

Abstract
Cryptococcus neoformans is an opportunistic fungal pathogen that causes meningoencephalitis. Its cell wall is composed of glucans, proteins, chitin, and chitosan. Multiple genetic approaches have defined a chitosan-deficient syndrome that includes slow growth and decreased cell integrity. Here we demonstrate chitosan is necessary for virulence and persistence in the mammalian host.
TEXT
Cryptococcus neoformans is an opportunistic fungal pathogen that causes cryptococcosis in immunocompromised individuals. The incidence of cryptococcosis is rising in direct proportion to the spread of human immunodeficiency virus (for review see, Heitman et al. [10]). It is estimated that worldwide, over 1 million people develop life-threatening cryptococcal meningitis and that an estimated 700,000 die every year from cryptococcosis (18). Current antifungal therapies for treatment of cryptococcosis are inadequate: amphotericin B is effective, but toxic; fluconazole is fungistatic, having high relapse rates; flucytosine (5-FC) incurs frequent resistance (19–21, 25). Components of the fungal cell wall are attractive antifungal drug targets because they are absent in the host. The echinocandins, for example, target β-(1,3)-glucan synthase, an enzyme essential for the synthesis of a major component in the fungal cell wall (13, 24). Echinocandins are well-tolerated by patients but are not effective against C. neoformans (14).
The cell wall of C. neoformans is an essential organelle that provides cellular structure and integrity. An indispensable component of this cell wall that contributes to its strength and integrity is chitin (1). Chitin is a linear polymer of β-(1,4)-linked N-acetylglucosamine (GlcNAc) synthesized by the chitin synthase/regulator (Chs/Csr) system. Chitin can then be enzymatically deacetylated to chitosan by chitin deacetylases (EC 3.5.1.41). The cell wall of C. neoformans contains polymers of both chitin and chitosan, and the quantity and ratio can be dynamic depending on the growth phase (4, 5). In C. neoformans, only the isoforms Chs3 and Csr2 produce the majority of vegetative cell wall chitin, which is converted to chitosan (despite there being eight chitin synthase genes [CHS] and three synthase regulator genes [CSR]) (5). C. neoformans also expresses three chitin deacetylases, Cda1, Cda2, and Cda3, that can each produce chitosan. Strains of C. neoformans deleted for CHS3, CSR2, or all three chitin deacetylases are deficient in chitosan production and have a common set of phenotypes, including sensitivity to a variety of cell wall inhibitors. This indicates that chitosan is essential for the proper maintenance of cell wall integrity in C. neoformans, and Chs3, Csr2, and the chitin deacetylases are the main proteins that contribute to its formation (4, 5). Chitosan deficiency also affects two virulence factors of C. neoformans, melanin and capsule. The chs3Δ, csr2Δ, and cda1Δ cda2Δ cda3Δ strains “leak” melanin, and under certain conditions their capsule is enlarged and less uniform (4, 5). These three strains, chs3Δ, csr2Δ, and cda1Δ cda2Δ cda3Δ, represent three distinct deletions of proteins with different activities, which independently lead to chitosan deficiency.
We hypothesized that chitosan would be an important structural component for C. neoformans growth in the mammalian host, based on the impaired cell wall observed in chitosan-deficient mutants in vitro. In this report we show that chitosan is produced by Cryptococcus during murine infection and that strains deficient in chitosan due to mutations in three distinct genetic processes are avirulent and are rapidly cleared by the mammalian host. For each of the chitosan-deficient lines, at least two independently derived isolates were obtained and tested. The data were similar for the two independently derived isolates in all three lines; therefore, data shown here for each chitosan-deficient strain were derived from one of the isolates.
C. neoformans produces chitosan during in vivo growth.
In vitro analyses indicated that the cell wall integrity of strains that produce little to no chitosan is compromised, but the strains are still viable (4, 5). This suggests that chitosan is not essential for in vitro growth; however, we postulated that the loss of cell wall chitosan could impact the ability of C. neoformans to survive within the host. To begin addressing this question, we considered that if chitosan is important for growth in vivo, then the celluar content of it would be similar to amounts measured for cultures grown in vitro. Therefore, we first established whether chitosan was produced by C. neoformans during growth in the mammalian host. Mice were inoculated intranasally with 106 wild-type (KN99) cells, and lungs from each mouse were harvested 16 days postinfection (p.i.), homogenized in phosphate-buffered saline, and then extracted in 1 M KOH at 80°C for 1.5 h. Deacetylation of chitin under the conditions used for alkaline extraction was not detectable in control experiments (data not shown). Pellets containing the cryptococci were collected by centrifugation from each lung extract along with small amounts of lung material. The pellets were suspended in 1 ml 0.1 M KOH. A 10-μl aliquot was removed, stained with Solophenyl flavine (11) to facilitate counting using a hemacytometer when viewed by fluorescence microscopy (fluoroscein isothiocyanate filters). Each sample, containing approximately 2 × 108 cryptococci, was divided in two, and the chitosan in one half was converted to chitin by acetylation with acetic anhydride and the other half remained untreated. The chitin in both halves was measured, and the chitosan amount was calculated as the difference between the two measurements as previously described (4). The chitosan and chitin data, normalized to the number of cryptococcus cells from each sample, were found to be comparable to KN99 cultured in yeast extract-peptone-dextrose (YPD) at 30°C for 1 day and 3 days (Fig. 1). Chitin and chitosan were not detected from uninfected lungs (data not shown). After 16 days in the host lung, C. neoformans cell number increased by 200-fold (about 8 doublings). Chitin and chitosan levels are dynamic during growth in YPD (Fig. 1), so direct comparisons to any specific in vitro condition are difficult (5). These data demonstrated that chitosan was produced by C. neoformans during in vivo growth and implied that it may be important during infection of the mammalian host.
Fig. 1.

Chitosan and chitin levels from KN99 recovered from mouse lungs 16 days p.i. Chitosan and chitin levels from mouse lungs were compared to levels measured for KN99 grown in YPD at 30°C for 1 day and 3 days from a starting optical density at 600 nm of 0.10. Error bars indicate standard deviations for averaged measurements from four mice, and five 1-day YPD and four 3-day YPD cultures.
Strains with reduced chitosan grow slowly at 37°C.
The ability to grow at host temperature is a feature indicative of a successful mammalian pathogen. Previous reports on the three chitosan-deficient strains, chs3Δ, csr2Δ, and cda1Δ cda2Δ cda3Δ, indicated that chs3Δ and csr2Δ are temperature sensitive when grown in vitro and that cda1Δ cda2Δ cda3Δ is not (4, 5). However, these experiments were performed on solid media. The inability of the chs3Δ and csr2Δ strains to grow on a plate may have indicated that the cells were dead or that they had entered a state of stasis, in which they were still viable but unable to divide. To distinguish between these two possibilities, we investigated the growth rate of the chitosan-deficient strains in liquid yeast nitrogen base with 2% glucose (YNB) medium, pH 4, at either 30°C or 37°C. Strains deficient in cellular chitosan, chs3Δ, csr2Δ, and cda1Δ cda2Δ cda3Δ, grew more slowly than the wild type at both temperatures and had a greater growth defect at 37°C. They experienced a long lag phase compared to the wild type, anywhere between 12 h and 24 h at both temperatures, before undergoing a doubling event over the next 24-hour period (Fig. 2). These data demonstrate that although the chitosan-deficient strains grow more slowly, they are viable at 37°C and will grow at the mammalian host temperature.
Fig. 2.

Growth curves of strains in liquid YNB at 30°C (A) or 37°C (B). The inset of panel B shows csr2Δ, chs3Δ, cda1Δ cda2Δ cda3Δ, and trx1Δ strains on an expanded y axis. Strains are listed on the right side of panel A. The x axis is in hours. Data consist of three bioreplicates, each with two technical replicates, with standard errors shown.
Lack of cellular chitosan attenuates virulence.
A popular hypothesis is that strains that grow slowly at host temperature will be avirulent. However, Missall et al. observed that a strain deleted for a thioredoxin, TRX1, that grows slowly at 37°C was still virulent (15). We compared the growth rate of the trx1Δ mutant at both 30°C and 37°C in YNB and found that it was very similar to what we observed with the three chitosan-deficient strains (Fig. 2). These data suggest that slow growth at 37°C does not necessarily predict avirulence and that testing the chitosan-deficient strains for virulence might provide valuable insights.
Our initial hypothesis was that chitosan would be important for virulence, based on the in vitro cell wall sensitivities of the deficient strains and on the observation that wild-type C. neoformans produces chitosan during infection. To determine if loss of cellular chitosan affects virulence, we used an inhalation mouse model as previously described (8, 15) with the following exceptions; mice (CBA/J; Jackson Laboratories) were inoculated with 1 × 105 cells per strain tested and sacrificed when they reached 75% of their original body weight. All mice inoculated with the wild-type strain became ill and were sacrificed by 19 days p.i. None of the mice infected with the chitosan-deficient strains showed loss of weight or displayed symptoms of illness throughout the 60-day experiment, supporting the hypothesis that chitosan was necessary for virulence (Fig. 3). A second, independently generated mutant line for each of the deletion strains was also tested in mice, with identical results (data not shown). Taken together, these data indicated that chitosan is necessary for the virulence of C. neoformans.
Fig. 3.
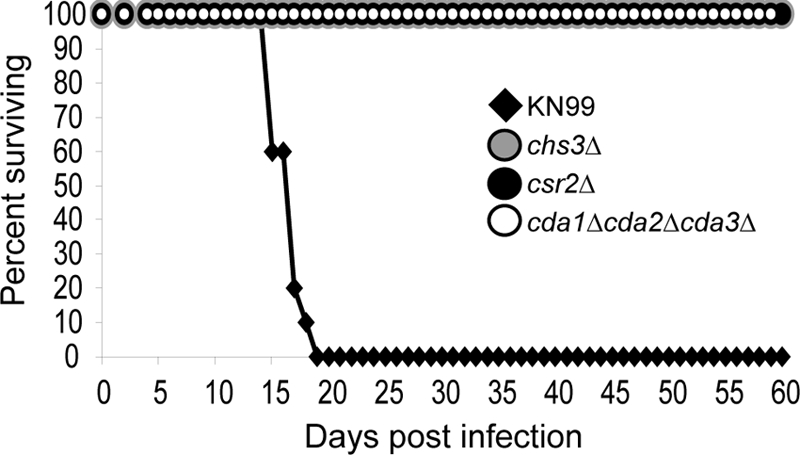
Virulence of chitosan-deficient strains. Strains are listed. Data represent results for two independent isolates per deletion strain.
Chitosan-deficient strains are quickly eradicated from the lungs.
We sought to determine if the Cryptococcus strains were avirulent, because they were more susceptible to killing and clearance by the host. To test this, four mice were infected with two independently generated strains of each mutation as described above, and lung tissue harvested at 2 h, 8 h, 24 h, 48 h, 7 days, and 15 days p.i. For the determination of CFU, one lung from each mouse was placed in 2.5 ml of 1× phosphate-buffered solution (pH 7), homogenized, serially diluted, and plated onto YPD supplemented with 100 μg/ml streptomycin and ampicillin. The second lung of each mouse was preserved for histology preparation in 5 ml of 10% buffered formalin (see below). We found that the fungal burden of the trx1Δ strain, which grows slowly at 37°C, was similar to its wild-type (H99) progenitor. Only at very early time points (2 to 24 h) were live cells recovered from mice infected with chitosan-deficient strains, and no CFU were recovered from tissue infected with these strains past 24 h p.i. (Fig. 4A). These data demonstrated that chitosan is necessary for the viability of Cryptococcus within the host and suggest that it is important for the establishment of a successful infection.
Fig. 4.

Fungal burden over time for selected strains. Data are from four mice per strain. Strains are listed to the right of each graph. nd, not determined; a dash indicates no live cells were recovered. (A) *, significant difference between deletion strains and wild type at 2 h (chs3Δ, P = 0.009; csr2Δ, P = 0.010; cda1Δ cda2Δ cda3Δ, P = 0.003); P < 0.001 for the 8-h and 24-h time points based on one-way analysis of variance with Dunnett's post hoc test. Data represent one of two independent isolates for each chitosan-deficient strain after the 24-h time point. Standard errors are shown. (B) Fungal burden of the complemented strain. Wild-type and cda2Δ cda3Δ controls were included.
Chitosan-deficient strains are cleared faster than other avirulent strains.
Two well-established virulence factors of C. neoformans are its abilities to produce a polysaccharide capsule and the pigment melanin (7, 22). Both of these virulence factors are associated with the cell wall. The deletions cap59Δ (an acapsular strain) and lac1Δ (a strain lacking melanin production) each lead to the complete loss of virulence of C. neoformans (7, 22). Given that the chitosan-deficient strains are also avirulent, we sought to determine if their clearance patterns were similar to the acapsular and melanin-deficient strains. The fungal burdens of mice infected with the chitosan-deficient strains were much lower than those of acapsular or melanin-deficient strains, as only cap59Δ and lac1Δ cells were recovered from infected tissue 48 h to 15 days p.i. (Fig. 4A). These data indicate that the loss of chitosan renders Cryptococcus more susceptible to killing by the host than does the loss of capsule or melanin.
Complementation of a chitosan-deficient strain restores fungal burden to wild-type levels.
To date, we have been unable to complement the chs3Δ and csr2Δ strains. Several attempts were made to biolistically reintroduce a wild-type copy of CHS3 or CSR2 into their respective deletion strains. In addition to our normal transformation procedures, we tried two modifications, including reduction in the pressure at which the DNA-covered gold particles were delivered and inclusion of sorbitol in the transformation recovery plates. Furthermore, we tried to introduce the complementation DNA by using an electroporation method both with and without sorbitol in the recovery plates (9). No colonies were ever recovered from any of these transformation attempts, while control strains generally produced hundreds of colonies. This indicated that the cell walls of the chs3Δ and csr2Δ strains were compromised to a point that they could not survive any of the transformation procedures.
Interestingly, the cda1Δ cda2Δ cda3Δ strain was transformable, and we were able to complement it by integrating a wild-type copy of CDA1 into the cda1 locus. The cda1Δ cda2Δ cda3Δ makes more chitin than either the chs3Δ or csr2Δ strain (4), and this additional chitin may provide enough structural integrity to the cell wall to permit transformation (4).
A cda2Δ cda3Δ strain, which still contains a wild-type copy of CDA1 and makes near-wild-type levels of chitin and chitosan (4), had a clearance pattern similar to the wild type and was used as a positive control for the cda1Δ::CDA1 cda2Δ cda3Δ strain. The fungal burden of cda1Δ::CDA1 cda2Δ cda3Δ was similar to both cda2Δ cda3Δ and wild-type strains (Fig. 4B). These data suggest that the complete lack of chitosan is necessary for the rapid killing of C. neoformans in the mammalian host.
The host readily cleared nonviable cells of chitosan-deficient strains from lung tissue.
Because the fungal burden assay provides only live cell counts, we determined if the nonviable cells were also being readily cleared from the host. Lung tissue was harvested for histology as described above. Histology preparations of stepwise, longitudinal lung sections stained with Grocott's methenamine silver (GMS) were made by the Department of Pathology core facility at Washington University School of Medicine (St. Louis, MO). We observed cells in all samples up to 48 h p.i. (Fig. 5). The chitosan-deficient strains showed decreased cell numbers after 8 h, suggesting that clearance of the nonviable cells was occurring.
Fig. 5.

Average cryptoccocal cell numbers in longitudinal lung sections. Cryptoccocal cells in three stepwise lung sections from each mouse were stained with Grocott's methenamine silver (GMS). All cells in lung sections were counted by viewing with a light microscope. The number of cells for time points 8 to 48 h of each strain were calculated as a percentage of cells counted for each respective 2-h time point. Error bars indicate standard deviations between the four mice (12 lung sections) in each group.
Chitosan is likely critical for virulence of C. neoformans.
Our results are consistent with and suggestive of the notion that chitosan-deficient cell walls make fungal cells more vulnerable to host defense mechanisms, and they indirectly suggest a role for this cell wall component in virulence. The result that chitosan-deficient mutants with genetic defects in three distinct proteins were all avirulent and rapidly killed by the host lends strength to the argument that chitosan deficiency was directly responsible. It is possible that destabilization of the cell wall due to lack of chitosan renders C. neoformans highly susceptible to host killing and clearance (23). Alternatively, a lack of chitosan could expose other pathogen-associated molecular patterns (PAMPs) and increase the host response to the mutant Cryptococcus. Finally, host chitinases are induced by C. neoformans and other fungal pathogens, and converting chitin to chitosan may be a mechanism to protect the fungus from detrimental effects of chitinases.
Proteins that produce chitosan may be good antifungal drug targets.
This is the first report to demonstrate the importance of cell wall chitosan for virulence of a human fungal pathogen. Other opportunistic fungal pathogens have been reported to produce chitosan. Aspergillus fumigatus produces chitosan in its mycelia and conidia (16), while Mucor rouxii generates significant amounts of chitosan in the cell wall of structures found throughout its life cycle (3, 6, 12, 17, 26). Additionally, a chitin deacetylase gene was shown to be highly transcribed in the yeast phase of Paracoccidioides brasiliensis (2). It has yet to be determined if loss of chitosan will have the same effect on other pathogenic fungi as it does on C. neoformans. Many fungal pathogens produce chitosan in some phase of their life cycles, but here chitosan has been shown to be essential for cryptococcal virulence. We postulate that the proteins that generate chitosan could prove to be excellent antifungal drug targets.
Acknowledgments
We thank Kim Gerik and Marshall Michener for critical reading of the manuscript, Chrono Lee for help with measuring chitin and chitosan from mice, and Woei Lam, Kim Gerik, Rajendra Upadhya, Nicole Gilbert, Marshall Michener, Maureen Donlin, and Jacob Gyore for assistance with the animal experiments.
This work was supported by NIH NIAID grants R01AI072195 and R01AI050184 to J.K.L.
Footnotes
Published ahead of print on 22 July 2011.
REFERENCES
- 1. Adams D. J. 2004. Fungal cell wall chitinases and glucanases. Microbiol. 150:2029–2035 [DOI] [PubMed] [Google Scholar]
- 2. Andrade R., et al. 2006. Cell organisation, sulphur metabolism and ion transport-related genes are differentially expressed in Paracoccidioides brasiliensis mycelium and yeast cells. BMC Genomics 7:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arcidiacono S., Kaplan D. L. 1992. Molecular weight distribution of chitosan isolated from Mucor rouxii under different culture and processing conditions. Biotech. Bioeng. 39:281–286 [DOI] [PubMed] [Google Scholar]
- 4. Baker L. G., Specht C. A., Donlin M. J., Lodge J. K. 2007. Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryot. Cell 6:855–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Banks I. R., et al. 2005. A chitin synthase and its regulator protein are critical for chitosan production and growth of the fungal pathogen Cryptococcus neoformans. Eukaryot. Cell 4:1902–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bartnicki-Garcia S. 1968. Cell wall chemistry, morphogenesis, and taxonomy of fungi. Annu. Rev. Microbiol. 22:87–108 [DOI] [PubMed] [Google Scholar]
- 7. Chang Y. C., Kwon-Chung K. J. 1994. Complementation of a capsule-deficient mutation of Cryptococcus neoformans restores its virulence. Mol. Cell. Biol. 14:4912–4919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cox G. M., Mukherjee J., Cole G. T., Casadevall A., Perfect J. R. 2000. Urease as a virulence factor in experimental cryptococcosis. Infect. Immun. 68:443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gerik K. J., Bhimireddy S. R., Ryerse J. S., Specht C. A., Lodge J. K. 2008. Protein kinase C1 (PKC1) is essential for protection against both oxidative and nitrosative stress, cell integrity, and normal manifestation of virulence factors in the pathogenic fungus Cryptococcus neoformans. Eukaryot. Cell 7:1685–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heitman J., Kozel T., Kwon-Chung J., Perfect J., Casadevall A. 2011. Cryptococcus: from human pathogen to model yeast, p. 620 ASM Press, Washington D. C [Google Scholar]
- 11. Hoch H. C., Galvani C. D., Szarowski D. H., Turner J. N. 2005. Two new fluorescent dyes applicable for visualization of fungal cell walls. Mycologia 97:580–588 [DOI] [PubMed] [Google Scholar]
- 12. Kafetzopoulos D., Martinou A., Bouriotis V. 1993. Bioconversion of chitin to chitosan: purification and characterization of chitin deacetylase from Mucor rouxii. Proc. Natl. Acad. Sci. U. S. A. 90:2564–2568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kauffman C. A. 2006. Clinical efficacy of new antifungal agents. Curr. Opin. Microbiol. 9:483–488 [DOI] [PubMed] [Google Scholar]
- 14. Krishnarao T., Galgiani J. 1997. Comparison of the in vitro activities of the echinocandin LY303366, the pneumocandin MK-0991, and fluconazole against Candida species and Cryptococcus neoformans. Antimicrob. Agents Chemother. 41:1957–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Missall T. A., Lodge J. K. 2005. Function of the thioredoxin proteins in Cryptococcus neoformans during stress or virulence and regulation by putative transcriptional modulators. Mol. Microbiol. 57:847–858 [DOI] [PubMed] [Google Scholar]
- 16. Mouyna I., Fontaine T. 2009. Cell wall of Aspergillus fumigatus: a dynamic structure, p. 169–183 In Latge J. P., Steinbach W. J. (ed.), Aspergillus fumigatus and aspergillosis. ASM Press, Washington, DC [Google Scholar]
- 17. Orlowski M. 1991. Mucor dimorphism. Microbiol. Rev. 55:234–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park B. J., et al. 2009. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23:525–530 [DOI] [PubMed] [Google Scholar]
- 19. Paugam A., et al. 1994. Increased fluconazole resistance of Cryptococcus neoformans isolated from a patient with AIDS and recurrent meningitis. Clin. Infect. Dis. 19:975–976 [DOI] [PubMed] [Google Scholar]
- 20. Powderly W. G., et al. 1995. A randomized trial comparing fluconazole with clotrimazole troches for the prevention of fungal infections in patients with advanced human immunodeficiency virus infection. N. Engl. J. Med. 332:700–705 [DOI] [PubMed] [Google Scholar]
- 21. Saag M. S., et al. 2000. Practice guidelines for the management of cryptococcal disease. Infectious Diseases Society of America. Clin. Infect. Dis. 30:710–718 [DOI] [PubMed] [Google Scholar]
- 22. Salas S., Bennett J., Kwon-Chung K., Perfect J., Williamson P. 1996. Effect of the laccase gene CNLAC1 on virulence of Cryptococcus neoformans. J. Exp. Med. 184:377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vicencio A. G., et al. 2008. Pulmonary cryptococcosis induces chitinase in the rat. Respir. Res. 9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Walsh T. J. 2002. Echinocandins: an advance in the primary treatment of invasive candidiasis. N. Engl. J. Med. 347:2070–2072 [DOI] [PubMed] [Google Scholar]
- 25. White M. H., Armstrong D. 1994. Cryptococcosis. Infect. Dis. Clin. North Am. 8:383–398 [PubMed] [Google Scholar]
- 26. White S. A., Farina P. R., Fulton I. 1979. Production and isolation of chitosan from Mucor rouxii. Appl. Environ. Microbiol. 38:323–328 [DOI] [PMC free article] [PubMed] [Google Scholar]