novel molecular target that is essential for the growth of S. aureus, including all drug-resistant variants. Consequently, cross-resistance to MUT056399 and the antibacterial agents currently used in clinical practice to treat staphylococcal infections is unlikely to occur.
ACKNOWLEDGMENTS
We thank E. Malacain, S. Floquet, A. Walton, and F. Faivre for excellent technical assistance.
Part of this study was supported by a grant for the MODEXA project from the Medicen Paris Region competitiveness cluster.
Footnotes
Published ahead of print on 8 August 2011.
REFERENCES
- 1. Altabe S., Lopez P., De Mendoza D. 2007. Isolation and characterization of unsaturated fatty acid auxotrophs of Streptococcus pneumoniae and Streptococcus mutans. J. Bacteriol. 189:8139–8144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andes D., Craig W. A. 2003. Pharmacodynamics of the new des-f(6)-quinolone garenoxacin in a murine thigh infection model. Antimicrob. Agents Chemother. 47:3935–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Appelbaum P. C. 2006. MRSA—the tip of the iceberg. Clin. Microbiol. Infect. 12(Suppl. 2):3–10 [DOI] [PubMed] [Google Scholar]
- 4. Appelbaum P. C. 2007. Reduced glycopeptides susceptibility in methicillin-resistant Staphylococcus aureus (MRSA). Int. J. Antimicrob. Agents 30:398–408 [DOI] [PubMed] [Google Scholar]
- 5. Balemans W., et al. 2010. Essentiality of FASII pathway for Staphylococcus aureus. Nature 463(7279):E3; discussion E4 [DOI] [PubMed] [Google Scholar]
- 6. Brinster S., et al. 2009. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature 458:83–86 [DOI] [PubMed] [Google Scholar]
- 7. Bryskier A., et al. 2010. MUT056399: mode of action and mechanisms of resistance, poster F1-832. 50th ICAAC, Boston, MA [Google Scholar]
- 8. Campbell J. W., Cronan J. E. 2001. Bacterial fatty acid biosynthesis: targets for antibacterial drug discovery. Annu. Rev. Microbiol. 55:305–332 [DOI] [PubMed] [Google Scholar]
- 9. Clinical and Laboratory Standards Institute 1999. Methods for determining bactericidal activity of antimicrobial agents, M26-A. Vol. 19, no. 18 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 10. Clinical and Laboratory Standards Institute 2006. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, M7-A7. Vol. 26, no. 2 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 11. Clinical and Laboratory Standards Institute 2007. Methods for antimicrobial susceptibility testing of anaerobic bacteria. Approved standard, 7th ed., M11-A7 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 12. Clinical and Laboratory Standards Institute 2003. Susceptibility testing of mycobacteria, nocardiae, and other aerobic actinomycetes, M24A. Approved standard. Clinical and Laboratory Standards Institute, Wayne, PA: [PubMed] [Google Scholar]
- 13. Clinical and Laboratory Standards Institute 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts, M27-A3. Approved standard, 3rd ed Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 14. Clinical and Laboratory Standards Institute 2006. Methods for antimicrobial dilution and disk susceptibility testing of infrequently isolated or fastidious bacteria, M45A. Approved guideline. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 15. Dangor Y., Ballard R. C., Miller S. D., Koornhof H. J. 1990. Antimicrobial susceptibility of Haemophilus ducreyi. Antimicrob. Agents Chemother. 34:1303–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De Lassence A., et al. 2006. Control and outcome of a large outbreak of colonization and infection with glycopeptide-intermediate Staphylococcus aureus in an intensive care unit. Clin. Infect. Dis. 42:170–178 [DOI] [PubMed] [Google Scholar]
- 17. Drew R. H. 2007. Emerging options for treatment of invasive, multidrug-resistant Staphylococcus aureus infections. Pharmacotherapy 27:227–249 [DOI] [PubMed] [Google Scholar]
- 18. Escaich S., et al. 2009. MUT056399 FabI inhibitor: a new antibacterial candidate against Staphylococcus aureus, poster F1-2012. 49th ICAAC, San Francisco, CA [Google Scholar]
- 19. Gross M., et al. 2003. Pharmacology of novel heteroaromatic polycycle antibacterials. Antimicrob. Agents Chemother. 47:3448–3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heath R. J., White S. W., Rock C. O. 2001. Lipid biosynthesis as a target for antibacterial agents. Prog. Lipid Res. 40:467–497 [DOI] [PubMed] [Google Scholar]
- 21. Heath R. J., White S. W., Rock C. O. 2002. Inhibitors of fatty acid synthesis as antimicrobial chemotherapeutics. Appl. Microbiol. Biotechnol. 58:695–703 [DOI] [PubMed] [Google Scholar]
- 22. Ji Y., et al. 2004. Validation of antibacterial mechanism of action using regulated antisense RNA expression in Staphylococcus aureus. FEMS Microbiol. Lett. 231:177–184 [DOI] [PubMed] [Google Scholar]
- 23. Julian K., et al. 2007. Characterization of a daptomycin-nonsusceptible vancomycin-intermediate Staphylococcus aureus strain in a patient with endocarditis. Antimicrob. Agents Chemother. 51:3445–3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karlowsky J. A., Kaplan N., Hafkin B., Hoban D. J., Zhanel G. G. 2009. AFN-1252, a FabI inhibitor, demonstrates a Staphylococcus-specific spectrum of activity. Antimicrob. Agents Chemother. 53:3544–3548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klein E., Smith D. L., Laxminarayan R. 2007. Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States, 1999-2005. Emerg. Infect. Dis. 13:1840–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Levy C. W., et al. 1999. Molecular basis of triclosan activity. Nature 398:383–384 [DOI] [PubMed] [Google Scholar]
- 27. Ling L. L., et al. 2004. Identification and characterization of inhibitors of bacterial enoyl-acyl carrier protein reductase. Antimicrob. Agents Chemother. 48:1541–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu H., Tonge P. J. 2008. Inhibitors of FabI, an enzyme drug target in the bacterial fatty acid biosynthesis pathway. Acc. Chem. Res. 41:11–20 [DOI] [PubMed] [Google Scholar]
- 29. Marrakchi H., et al. 2003. Characterization of Streptococcus pneumoniae enoyl-(acyl-carrier protein) reductase (FabK). Biochem. J. 370:1055–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marrakchi H., Zhang Y. M., Rock C. O. 2002. Mechanistic diversity and regulation of type II fatty acid synthesis. Biochem. Soc. Trans. 30:1050–1055 [DOI] [PubMed] [Google Scholar]
- 31. Massengo-Tiassé R. P., Cronan J. E. 2009. Diversity in enoyl-acyl carrier protein reductases. Cell. Mol. Life Sci. 66:1507–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moir D. T. 2005. Identification of inhibitors of bacterial enoyl-acyl carrier protein reductase. Curr. Drug Targets Infect. Disord. 5:297–305 [DOI] [PubMed] [Google Scholar]
- 33. Park H. S., et al. 2007. Antistaphylococcal activities of CG400549, a new bacterial enoyl-acyl carrier protein reductase (FabI) inhibitor. J. Antimicrob. Chemother. 60:568–574 [DOI] [PubMed] [Google Scholar]
- 34. Payne D. J., Warren P. V., Holmes D. J., Ji Y., Lonsdale J. T. 2001. Bacterial fatty-acid biosynthesis: a genomics-driven target for antibacterial drug discovery. Drug Discov. Today 6:537–544 [DOI] [PubMed] [Google Scholar]
- 35. Rock C., Frank M., Parsons J. Metabolic basis for the susceptibility of Gram-positive bacteria to fatty acid synthesis inhibitors, poster F2-864. 50th ICAAC, Boston, MA [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saito J., et al. 2008. Crystal structure of enoyl-acyl carrier protein reductase (FabK) from Streptococcus pneumoniae reveals the binding mode of an inhibitor. Protein Sci. 17:691–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sampson P. B., et al. 2009. Spiro-naphthyridinone piperidines as inhibitors of S. aureus and E. coli enoyl-ACP reductase (FabI). Bioorg. Med. Chem. Lett. 19:5355–5358 [DOI] [PubMed] [Google Scholar]
- 38. Schmid N. B., Kaplan N. 2004. Reduced triclosan susceptibility in methicillin resistant Staphylococcus epidermidis. Antimicrob. Agents Chemother. 48:1397–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sivaraman S., et al. 2004. Inhibition of the bacterial enoyl reductase FabI by triclosan: a structure-reactivity analysis of FabI inhibition by triclosan analogues. J. Med. Chem. 47:509–518 [DOI] [PubMed] [Google Scholar]
- 40. Xu H., et al. 2008. Mechanism and inhibition of saFabI, the enoyl reductase from Staphylococcus aureus. Biochemistry 47:4228–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang Y. M., Marrakchi H., White S. W., Rock C. O. 2003. The application of computational methods to explore the diversity and structure of bacterial fatty acid synthase. J. Lipid Res. 44:1–10 [DOI] [PubMed] [Google Scholar]
- 42. Zhang Y. M., Rock C. O. 2008. Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 6:222–232 [DOI] [PubMed] [Google Scholar]
- 43. Zhang Y.-M., White S. W., Rock C. O. 2006. Inhibiting bacterial fatty acid synthesis. J. Biol. Chem. 281:17541–17544 [DOI] [PubMed] [Google Scholar]
- 44. Zheng C. J., Sohn M. J., Kim W. G. 2009. Vinaxanthone, a new FabI inhibitor from Penicillium sp. J. Antimicrob. Chemother. 63:949–953 [DOI] [PubMed] [Google Scholar]