

Abstract
Despite several attempts to develop an effective prophylactic vaccine for HSV-2, all have failed to show efficacy in the clinic. The most recent of these failures was the GlaxoSmithKline (GSK) subunit vaccine based on the glycoprotein gD with the adjuvant monophosphoryl lipid A (MPL). In a phase 3 clinical trial, this vaccine failed to protect from HSV-2 disease, even though good neutralizing antibody responses were elicited. We aimed to develop a superior, novel HSV-2 vaccine containing either gD or gB alone or in combination, together with the potent adjuvant CpG oligodeoxynucleotides (CPG). The immunogenic properties of these vaccines were compared in mice. We show that gB/CPG/alum elicited a neutralizing antibody response similar to that elicited by gD/CPG/alum vaccine but a significantly greater gamma interferon (IFN-γ) T cell response. Furthermore, the combined gB-gD/CPG/alum vaccine elicited significantly greater neutralizing antibody and T cell responses than gD/MPL/alum. The efficacies of these candidate vaccines were compared in the mouse and guinea pig disease models, including a novel male guinea pig genital disease model. These studies demonstrated that increased immune response did not correlate to improved protection. First, despite a lower IFN-γ T cell response, the gD/CPG/alum vaccine was more effective than gB/CPG/alum in mice. Furthermore, the gB-gD/CPG/alum vaccine was no more effective than gD/MPL/alum in mice or male guinea pigs. We conclude that difficulties in correlating immune responses to efficacy in animal models will act as a deterrent to researchers attempting to develop effective HSV vaccines.
INTRODUCTION
Herpes simplex virus type 2 (HSV-2) commonly causes infections of the genital area. The virus is sexually transmitted, and infection leads to a primary disease characterized by painful genital lesions. HSV-2 is able to transit via sensory nerves by retrograde axonal transport from the site of primary infection to local ganglia, where it establishes latency (20) until reactivation occurs and recurrent disease is manifested. Worldwide, 16% of the population within the 15 to 49 year age group are living with HSV-2 (19). Clinical manifestations of HSV-2 are not usually life threatening but can cause serious complications in infants and immune-compromised hosts and are also believed to increase susceptibility to the acquisition of HIV (6); asymptomatic shedding is also common (16). Current treatment includes antiviral therapy with acyclovir, valacyclovir, or famciclovir, which reduce symptoms but do not cure HSV-2 infection, making the virus an important target for vaccine development.
Several attempts have been made to develop an effective prophylactic vaccine (5); however, to date, none has shown efficacy in humans (26). A subunit vaccine consisting of glycoprotein D (gD) adjuvanted with monophosphoryl lipid A (MPL) and alum developed by GlaxoSmithKline (GSK) demonstrated significant protection from the acquisition of disease in HSV-1-seronegative women in two phase 3 clinical trials (28), but not in other populations. However, a more recent phase 3 study of this vaccine was not able to recapitulate these results and did not show significant protection of HSV-1-seronegative women, and development of this candidate has been terminated (http://www.niaid.nih.gov/news/newsreleases/2010/Pages/Herpevac.aspx). This is a major setback to HSV vaccinology, and novel methods to develop and assess superior vaccines that can consistently protect the broad population are therefore urgently needed.
There is mounting evidence that an HSV-2 vaccine needs not only to induce a strong neutralizing antibody (Ab) response, but also to generate robust T cell immunity. First, more severe HSV-2 infections are seen in HIV-positive individuals than in HIV-negative individuals (24). Also, observations of developing disease lesions in humans show that CD8 T cells infiltrate the site with kinetics associated with regression (33). In addition, studies in mice have demonstrated that HSV-specific T cell responses can control virus reactivation at the level of the ganglia (18) and reduce disease at the mucosal site (21). Lastly, HSV-specific CD4 and CD8 T cell responses have been found in HSV-2-seronegative human subjects with known exposure to HSV-2, implying a protective role for T cell immunity (25).
There are a number of ways to enhance the T cell component of a vaccine. One is to include antigens known to contain dominant T cell epitopes. Envelope protein gB has been shown to induce strong T cell responses in humans (15) (as well as neutralizing antibody [1]). In small-animal models, gB vaccination induced protection from HSV-2 challenge (14). Also, CD8 T cell responses to gB have been shown to control latency and prevent reactivation from dorsal root ganglia in mice infected with HSV-2 (11). However, when a gB-gD vaccine (adjuvanted with MF59) was tested in clinical trials, it failed to protect men or women from HSV infection and showed no obvious effect on the course of disease in subjects who were subsequently infected (2). The observation that MF59-adjuvanted vaccine elicited high-titer and neutralizing antibody but did not protect indicates that an adjuvant capable of also inducing strong Th1 T cell response, such as CpG oligodeoxynucleotides (CPG), may be required. CPG is a novel TLR9 agonist which can act as a powerful vaccine adjuvant. Data obtained with hepatitis B surface antigen (HBsAg) showed that CPG/alum induced more rapid and more potent antibody (4, 30) and T cell (4) responses than MPL/alum in human volunteers.
In the current study, we investigated whether HSV-2 vaccine efficacy could be improved by inclusion of antigens and adjuvant with superior T cell and neutralizing antibody properties. We show that by using a more potent adjuvant (CPG) in combination with the inclusion of an additional antigen (gB) we could elicit broader and more potent antibody and T cell responses in mice that were greater than those induced by the recent failed clinical candidate, gD/MPL adjuvant. We developed a novel disease model in male guinea pigs and tested the various vaccine candidates in male and female guinea pig and female mouse models. We show that gD/CPG/alum is more protective than gB/CPG/alum despite gD generating a similar antibody response and an inferior T cell response. We also show that the gB-gD/CPG/alum vaccine, which generated the greatest immune responses, was no more protective than gD/MPL/alum in the models. We conclude that immunity and protection do not correlate in the available HSV-2 disease models and that this needs to be addressed if researchers are to invest the significant resources needed to develop an effective HSV vaccine.
MATERIALS AND METHODS
Vaccines.
The recombinant glycoprotein antigens gB and gD (which lacked the carboxyl-terminal membrane anchor) were transiently expressed in HEK293 cells, and the secreted proteins were harvested by batch centrifugation. The proteins were purified from the supernatant by Ni-nitrilotriacetic acid (NTA) affinity chromatography utilizing the proteins' engineered C-terminal hexahistidine tag. This tag was proteolytically cleaved from the final protein by incubation with thrombin targeted to an engineered thrombin cleavage site (gD only), and both were separated from the protein of interest by benzamidine ion exchange and further Ni-NTA chromatography, respectively. Endotoxin levels were reduced using Mustang E (Pall) chromatography filters, if required, prior to buffer exchange into Dulbecco's phosphate-buffered saline (DPBS) and 0.2-μm filtration.
Mice or guinea pigs were immunized with equivalent amounts (by weight) of glycoprotein. Since the truncated recombinant gD protein is approximately half the molecular weight of the recombinant gB protein, vaccines contained, mole for mole, approximately twice as much gD protein. Each mouse or guinea pig received recombinant glycoprotein formulated with 100 μg of a class B CPG (Pfizer Inc.) or 50 μg MPL (Invivogen) and 70 μg aluminum hydroxide gel (Alhydrogel “85” 2%; Brenntag Biosector, Frederikssund, Denmark). In dose-ranging immunogenicity studies, mice were vaccinated with 1, 10, or 40 μg of total protein. In challenge studies, animals were vaccinated with 20 μg of total protein. The animals were immunized by intramuscular injection (50 μl) into the tibialis anterior muscle on a prime/boost schedule of days 0 and 30 for mice; days 0, 30, 51, and 79 for female guinea pigs; and days 0 and 28 for male guinea pigs (immunized with a 110-μl volume). In challenge studies, animals were infected 2 weeks after the last vaccine boost.
Animals.
Six- to 8-week-old female BALB/c mice (Charles River, United Kingdom) and male and female Hartley guinea pigs (Charles River, Canada) weighing ∼250 to 410 g were used in the studies. All experiments were carried out in compliance with United Kingdom or Canadian legislation and were subject to local ethical review.
HSV-2 mouse challenge models. (i) Genital model.
Six or 7 days prior to infection, mice were injected subcutaneously with 2 mg of progesterone (Depo-provera; Pharmacia). On the day of infection, 20 μl of HSV-2 strain 333 (104 or 105 PFU; a gift from S. Efstathiou, Cambridge, United Kingdom) was instilled into the vagina. The mice were then weighed and scored daily for disease in the genital area by operators who were blinded to the identity of the treatment groups. Scoring was as follows: 0, no apparent infection; 1, slight redness; 2, redness and swelling; 3, severe redness/swelling, including involvement of the surrounding area; 4, ulceration and or hair loss; 5, generalized signs of severe disease. Any mice approaching a score of 5 were sacrificed.
(ii) Intranasal model.
Mice were lightly anesthetized using isoflurane, and 50 μl of 106 PFU HSV-2 strain 333 was slowly pipetted onto the tip of the nose. The mice were kept on their backs until complete recovery from anesthesia. Following infection, the mice were weighed and scored daily by operators (who were blinded to the identity of the treatment groups) using the following clinical scores: 0, no apparent infection; 1, piloerection and/or breathing difficulties; 2, reduced movement/dull squinting eyes; 3, sores on eyes, ocular discharge, and/or hunched; 4, exacerbation or additional symptoms from grade 3; 5, severe respiration difficulties; 6, paralysis. Any mice approaching a score of 5 were sacrificed.
HSV-2 guinea pig genital challenge model. (i) Female guinea pig model.
Immediately prior to virus inoculation, the vagina was swabbed using a sterile applicator (Starplex Scientific, Etobicoke, Ontario, Canada) moistened with sterile phosphate-buffered saline (PBS) to rupture the vaginal closure membrane. The guinea pigs were then inoculated intravaginally with ∼106 PFU HSV-2 (MS strain; ATCC [Manassas, VA] VR-540) in a volume of 0.1 ml. Following virus inoculation, the animals were weighed and monitored daily for evidence of primary disease. Virus-induced lesions were scored by a single investigator for 15 days after inoculation, using a method described previously (13). In brief, primary genital skin disease was quantified by use of a lesion score scale as follows: 0, no disease; 1, redness or swelling of external vagina; 2, a few small vesicles; 3, several large vesicles; and 4, several large ulcers with maceration. Clinical features of the disease, such as urinary retention, hind limb paralysis, and mortality, were monitored daily.
(ii) Male guinea pig model.
One day prior to inoculation, the lower abdomen and hind legs of the guinea pig were clipped and chemically depilated. Depilation was performed on the area when necessary throughout the study. The animals were challenged on day 42 with 105 PFU HSV-2 (MS strain) applied to the scarified tissue of the penis in a fixed volume of 0.01 ml for 2 min. Scoring of the lesions was performed daily using the following scale: 0, no disease; 1, redness and/or swelling of the genital area or unable to protrude penis; 2, 1 to 5 small lesions; 3, greater than 5 small lesions or a few large lesions; 4, confluent lesions with ulcer formation.
Statistical analysis of challenge model data.
For the challenge models, linear regression was performed on the day 5 to 14 weight data from each animal using ordinary least squares. The slope of the linear regression indicates the rate of change in body weight (g) over time for each mouse. The calculated rates were analyzed using a linear mixed model that investigated differences between the challenge doses and antigens. Individual comparisons of the antigens to the naïve or adjuvant-only groups were quantified at each challenge dose through the average rate of weight loss, differences in weight loss, 95% confidence intervals, and P values (a subset are reported). A P value of <0.05 was considered significant for all of the statistical comparisons. All analyses were performed using SAS v9.2.
Viral antibody neutralization assay.
Baby hamster kidney cells (BHK-21; ATCC CCL-10) in minimal essential medium (MEM) supplemented with fetal bovine serum, 200 mM l-Glu, and 10,000 U penicillin/streptomycin (Invitrogen, Paisley, United Kingdom) were seeded onto 96-well tissue culture plates and incubated overnight at 37°C and 5% CO2. Heat-inactivated serum samples plus 10 hemolytic units (HU)/ml guinea pig complement (Sigma, United Kingdom) and an assay control (anti-HSV-1/2 monoclonal antibody [MAb] MCA404G; Serotec, Oxford, United Kingdom) were diluted in supplemented MEM and incubated with HSV-2 strain HG52 (European Centre for Animal Cell Cultures, United Kingdom) on a shaker for 60 min at 37°C and 70 rpm. An aliquot of this suspension was transferred onto confluent BHK-21 cells and incubated for 96 h at 37°C and 5% CO2. Cell viability was determined by measuring the absorbance at 492 nm following addition of CellTiter 96 AQueous One Solution Cell Proliferation Assay Reagent (Promega, Southampton, United Kingdom). Data are presented as the serum dilution factor required to reduce viral neutralization efficiency to 50% compared with BHK-21 cells that were not exposed to HSV-2. Statistical significance tests were performed with LabStats, a custom-built software package run in Excel.
Ex vivo IFN-γ ELISpot assay.
Peptides (CPC Scientific) representing selected sequences of gD and gB that had previously been demonstrated to contain T cell epitopes (data not shown) were synthesized as 15-mers, quality assessed, and formulated into pools representing each protein. The number of peptides per pool ranged from 10 to 24, and the concentration of dimethyl sulfoxide (DMSO) in the assay was kept below 0.25%. An enzyme-linked immunospot (ELISpot) assay was performed following the manufacturer's instructions. Briefly, 96-well polyvinylidene difluoride-backed plates (MSIP S 45; Millipore, Bedford, MA) were coated with 7.5 μg/ml of anti-IFN-γ MAb AN18 (Mabtech, Sweden) overnight at 4°C. The plates were then washed 6 times with PBS and blocked with RPMI plus glutamax-supplemented penicillin/streptomycin, MEM nonessential amino acids (NEAA), and 10% heat-inactivated fetal calf serum (FCS) (R10) for 4 h. Splenocytes were recovered, washed 3 times, and resuspended in R10. The splenocytes were added to the plates in 100 μl R10/well. The input cell numbers were 5 × 105/well in duplicate wells. For assays performed in parallel with limiting-dilution analyses (LDAs), duplicate wells with 2.5 × 105 and 1.25 × 105 splenocytes/well were used. Peptides were added at a final concentration of 2 μg/ml/peptide in R10, with a 100-μl volume per well. The assay mixtures were incubated for 16 h at 37°C and 5% CO2 and arrested by shaking off the contents and washing them 6 times with PBS-0.05% Tween 20 (Sigma Chemical Co.). Next, 100 μl of 1-μg/ml biotinylated anti-IFN-γ MAb R4-6AZ-biotin (Mabtech, Sweden) was added. After 2 h of incubation, the plates were washed six more times, a 1:1,000 dilution of streptavidin-alkaline phosphatase conjugate (Mabtech) was added to the wells, and the plates were incubated at room temperature in the dark for a further 1 h. Next, the wells were again washed 6 times, and 100 μl of BCIP (5-bromo-4-chloro-3-indolylphosphate)/nitroblue tetrazolium (NBT) substrate (Mabtech) was added. After approximately 10 min, the colorimetric reaction was terminated by washing with tap water, and the plates were air dried. ELISpot plates were evaluated with an automated ELISpot counter (AID-GmbH, Germany). The settings for intensity and spot size were predefined, and the same settings were used throughout.
MSD assay.
Briefly, splenocytes were recovered, washed 3 times, and resuspended in R10. The splenocytes were added to the plates in 100 μl R10/well. The input cell numbers were 5 × 105/well in duplicate wells. Peptides were added at a final concentration of 2 μg/ml/peptide in R10 at 100-μl volume per well. The assay mixtures were incubated for 72 h at 37°C and 5% CO2, and then 100 μl of supernatant was harvested and the levels of IFN-γ were quantified using a single-plex mesoscale diagnostic (Meso Scale Discovery) murine IFN-γ detection kit (MSD, MD) according to the manufacturer's instructions.
RESULTS
Immunological comparison of HSV-2 antigens with CPG or MPL adjuvant.
The immunogenicities of gD and gB proteins (delivered separately or combined) formulated with CPG/alum was assessed in mice 28 days postvaccination and compared to the immune response induced by gD with MPL/alum adjuvant. The gB-gD/CPG/alum vaccine induced the highest neutralizing antibody response at all dose levels, significantly higher than that induced by the individual antigens alone (P = <0.0001) (Fig. 1). The gD/MPL/alum vaccine was the least effective at inducing neutralizing antibody at all doses (P = <0.001 versus gB-gD/CPG/alum at the 40-μg dose, and P = 0.0067 versus gD or gB/CPG/alum at the 40-μg dose.
Fig. 1.
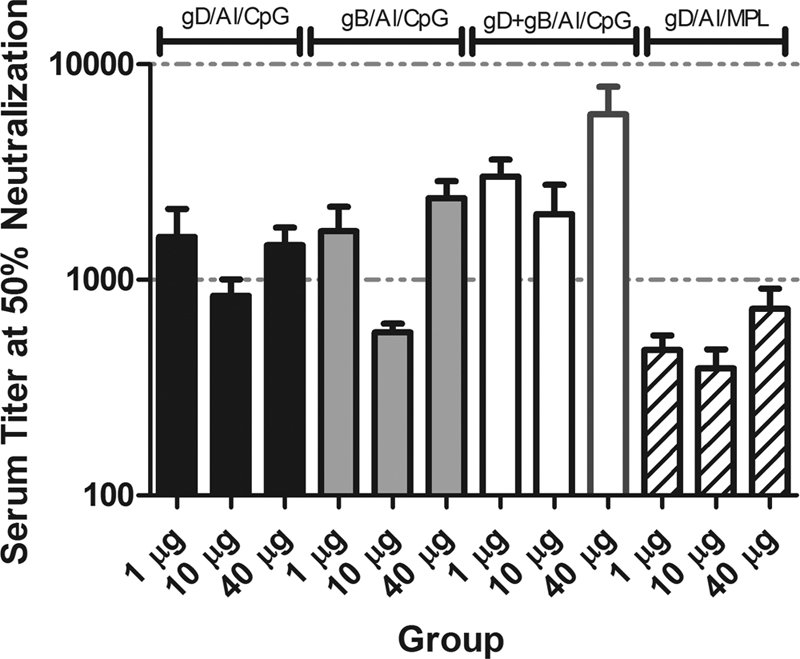
Neutralizing activity of serum in BALB/c mice. The animals were vaccinated with different concentrations of gD/CPG/alum (Al), gB/CPG/alum, gB-gD/CPG/alum, or gD/MPL/alum (the concentration is the sum of equal quantities of each antigen). The error bars indicate 1 standard deviation.
The IgG subclasses induced by the antigens were analyzed. It is known that IgG1 is a Th2-associated antibody and IgG2a is a Th1-associated antibody (29). gB/CPG/alum induced the most Th1-biased response (mean IgG2a/IgG1 ratio change, 0.9 to 1.0) compared to gD/CPG/alum (mean IgG2a/IgG1 ratio change, 0.1 to 0.2). gD/MPL elicited the most Th2-biased antibody response (mean IgG2a/IgG1 ratio changes, 0.02, 0.04, and 0.05 at 1-, 10-, and 40-μg doses, respectively).
T cell responses in mice were assessed in spleen cells by measuring the production of IFN-γ by ELISpot and MSD following in vitro stimulation with gD- or gB-specific peptides. Negative unstimulated controls for all tests were below 30 SFC/5 × 105 cells, with an average below 10 spot-forming units (SFC)/5 × 105 cells. All tests had positive-control responses above 200 SFC/5 × 105 cells (data not shown). As shown in Fig. 2, T cells from mice vaccinated with either 10 or 40 μg of gB/CPG/alum induced a significantly stronger IFN-γ response than those from mice vaccinated with gD/CPG/alum (P = 0.0153 for the 10-μg concentration; P = 0.0045 for the 40-μg concentration).
Fig. 2.

(A and B) Numbers of T cells secreting IFN-γ in response to gD protein (A) or gB protein (B). (C) Amount of IFN-γ secreted by T cells as measured by MSD. Six- to 8-week-old BALB/c mice were vaccinated with different doses of the HSV-2 antigen gD with either CPG/alum or MPL/alum or gB-gD/CPG/alum (the concentration is the sum total of equal quantities of both antigens). The error bars indicate standard deviations.
Combining gD with gB in the same vaccine induced gD-specific IFN-γ T cell responses similar to those induced by vaccinating with gD alone. However, the gD/gB combined vaccine appeared to induce a lower IFN-γ ELISpot response to gB protein than vaccination with gB alone. T cells from mice vaccinated with gD/MPL/alum produced significantly less IFN-γ than was measured in other vaccine groups (P = <0.001 across all doses of gB and gD). These data are supported by MSD data measuring the concentration of IFN-γ secreted into the supernatants in the ELISpot assay (Fig. 2C). Thus, both ELISpot data, measuring the frequency of IFN-γ-producing T cells, and MSD data, measuring the total amount of IFN-γ produced, indicated the following ranking: gB-gD/CPG/alum  gB/CPG/alum > gD/CPG/alum > gD/MPL/alum.
gB/CPG/alum > gD/CPG/alum > gD/MPL/alum.
Comparison of gD and gB efficacies in mouse and guinea pig genital models.
Having established that gB induced a neutralizing antibody response similar to but a T cell response superior to those of gD when formulated with CPG/alum, the efficacious properties of the two antigens were assessed in mouse and guinea pig HSV-2 genital models. The course of primary infection in control mice or guinea pigs was very similar to that previously reported by other groups (9, 27), i.e., lesions generally appeared on the external genitalia of animals within 4 to 5 days after viral inoculation (Fig. 3 and 4). In the mouse genital model, control groups had significant weight loss (mean, −0.62 g/day) and mortality (75% in the 104-PFU challenge group and 100% in the 105-PFU challenge group). Animals immunized with gB or gD and challenged with the lower virus challenge dose (104 PFU/mouse) were completely protected from disease symptoms with no weight loss and no mortality (Fig. 3A and C). Similar results were seen for the gD-vaccinated mice challenged with the higher viral dose (105 PFU/mouse) (Fig. 3B). However, mice vaccinated with gB and inoculated with the higher viral challenge were not totally protected from disease (Fig. 3D) and had 25% mortality. These mice had a delayed onset of disease progression (day 10) compared to the control groups (day 5) and showed better protection from weight loss than the control animals (P = <0.0001). Similar results were seen in the female guinea pig genital model (Fig. 4). Animals immunized with gD/CPG/alum had significantly reduced mean clinical scores, no weight loss, and no mortality compared to naive animals, which had significant weight loss (mean, −12.47 g/day and 43% mortality). Animals immunized with gB/CPG/alum had a delayed onset of disease progression and reduced weight loss (mean, −2.02 g/day and no mortality). These data show that despite the higher T cell response seen with gB/CPG/alum, the level of protection was less than that induced by gD/CPG/alum.
Fig. 3.

Protective efficacy of HSV-2 gB and gD antigens in a mouse genital model. Six- to 8-week-old female BALB/c mice were vaccinated by intramuscular injection with either 20 μg of gD (A and B) or 20 μg of gB (C and D) protein on days 0 and 28. On day 62, the mice were infected via the intravaginal route with either 104 PFU (A and C) or 105 PFU (B and D) of HSV-2 333. Following infection, the mice were scored daily for 14 days. The error bars indicate standard deviations.
Fig. 4.
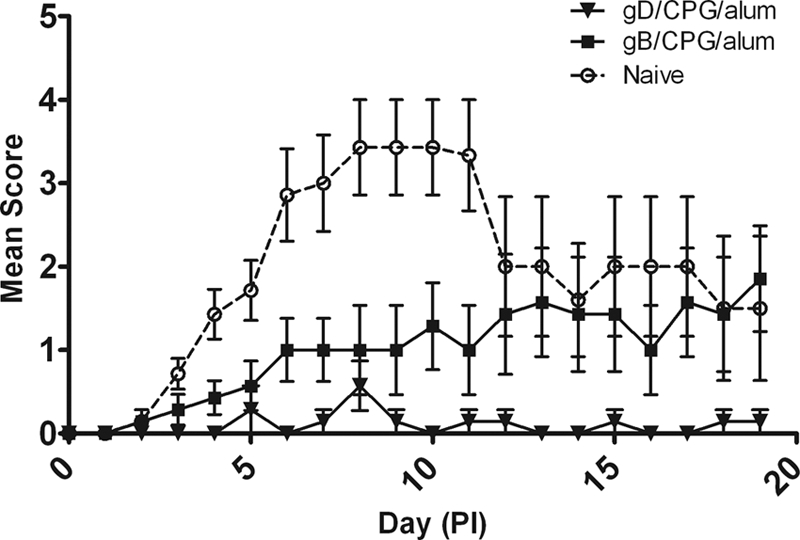
Protective efficacies of HSV-2 gB and gD antigens in a guinea pig HSV-2 model. Female Hartley guinea pigs were immunized by intramuscular injection on days 0, 30, 51, and 79 with 20 μg gB or 20 μg HSV gD in combination with CPG/alum or DPBS (Naive). PI, postinfection. The error bars indicate standard deviations.
Efficacy of gB-gD/CPG/alum vaccine compared with gD/MPL/alum vaccine in novel HSV-2 mouse and guinea pig models.
The relative efficacies of gB-gD/CPG/alum and gD/MPL/alum were compared in two different HSV-2 challenge models, a herpes encephalitis model, where the virus is delivered by intranasal infection (2), and a novel male guinea pig genital model. In these models, we assessed whether the superior immunogenicity induced by the gB-gD/CPG/alum vaccine could improve upon the protection afforded by the gD/MPL/alum vaccine.
In the mouse encephalitis model, the course of disease in naive control mice was similar to that reported elsewhere (10). Naive mice began to show clinical signs of illness at around 4 days postinfection (Fig. 5A), with piloerection, lesions on the eye, decreased bodyweight, and often hind limb paralysis, at which point they were humanely culled. Mortality in this group was observed to be 62.5% (Table 1). Mice vaccinated with gB-gD/CPG/alum showed complete protection from disease symptoms (Fig. 5), including significant weight gain (Table 1). However, in this model, mice vaccinated with gD/MPL/alum also showed complete protection, which was not statistically different from that with the gB-gD/CPG/alum vaccine.
Fig. 5.

Protective efficacies of HSV-2 gB and gD antigens in mouse and male guinea pig HSV-2 models. (A) Six- to 8-week-old female BALB/c mice were vaccinated with either 20 μg of gD/CPG/alum, 20 μg of gD/MPL/alum, or a combination of gB-gD/CPG/alum (10 μg of each protein) on days 0 and 28. On day 62, the mice were infected via the intravaginal route with 106 PFU of HSV-2 333. Following infection, the mice were scored daily for 14 days. (B) Male Hartley guinea pigs were immunized by intramuscular injection on days 0 and 28 with 10 μg gB and 10 μg gD in combination with CPG/alum or 20 μg gD in combination with MPL/alum. Following infection, the guinea pigs were scored daily for clinical signs of infection. The error bars indicate standard deviations.
Table 1.
Comparison of disease in mice vaccinated with gB-gD/CPG and gD/MPL and challenged with HSV-2a
| Antigen | Avg wt loss (g/day)b | P valuec | % Mortality (n = 10) | % Disease symptoms |
|---|---|---|---|---|
| Naive | −0.60 | 62.5 | ||
| CPG/alum | −0.71 | 0.5558 | 50.0 | 100 |
| MPL/alum | −0.73 | 0.5031 | 87.5 | 100 |
| gD/MPL/alum | +0.08 (gain) | 0.0005 | 0.00 | 12.5 |
| gB-gD/CPG/alum | +0.10 (gain) | 0.0004 | 0.00 | 0 |
Mice were immunized at weeks 0 and 4 and challenged with 106 PFU HSV-2 by the intranasal route at week 6.
Mean estimated slope of the weight loss between days 5 and 14 postchallenge.
P values were calculated on differences between weight loss in vaccinated and naive animals at a given HSV-2 dose.
In an effort to differentiate the immunologically superior gB-gD/CPG/alum vaccine from gD/MPL/alum, we developed a novel genital HSV-2 challenge model using male guinea pigs. Control animals inoculated with 105 PFU HSV developed multiple vesicular lesions by day 4, with individual peak scores between days 4 and 9. The mortality rate in this group was 50%, Three of 6 animals developed urinary retention, and 3 of 6 animals had hind limb paralysis. As shown in Fig. 5B, even within the vaccinated groups, guinea pigs developed genital skin lesions following challenge with HSV-2. However, the lesions were less severe in immunized animals than in controls, indicating that both vaccines had induced partial protection in this model. In addition, there was no mortality in immunized animals, nor was any incidence of hind limb paralysis or urinary retention recorded (Tables 2 and 3).
Table 2.
Incidence of clinical features in male guinea pigsa
| Clinical feature | Naïve incidence (%) | gD/gB/CPG/alum incidence (%) | gD/MPL/alum incidence (%) |
|---|---|---|---|
| Genital skin lesion | 6/6 (100) | 6/6 (100) | 6/6 (100) |
| Urinary retention | 3/6 (50) | 0/6 (0) | 0/6 (0) |
| Hind limb paralysis | 3/6 (50) | 0/6 (0) | 0/6 (0) |
Following penile scarification and infection, clinical scores were monitored daily.
Table 3.
Weight loss and mortality in male guinea pigs vaccinated with gB+gD/CPG/alum or gD/MPL/alum and challenged with HSV-2a
| Antigen | Avg wt loss (g/day)a | P valuec | % Mortality (n = 6) |
|---|---|---|---|
| Naive | −9.19 | 50 | |
| gB-gD/CPG/alum | 3.05 | <0.0001 | 0 |
| gD/MPL/alum | 3.06 | <0.0001 | 0 |
Male guinea pigs (n = 6) were immunized on days 0 and 28. Two weeks after the last boost, the guinea pigs were challenged with105 PFU of HSV-2.
Mean estimated slope of the weight loss between days 5 and 14 postchallenge.
P values were calculated on differences between weight loss in vaccinated and naive animals at a given HSV-2 dose.
DISCUSSION
A vaccine for HSV-2 genital herpes would be of great health benefit but has remained elusive. Most attempts to develop an HSV-2 vaccine have focused on the surface glycoproteins gD and gB, but although these have proved capable of eliciting neutralizing Ab in humans, they have not been able to prevent infection or disease when tested in large-scale clinical trials (5). Recently, in a large phase 3 clinical trial, the GSK vaccine gD/MPL/alum failed to show protection in HSV-1 seronegative women (http://www.niaid.nih.gov/news/newsreleases/2010/Pages/Herpevac.aspx) despite data from previous trials suggesting efficacy in this small subpopulation was achievable (GlaxoSmithKline press release).
The gD/MPL/alum vaccine had previously been shown to provide protection in preclinical models of HSV-2 (3), and we were interested to assess the relationship between immunogenicity and protection for HSV-2 vaccines. We set out to assess the immunogenicity and protection afforded by gD/MPL/alum and to determine whether these could be improved by the addition of gB or the replacement of the adjuvant with CPG/alum. In addition to testing in the standard HSV-2 models in female mice and guinea pigs, we also developed a novel disease model using male guinea pigs, as males represent an important segment of the at-risk human population.
Comparing vaccines in preclinical immunogenicity studies in BALB/c mice, we determined the following ranking for inducing neutralizing antibody responses in mice: gB-gD/CPG/ alum > gD/CPG/alum gB/CPG/alum > gD/MPL/alum. Regarding IFN-γ T cell responses, the ranking was as follows: gB/CPG/alum > gB-gD/CPG/alum > gD/CPG/alum > gD/MPL/alum. This observation raises the possibility that gD may be inhibitory for gB, highlighting the need to evaluate multivalent HSV-2 vaccines containing a wide spectrum of antigens that can simultaneously induce multiple arms of the immune system. Also, care needs to be exercised when interpreting immunogenicity data for gB and gD proteins in a particular mouse strain. For example, different results may be obtained using C57BL/6 mice, especially considering that gB contains a very potent cytotoxic T lymphocyte (CTL) epitope in these mice (31).
Nevertheless, when adjuvanted with CPG, gD, and gB, whether individually or combined, were superior vaccines to gD/MPL, emphasizing the critical role the adjuvant plays in determining vaccine immunogenicity. CPG is a highly active TLR9 agonist capable of generating higher levels of neutralizing antibody and superior T cell responses in mice compared to MPL, which is a TLR4 agonist (4, 7, 32). TLRs link innate and adaptive immunity; after recognition of their ligands, all TLRs (except TLR3) signal through the adaptor protein MyD88, leading to the activation of NF-κB and consequently the induction of many genes involved in immunity (17). However, TLR4 signaling also occurs independently of MyD88 via the adaptor protein TRIF, which leads to the induction of type I interferon. TLR9 is also capable of activating type I IFN, but in a MyD88-dependent manner. Thus, as highlighted by the current data, the choice of the TLR agonist to be used as an adjuvant is a crucial step in vaccine development.
The various vaccines were tested for efficacy in preclinical HSV-2 disease models. Despite the observation that gB/CPG/alum was able to induce a higher T cell response than gD/CPG/alum (and an equivalent neutralizing antibody response), the level of protection induced by gB/CPG/alum was less than that induced by gD/CPG/alum in both the female guinea pig and female mouse genital challenge models. In light of the immunogenicity data generated with gB, these efficacy results were surprising and represent a novel finding, as they suggest that superior immunogenicity does not correlate to superior protection. A difference in the protective abilities of gB and gD has been reported previously, using a mouse model of HSV-2-induced neurological disease (7). In this case, gB (plus alum only)-immunized mice were not protected and showed a mortality rate of 60 to 90%, while significant protection against HSV-2 challenge was observed with gD immunization. However, in this study, immunological endpoints were not measured (other than specific antibody levels by immune precipitation) and so could not be correlated to disease protection.
Since our vaccines gave complete protection from disease in the standard female genital models, we developed two new challenge models in which we tried to differentiate the efficacies of gB-gD/CPG/alum and gD/MPL/alum. In the murine intranasal model, both vaccines gave almost complete protection, and it was not possible to differentiate between the two, even with a very high virus challenge. Perhaps differentiation between the protective capacities of the two vaccines might have been possible if lower, immunologically suboptimal doses had been tested.
In a novel male genital model, gD/MPL/alum failed to show complete protection from disease, a result similar to that seen in clinical trials. The suboptimal level of protection induced by gD/MPL/alum provided a clinical window in which superior gB-gD/CPG/alum vaccine protection could be tested. However, in the experiment, gB-gD/CPG/alum could not be differentiated from the gD/MPL/alum vaccine. It provided a level of efficacy similar to that of the gD/MPL/alum vaccine, despite eliciting higher neutralizing antibody and T cell responses (as measured in the mouse immunogenicity experiments).
The failure of gD/MPL/alum to protect in the clinic and of the immunologically superior gB-gD/CPG/alum to protect in the male genital guinea pig model could imply that subunit vaccines induce too narrow an immune response to be efficacious in HSV-2. Some have argued that alternative approaches that provide exposure to a wide spectrum of immunogens may more accurately induce an immune response that correlates with protection. However, while nonreplicating virus vaccines have their limitations, clinical efficacy has recently been reported with a cytomegalovirus (CMV) glycoprotein vaccine (23), so a precedent has been set in the herpes field for an efficacious subunit vaccine.
Overall we conclude that gB-gD/CPG/alum is more immunogenic in mice than gD/MPL/alum. However, the level of protection seen in the female and male HSV-2 models was not correlated to differences in circulating neutralizing antibody or IFN-γ T cell responses, endpoints regarded as industry standard evaluations. It is possible that the correlate(s) of protection in these models was an immunological endpoint that we did not measure, for example, cytotoxic T cells, NK cells, mucosal IgA, multifunctional T cells, or regulatory T cells. It is unlikely to be a component of the innate, nonspecific immune response, since control mice receiving adjuvant only, containing either the TLR4 agonist MPL or the TLR9 agonist CPG, showed no evidence of protection from HSV challenge. The evidence in the literature suggests that neutralizing antibody has a major role to play in protection from HSV-2 disease (8, 22). The role for IFN-γ T cells in protection from disease is equally compelling. Protection was severely compromised by T cell depletion of immune mice (8) or in the absence of IFN-γ (12), suggesting that IFN-γ may be an important correlate of protection in mice.
We conclude that until there is a better understanding of the relationship between immunity and protection and improved translation of this protection from preclinical models into patients, vaccine developers will hesitate to produce further vaccines for HSV-2 disease.
ACKNOWLEDGMENT
This work was wholly supported by Pfizer Global Research and Development.
Footnotes
Published ahead of print on 18 August 2011.
REFERENCES
- 1. Ashley R., Benedetti J., Corey L. 1985. Humoral immune response to HSV-1 and HSV-2 viral proteins in patients with primary genital herpes. J. Med. Virol. 17:153–166 [DOI] [PubMed] [Google Scholar]
- 2. Blaney J. E., Jr., et al. 1998. Immunization with a single major histocompatibility complex class I-restricted cytotoxic T-lymphocyte recognition epitope of herpes simplex virus type 2 confers protective immunity. J. Virol. 72:9567–9574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bourne N., et al. 2003. Herpes simplex virus (HSV) type 2 glycoprotein D subunit vaccines and protection against genital HSV-1 or HSV-2 disease in guinea pigs. J. Infect. Dis. 187:542–549 [DOI] [PubMed] [Google Scholar]
- 4. Cooper C. L., et al. 2005. CPG 7909 adjuvant improves hepatitis B virus vaccine seroprotection in antiretroviral-treated HIV-infected adults. AIDS 19:1473–1479 [DOI] [PubMed] [Google Scholar]
- 5. Corey L., et al. 1999. Recombinant glycoprotein vaccine for the prevention of genital HSV-2 infection: two randomized controlled trials. Chiron HSV Vaccine Study Group. JAMA 282:331–340 [DOI] [PubMed] [Google Scholar]
- 6. Corey L., Wald A., Celum C. L., Quinn T. C. 2004. The effects of herpes simplex virus-2 on HIV-1 acquisition and transmission: a review of two overlapping epidemics. J. Acquir. Immune Defic.Syndr. 35:435–445 [DOI] [PubMed] [Google Scholar]
- 7. Dix R. D., Mills J. 1985. Acute and latent herpes simplex virus neurological disease in mice immunized with purified virus-specific glycoproteins gB or gD. J. Med. Virol. 17:9–18 [DOI] [PubMed] [Google Scholar]
- 8. Dudley K. L., Bourne N., Milligan G. N. 2000. Immune protection against HSV-2 in B-cell-deficient mice. Virology 270:454–463 [DOI] [PubMed] [Google Scholar]
- 9. Gillgrass A. E., et al. 2005. Estradiol regulates susceptibility following primary exposure to genital herpes simplex virus type 2, while progesterone induces inflammation. J. Virol. 79:3107–3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haynes J. R., et al. 2006. Potent protective cellular immune responses generated by a DNA vaccine encoding HSV-2 ICP27 and the E. coli heat labile enterotoxin. Vaccine 24:5016–5026 [DOI] [PubMed] [Google Scholar]
- 11. Hoshino Y., et al. 2007. Rates of reactivation of latent herpes simplex virus from mouse trigeminal ganglia ex vivo correlate directly with viral load and inversely with number of infiltrating CD8+ T cells. J. Virol. 81:8157–8164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson A. J., et al. 2010. Herpes simplex virus (HSV)-specific T cells activated in the absence of IFN-gamma express alternative effector functions but are not protective against genital HSV-2 infection. J. Reprod. Immunol. 84:8–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kern E. R., et al. 1978. Treatment of experimental herpesvirus infections with phosphonoformate and some comparisons with phosphonoacetate. Antimicrob. Agents Chemother. 14:817–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kino Y., et al. 1986. Immunogenicity of purified glycoprotein gB of herpes simplex virus. Arch. Virol. 89:69–80 [DOI] [PubMed] [Google Scholar]
- 15. Koelle D. M., et al. 1994. Antigenic specificities of human CD4+ T-cell clones recovered from recurrent genital herpes simplex virus type 2 lesions. J. Virol. 68:2803–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koelle D. M., Wald A. 2000. Herpes simplex virus: the importance of asymptomatic shedding. J. Antimicrob. Chemother. 45(Suppl. T3):1–8 [DOI] [PubMed] [Google Scholar]
- 17. Lahiri A., Das P., Chakravortty D. 2008. Engagement of TLR signaling as adjuvant: towards smarter vaccine and beyond. Vaccine 26:6777–6783 [DOI] [PubMed] [Google Scholar]
- 18. Liu T., et al. 2000. CD8(+) T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J. Exp. Med. 191:1459–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Looker K. J., Garnett G. P., Schmid G. P. 2008. An estimate of the global prevalence and incidence of herpes simplex virus type 2 infection. Bull. World Health Organ. 86:805–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Margolis T. P., et al. 2007. Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J. Virol. 81:1872–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morrison L. A., Knipe D. M. 1997. Contributions of antibody and T cell subsets to protection elicited by immunization with a replication-defective mutant of herpes simplex virus type 1. Virology 239:315–326 [DOI] [PubMed] [Google Scholar]
- 22. Morrison L. A., Zhu L., Thebeau L. G. 2001. Vaccine-induced serum immunoglobin contributes to protection from herpes simplex virus type 2 genital infection in the presence of immune T cells. J. Virol. 75:1195–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pass R. F., et al. 2009. Vaccine prevention of maternal cytomegalovirus infection. N. Engl. J. Med. 360:1191–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Posavad C. M., et al. 1997. Severe genital herpes infections in HIV-infected individuals with impaired herpes simplex virus-specific CD8+ cytotoxic T lymphocyte responses. Proc. Natl. Acad. Sci. U. S. A. 94:10289–10294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Posavad C. M., et al. 2010. Detailed characterization of T cell responses to herpes simplex virus-2 in immune seronegative persons. J. Immunol. 184:3250–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stanberry L. R. 1998. Herpes. Vaccines for HSV. Dermatol. Clin. 16:811–816 [DOI] [PubMed] [Google Scholar]
- 27. Stanberry L. R., Kern E. R., Richards J. T., Abbott T. M., Overall J. C., Jr 1982. Genital herpes in guinea pigs: pathogenesis of the primary infection and description of recurrent disease. J. Infect. Dis. 146:397–404 [DOI] [PubMed] [Google Scholar]
- 28. Stanberry L. R., Rosenthal S. L. 2005. Progress in vaccines for sexually transmitted diseases. Infect. Dis. Clin. N. Am. 19:477–490 [DOI] [PubMed] [Google Scholar]
- 29. Street N. E., Mosmann T. R. 1991. Functional diversity of T lymphocytes due to secretion of different cytokine patterns. FASEB J. 5:171–177 [DOI] [PubMed] [Google Scholar]
- 30. Thoelen S., De Clercq N., Tornieporth N. 2001. A prophylactic hepatitis B vaccine with a novel adjuvant system. Vaccine 19:2400–2403 [DOI] [PubMed] [Google Scholar]
- 31. Wallace M. E., Keating R., Heath W. R., Carbone F. R. 1999. The cytotoxic T-cell response to herpes simplex virus type 1 infection of C57BL/6 mice is almost entirely directed against a single immunodominant determinant. J. Virol. 73:7619–7626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weeratna R. D., et al. 2000. CpG DNA induces stronger immune responses with less toxicity than other adjuvants. Vaccine. 18:1755–1762 [DOI] [PubMed] [Google Scholar]
- 33. Zhu J., et al. 2007. Virus-specific CD8+ T cells accumulate near sensory nerve endings in genital skin during subclinical HSV-2 reactivation. J. Exp. Med. 204:595–603 [DOI] [PMC free article] [PubMed] [Google Scholar]