

Abstract
2-Oxoglutarate is located at the junction between central carbon and nitrogen metabolism, serving as an intermediate for both. In nitrogen metabolism, 2-oxoglutarate acts as both a carbon skeletal carrier and an effector molecule. There have been only sporadic reports of its internal concentrations. Here we describe a sensitive and accurate method for determination of the 2-oxoglutarate pool concentration in Escherichia coli. The detection was based on fluorescence derivatization followed by reversed-phase high-pressure liquid chromatography separation. Two alternative cell sampling strategies, both of which were based on a fast filtration protocol, were sequentially developed to overcome both its fast metabolism and contamination from 2-oxoglutarate that leaks into the medium. We observed rapid changes in the 2-oxoglutarate pool concentration upon sudden depletion of nutrients: decreasing upon carbon depletion and increasing upon nitrogen depletion. The latter was studied in mutants lacking either of the two enzymes using 2-oxoglutarate as the carbon substrate for glutamate biosynthesis. The results suggest that flux restriction on either reaction greatly influences the internal 2-oxoglutarate level. Additional study indicates that KgtP, a 2-oxoglutarate proton symporter, functions to recover the leakage loss of 2-oxoglutarate. This recovery mechanism benefits the measurement of cellular 2-oxoglutarate level in practice by limiting contamination from 2-oxoglutarate leakage.
INTRODUCTION
In enteric bacteria, 2-oxoglutarate (2OG) conjoins the two most important central metabolism pathways, being at the junction between the tricarboxylic acid (TCA) cycle and central nitrogen metabolism. Unlike central carbon metabolism, central nitrogen metabolism is rather compact and consists of only three enzymes (42), glutamine synthetase (GS; encoded by glnA), glutamate synthase (GOGAT; encoded by gltBD), and glutamate dehydrogenase (GDH; encoded by gdhA), and three metabolites, glutamine (Gln), glutamate (Glu), and 2OG (see Fig. S1 in the supplemental material). NH4+, the preferred nitrogen source for cell growth, is assimilated through GS and GDH. 2OG from the TCA cycle serves as the sole carbon skeletal substrate, generating Glu through GOGAT and/or GDH and subsequently Gln through GS. Most of the total carbon that 2OG brings into nitrogen metabolism is recycled from the two central nitrogen intermediates: 2OG is regenerated after many transamination reactions from Glu, and Glu is regenerated after various amido transfer reactions from Gln. Almost all other cellular nitrogen is gained from these two types of nitrogen transfer reactions. Hence, 2OG has a dual identity: a central carbon intermediate and a nitrogen carrier.
2OG also functions as a regulatory effector. The most notable receptors of 2OG are PII family proteins (31, 38). There are two PII proteins in Escherichia coli, GlnB and its paralog, GlnK (6, 53). GlnB is one key player in nitrogen regulation, participating in two branched regulatory cascades that control the activity and expression of GS. In vitro studies have shown that the function of GlnB is regulated directly by 2OG and ATP/ADP and indirectly by Gln (22–24, 50). Mutational and structural studies have further clarified the interaction between GlnB and 2OG (7, 25, 57). The major function of GlnK is to interact with and regulate the channel protein AmtB for NH4+ uptake (8, 9, 17, 18, 53). This interaction is also regulated by 2OG (13). Numerous reports have documented the regulatory effects of 2OG on PII proteins targeting a variety of enzymes and transcriptional factors in other bacteria and archaea (31). Beyond PII proteins, 2OG also acts directly on other diverse proteins (14, 32, 51, 54). Remarkably, they are all nitrogen related, implying that 2OG is a sensory metabolite that relates to cellular nitrogen status.
This 2OG nitrogen-sensory view has been supported by several reports with measurements of 2OG pools in organisms other than enteric bacteria (12, 30, 37). On the other hand, Kustu and colleagues (21) have demonstrated that enteric bacteria perceive external nitrogen deficiency to be a limitation in the Gln pool (44). A similar relationship between the internal Gln concentration and external nitrogen deficiency is also found in fungi, algae, and higher plants (4, 16, 40). As 2OG possesses an identity of a central carbon intermediate whereas Gln is a central nitrogen intermediate, there has been a notion that 2OG and Gln serve as the carbon and nitrogen indicators, respectively, in enteric bacteria to regulate GS activity and expression (38). In vivo, there have been several reports of internal 2OG concentrations in E. coli concerning aspects other than its role in nitrogen regulation (3, 5, 34, 41, 48). However, the 2OG pool concentrations measured with different methods vary by more than 10-fold: from 0.2 up to 2.3 mM in cells grown in NH4+-glucose medium under similar batch culture conditions. A recent metabolomics study, employing a unique but unconventional filter-culturing methodology, has shown that the internal 2OG concentration decreases more than 10-fold in response to an NH4+ upshift (58).
To clarify the physiological role of 2OG in nitrogen regulation of enteric bacteria, a systematic in vivo study like that used to demonstrate the role of Gln (21) is desired. This demands a reliable and versatile method for determining the internal 2OG concentrations under a variety of growth conditions, which is the main focus of this study. We first adopted a detection method with high sensitivity and utilized this method in developing a sampling strategy. However, for accurate measurement of the pool concentration of a metabolite, sampling is usually more critical than detection. Thus, fast filtration was chosen as the method of choice for cell harvest. During the development of the sampling strategy, we noticed that cells leak 2OG into the medium. This first led to a subtraction strategy. We then observed that the 2OG pool is subject to fast depletion and accumulation in response to nutrient availability and cold treatment also triggers loss of 2OG. A wash-based sampling strategy was then established as an alternative. Using these strategies, we investigated the cellular leakage of 2OG and found that some strains recycle it through the function of KgtP, a known 2OG transporter (46, 47). Applications of the two alternative, filtration-based sampling strategies under different situations, as well as the physiological role of 2OG as a sensory molecule, will be discussed.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
NCM3722, an E. coli K-12 prototrophic strain (49), was used as the wild-type control. Mutants were isogenic derivatives of NCM3722: GDH-negative (GDH−) mutant FG1113 (ΔgdhA859::FRT), GOGAT-negative (GOGAT−) mutant FG1195 (ΔgltD841::FRT), and KgtP-negative (KgtP−) mutant FG1602 (ΔkgtP754::kan). All deletion alleles originated from the Keio deletion collection (2). SK2633 was a prototrophic Salmonella enterica serovar Typhimurium LT2 strain (10). The base medium N−C− was nitrogen and carbon free (10). The carbon source was 0.4% glycerol or glucose (wt/vol), and the nitrogen source was NH4Cl, provided at 20 mM unless otherwise specified. All batch cultures were grown aerobically at 37°C. A baffled flask was used for culturing and was shaken at 250 rpm, with the culture volume being no more than 15% of the flask volume.
Sample collection for measurement of internal, external, and total 2OG concentrations.
Cell filtration was carried out in a 37°C room, using a 10-place filtration manifold (Hoefer) and nylon membranes (25-mm HNWP disc with a 0.45-μm pore size; Millipore). The membrane was prewetted with wash medium. Dry membrane was used when cells were not washed during sampling. For cell harvest, 1 to 4 ml of culture containing no more than 1 ml × optical density at 600 nm (OD600) of cell mass was quickly filtrated under vacuum. Two milliliters of wash medium that equilibrated in the 37°C room was pipetted onto the membrane once or twice if wash was required. The membrane was immediately immersed in 1 ml of 0.8 M perchloric acid in an Eppendorf tube. After 30 s of vortexing, the tube was incubated at room temperature for ∼20 min, vortexed for another 30 s, and centrifuged at the maximum speed in a microcentrifuge for 4 min. After the filter and cell debris were discarded, the extract was frozen on dry ice and stored at −80°C.
For measurement of 2OG levels in medium, 1 ml of culture was passed through a 0.22-μm-pore-size syringe filter, and the filtrate was collected. One hundred microliters of filtrate was then mixed with an equal volume of 1.6 M perchloric acid. For total (internal plus external) 2OG amount, 100 μl of culture was mixed with an equal volume of 1.6 M perchloric acid and vigorously vortexed for 30 s. This direct acid extraction of cell culture served as a true no-harvest control in which the extract contains internal cellular plus external medium 2OG. Sample was frozen on dry ice and stored at −80°C.
2OG detection and quantification.
We modified a previously reported method developed for measuring 2-oxo acids in human serum (55). The process includes addition of external standard and reaction with a fluorescence reagent, followed by high-pressure liquid chromatography (HPLC) separation for quantification. Before the fluorescence derivatization, all frozen samples were thawed and centrifuged for 1 min. One volume of supernatant was mixed with 0.25 volume of external standard, 0.75 volume of HPLC-grade water, and 2 volumes of reagent mixture. The external standard was a mixture of 2-oxobutyric acid (2OB) and 2-oxovaleric acid (2OV), each at a concentration of 4 μM prepared in 0.8 M perchloric acid. The reagent mixture was freshly prepared by dissolving the fluorescence reagent 4,5-methylenedioxy-1,2-phenylenediamine (DMB; final concentration, 5 mM; Sigma) into a prefiltered solution (28 mM sodium dithionite, 1 M 2-mercaptoethanol, and 0.4 M hydrochloric acid). For 2OG quantification, at least one 2OG-2OB-2OV (1:1:1) standard was prepared for each batch of reactions. Reactions were carried out in a sealed tube at 95 to 100°C for 1 h. Derivatized sample was then filtered through an HPLC-grade microcentrifuge filter (0.2-μm-pore-size nylon filter; Costar) and stored at −80°C.
2-Oxo acid derivatives were separated by reversed-phase HPLC (55), using a Shimadzu Prominence HPLC system (the main modules were LC-20AB binary pump, SIL-10AF autosampler, and RF-10AxL fluorescence detector). Normally, 10 to 20 μl of sample was injected into an HPLC column. The column consisted of a C18 reversed-phase main column and its guard module (100 by 4.6 mm and 10 by 4.6 mm, respectively; Dynamax Microsorb AAA; Varian) and was maintained at 40°C during separation. Starting buffer A was 9 volumes of 20 mM K+-phosphate buffer (pH 7.0) mixed with 1 volume of buffer B, and elution buffer B was a mixture of acetonitrile and methanol (4:6, vol/vol). The flow rate was 1.0 ml/min. The elution was isocratic first with buffer A for 8 min, followed by a linear gradient to 55% buffer B during the next 12 min and isocratic at 55% buffer B for another 6 min before column reequilibration with buffer A. The fluorescence signal was monitored at an excitation wavelength of 370 nm and an emission wavelength of 445 nm.
The 2OB/2OV ratio of peak areas of the 2OG-2OB-2OV standard was used to check for any sign of abnormality of the 2OB/2OV ratio of samples. All the standards should be present in equal amounts with a variation of no more than 10% if the cellular 2OB or 2OV content was negligible (see Results). If so, the 2OG/2OV ratio was applied for normalization of 2OG amounts in samples. The 2OG pool concentration in mM internal concentration was converted from the unit of nanomoles per ml × OD600 cells as previously described (39).
Detection of kgtP expression.
The primer pair used for PCR was ACATATATGAGTGAAGTTGC and AAGGGAATACGCCATCCCCA. They were from the coding region of Salmonella serovar Typhimurium kgtP. Each primer contained one mismatch (underlined) with the corresponding sequence in E. coli kgtP. RNA extraction, reverse transcription, and PCR were carried out by using commercial reagents (RNeasy [Qiagen], random hexamers and ThermoScript reverse transcriptase [Invitrogen], GoTaq Green Master Mix [Promega]).
Other methods.
The NH4+ concentrations in medium were determined by the method of glutamate dehydrogenase from bovine liver (AA0100; Sigma). For comparison, internal Glu and Gln concentrations were measured as described previously (21, 39), employing both a standard no-harvest protocol and the sampling method detailed above for the 2OG pool.
RESULTS
2OG detection by fluorescence derivatization and HPLC separation.
Except for a couple of cases using mass spectrometry (3, 5), all reported measurements of the 2OG pool have been based on an assay employing glutamate dehydrogenase from bovine liver (34). The enzymatic method demands extraction of a sufficient amount of 2OG from cell culture with a sizable volume. This puts a limit on the speed of the sampling process, which could be a source of inaccuracy for determining 2OG pool concentrations. For a more sensitive detection, a method established for the determination of 2-oxo acid levels in human serum (55) was modified. Cellular extract was reacted with 4,5-methylenedioxy-1,2-phenylenediamine (DMB), resulting in a fluorescent derivative of 2OG. It was then separated by reversed-phase HPLC, and the 2OG amount was quantified. Several 2-oxo acids were tested as external standards for the normalization of 2OG amount. 2-Oxobutyric acid (2OB) and 2-oxovaleric acid (2OV) were chosen, as no other HPLC peak interfered with them. In all tested cell extracts, no detectable level of 2OB and 2OV was ever encountered.
HPLC peak separation and quantification were satisfactory, as examples of a chromatogram show (Fig. 1). The detection limit for 2OG, with a signal-to-noise ratio of 10, is ∼20 fmol in an injection volume of 10 μl. The noise is a tiny 2OG peak that is invisible in the blank at the scale shown in Fig. 1 and is possibly contamination from the reagents and/or water. It should be noted that this noise in the blank may increase up to 10-fold if HPLC-grade water for dilution of samples was left in a disposable plastic tube for more than a few days. The chemical basis of this phenomenon is unknown.
Fig. 1.
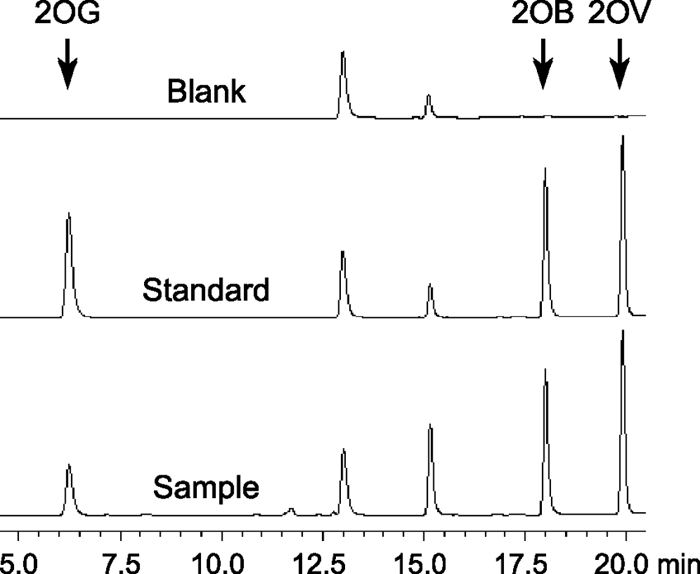
HPLC chromatograms of DMB derivatives of 2-oxo acids. Blank, HPLC-grade water (after derivatization reaction); standard, equal amounts of 2OG, 2OB, and 2OV; sample, cellular extract with the standard 2OB and 2OV. The two visible peaks in the blank overlap with those for some of other tested 2-oxo acids, possibly contamination from the reagents and/or water. Injection of water yields no fluorescence peak.
Cellular leakage of 2OG and a subtraction sampling strategy.
A central metabolite is normally subject to a high flux rate, so its pool concentration could be artificially changed upon cell manipulation even with a fast sampling process. A preferred practice to avoid any pool fluctuation before extraction is the so-called no-harvest or whole-broth method (5, 28); i.e., cell culture is directly mixed with methanol or acid without separation of cells from the medium. The Gln and Glu pool measurement that we routinely performed is based on such a sampling method (21, 39). However, this strategy is applicable only if no significant excretion occurs for the metabolite of interest. It has been reported that many central carbon metabolites, including 2OG, are present in growth media of E. coli and other bacteria (34, 48). We confirmed that E. coli cells indeed leak 2OG into the growth medium and subsequently quantified its concentration (Fig. 2 A). In an exponentially growing culture, we observed a linear accumulation of 2OG in the medium when the cell density was low (OD < 0.2). The external 2OG concentration then reached a plateau at ∼1 μM. This leakage could be characteristic of a weak acid such as 2-oxoglutaric acid (pKa=4.68), a passive phenomenon without involvement of a secretion system. The plateau at a high OD may be rationalized by a balance of this passive leakage with an active uptake mechanism (see below).
Fig. 2.

2OG leakage and steady-state pool concentration during exponential growth of E. coli. Wild-type strain NCM3722 was cultured in NH4Cl-glycerol medium. (A) 2OG concentrations in the medium were assayed from immediately after inoculation through the late exponential phase of growth. The growth curve is shown in the inset; the doubling time was 62 min. (B) Paired filter and medium samples were collected along the exponential phase of growth. Each pool value was the result of subtraction from paired samples.
The leakage made it unsuitable to directly apply the no-harvest strategy for quantification of the internal 2OG concentration. We initially reasoned that we might still obtain the internal 2OG concentration using a no-harvest approach. First, we would measure the total 2OG amount, which would include both the internal and external concentrations. We would then measure the external 2OG concentration. Finally, we would subtract this value from the total concentration to obtain the internal concentration. However, there is an error issue associated with the subtraction of two quantifications. Although various reported internal 2OG pool concentrations are in sub-mM to mM ranges, much higher than the external concentration, the relative internal and external amounts are close and even higher for the latter due to the tiny cell volume in comparison to the medium volume. A calculated estimate indicated that up to 90% of the total 2OG amount of a theoretical no-harvest sample is from external 2OG (see the no-harvest line in Fig. S2 in the supplemental material). Assuming a mere ±5% assay error, a subtraction may yield, in an extreme scenario, either a 2-fold overestimate or an underestimate with a value near zero: e.g., 100% ± 5% total value minus 90% ± 4.5% external value.
With consideration of the above, a modified subtraction strategy was tested. Rapid filtration with an immediate extraction of the filter was performed as a substitute for the no-harvest approach. The process was fast and lasted about 10 to 20 s. It was carried out in a 37°C room, and sufficient medium remained in contact with the cells on the filter. All of these minimized the disturbance of the cellular physiological state. We measured the medium volume retained by the filter by weight. The result was constant, ∼70 μl, with only a few percent variation, even with various amounts of cells. We limited the cell amount per filter at no more than 1 ml × OD600 since too many cells would slow the filtration speed. The process essentially served as a concentrating step that increased the ratio of the cell to culture volume. The filter extract gave a total 2OG amount from cells plus 70 μl medium. With a paired measurement of the 2OG medium concentration, subtraction led to a pool value. With the increased ratio of cell to medium volume, the medium 2OG fraction became smaller, so the potential error from the subtraction was minimized (see the filtration line in Fig. S2 in the supplemental material). Figure 2B shows the subtraction result of several data points from an exponentially growing culture. The internal 2OG concentration is constant at about an average of 0.6 mM and independent of cell density and external 2OG concentration, a characteristic of cells during steady-state growth.
Depletion and accumulation of 2OG pool upon nutrient shifts.
The subtraction strategy requires paired measurements. To simplify the method and cut the assay number in half, we explored a harvest strategy by fast filtration and wash. The wash step may simply eliminate the interference of external 2OG. Although the wash can be carried out in a few seconds, it introduces an uncertainty, as the manipulation may affect metabolic flux, thus altering the pool concentration. Because of the dual identity of 2OG as an intermediate, carbon and nitrogen contents in the wash solution are of the most concern. Some previous studies of 2OG pools involved a wash step with carbon- and nitrogen-free media or saline solution, but their effects were not tested (5, 48).
In order to minimize any potential temperature effect, cells were washed with warmed medium (equilibrated in the 37°C culture room). The carbon effect was tested first with a wash-down scheme (Fig. 3 A). With different glycerol concentrations in the wash medium, the pool value obtained was stable (with an average of 0.5 mM), until the glycerol concentration decreased below 0.1 mM. The glycerol-free wash resulted in an ∼50% drop in the internal 2OG concentration, even though there was only ∼15 s from the time that the wash medium touched the cells on the filter to the step of acid extraction. Cells take up glycerol by a facilitated diffusion mechanism and trap it first by the action of glycerol kinase with a Km of 10 μM (52). Although 2OG is located many steps down from the glycerol uptake and phosphorylation steps, the glycerol wash-down result indicates that the change of carbon influx can be felt almost immediately in the 2OG pool. This reaction could possibly be much shorter than the minimal 15-s handling time.
Fig. 3.

The 2OG pool is subject to a sudden decrease and increase upon nutrient shift. (A) Wild-type strain NCM3722 was inoculated in glycerol-NH4+ medium. Sampling was carried out during the exponential phase of growth. Wash medium contained different concentrations of glycerol from 43 mM (0.4% [wt/vol], as in the starting medium) down to zero. (B) The wild-type, GDH− (FG1113), and GOGAT− (FG1195) strains were inoculated in glycerol medium with 11.5 mM NH4+. Samples were collected during the exponential phase of growth, when the concentration of NH4+ left in the culture was 10 ± 0.2 mM. Wash medium contained different concentrations of NH4+ from 10 mM down to zero. (C) Data represented by open symbols are the same as those in panel B and plotted in a semilog scale. The closed symbols are from a washup experiment with the GOGAT− mutant. Cells were inoculated in glycerol medium with 3.5 mM NH4+. Samples were collected during the exponential phase of growth, when the concentration of NH4+ left in the culture was 2 ± 0.2 mM. The wash medium contained different concentrations of NH4+ from 2 mM up to 50 mM. All inoculations were started at an OD600 of ∼0.05, and samplings were performed at OD600s of between 0.35 and 0.40. For sample collection, 1 ml of cells was quickly filtered and washed twice with 2 ml medium. The time between the point when the wash medium contacted cells on the filter and the filter was immersed into acid for extraction was controlled to be 15 ± 2 s. The error bars in panels A and B are minimal and maximal values obtained in at least two independent experiments.
The nitrogen (NH4+) effect was tested next. A wash-down scheme was also applied in the experiment by using a starting material of wild-type cells in exponential growth when ∼10 mM NH4+ was left in the medium (WT in Fig. 3B and C). The wash medium contained NH4+ at a concentration ranging from a near perfectly matched 10 mM down to zero. The resulting 2OG pool concentration from the perfect wash was 0.5 mM. As the washing NH4+ concentration decreased, the pool concentration slowly drifted upwards, increasing by ∼40% (0.7 mM) with the 0.5 mM NH4+ wash and ∼5-fold (2.5 mM) when washed with the NH4+-free medium. The result is just the opposite of the carbon effect, demonstrating fast 2OG accumulation upon an NH4+ downshift.
The same wash-down experiments were performed with GDH− and GOGAT− mutants (Fig. 3B and C). E. coli can synthesize Glu through the action of either the GS/GOGAT cycle or GDH. Both 2OG and NH4+ are cosubstrates for the two pathways. The GDH− mutant (with only the GS/GOGAT pathway) showed almost the same pattern of 2OG pools as the wild type, except that the pool concentration was completely flat when using wash medium with >1 mM NH4+, so that it was slightly higher than that of the wild type. In contrast, the GOGAT− mutant (with only the GDH pathway) displayed a 3.5-fold increase in the pool concentration (from 1.1 to 3.9 mM) when the NH4+ concentration in the wash medium was changed from 10 mM down to 0.5 mM. Its pool concentration did not increase much further when cells were washed with media with lower NH4+ concentrations. There was less than a 25% difference between the washes with 0.5 and 0 mM NH4+. A reverse washup experiment was carried out using growing GOGAT− cells when ∼2 mM NH4+ remained in the culture (Fig. 3C). The measured 2OG pools traced well among washes with 2 to 10 mM NH4+ with those in the wash-down experiment and further declined with washes with higher NH4+ concentrations. The significance of these results will be discussed.
Temperature and time effects on the 2OG pool during filtration and wash.
Cold treatment of cells prior to metabolite extraction is a common practice to stop cellular metabolism, presumably freezing metabolite pools for accurate measurement. Therefore, we examined the temperature effect on the internal 2OG concentration. We utilized the observed NH4+ effect on the 2OG pool concentration of the wild type (Fig. 3B) and compared washes using either 37°C or 0°C medium (Fig. 4 A). The results from the 37°C wash were expected (compare the gray bars in Fig. 4A with those in Fig. 3B). The 0°C wash prevented the 2OG pool from increasing in response to low NH4+ concentrations. However, the treatment did not freeze the 2OG pool. The pool values of 0.2 to 0.3 mM from the cold treatment were significantly lower than the undisturbed internal concentration of 0.5 mM obtained from the perfect wash with 10 mM NH4+ (Fig. 3B). Thus, the cold treatment appears to be an error-prone strategy for determination of the 2OG pool concentration. The mechanism of 2OG decrease remains unknown.
Fig. 4.

Temperature and time effects on 2OG pool during filtration and wash. (A) Wild-type strain NCM3722 was inoculated in glycerol medium with 11.5 mM NH4+. Samples were collected during the exponential phase of growth, when the concentration of NH4+ left in the culture was 10 ± 0.2 mM. Wash medium equilibrated either in a 37°C culture room or in ice water contained different concentrations of NH4+. (B) Strain NCM3722 was inoculated in glycerol medium with 20 mM NH4+. Samples were collected during the exponential phase of growth, when the OD600 was between 0.3 and 0.4. Wash medium was identical to the starting medium, with 20 mM NH4+ and equilibrated at 37°C. N, normal handling speed of filtration and wash that lasted ∼15 s; N + 30", cells were held in a pipette tip for 30 s after the culture was withdrawn from the vessel and before filtration; N + 60", hold for 60 s. The error bars are minimal and maximal values for two different OD points.
Another concern was any potential change in internal 2OG concentration during the period between the withdrawal of cells from the culture vessel and the acid extraction, even though the sampling process was handled with the shortest possible duration of ∼15 s. One such potential change could be caused by the brief stop of culture aeration. We tested this possibility by holding cells in a pipette tip for up to 60 s before filtration, thus reversely increasing the handling duration. The result showed no change of the pool concentration with the prolonged sampling period (Fig. 4B), suggesting that the fast handling of 15 s should not cause a significant fluctuation of the internal 2OG concentration.
A simplified and conditional alternative for sample collection.
A harvest strategy by fast filtration and wash is desirable because of its simplicity. However, a matched perfect wash requires predetermination of the carbon and nitrogen concentrations at the time of sampling, which would be a cumbersome process. This is not necessary under many conditions when both carbon and nitrogen are in sufficient concentrations, as suggested by the results in Fig. 3. For example, the glycerol concentration is of lesser concern if it is in the mM range at the sampling time. For NH4+, 10 to 20 mM is sufficient for the wild type. Therefore, we did tests of cultures grown in medium with 20 mM NH4+. By using starting medium for washes, a stable and reproducible 2OG pool value was obtained for exponentially growing wild-type cells (Fig. 5). Even at a high cell density, when some NH4+ in the medium had been utilized, e.g., ∼16 mM remains at an OD600 of 0.8, as 1 mM yields an ∼0.2-OD600 cell mass, and washes with the starting 20 mM NH4+ medium did not cause a noticeable disturbance of the 2OG pool. The data average of 0.5 mM was similar to the subtraction result (Fig. 2B). For GDH− and GOGAT− mutants, the pool values obtained were also constant. Their averages of 0.9 and 0.8 mM, respectively, were larger than the average for the wild type. The internal 2OG concentration of wild-type cells using glucose as the carbon source was also determined. The data average of 0.4 mM is comparable to that in the earliest report on the internal 2OG concentration, which is 0.6 mM in an E. coli strain grown in another glucose-NH4+ medium (34), but not to some reported values obtained when carbon- and nitrogen-free washes were performed (5, 41). In addition, we measured the 2OG pool concentration of wild-type cells grown in a proline-glycerol medium (Fig. 5). Compared to NH4+, proline is a poor nitrogen source and the cells grow slowly (21, 39). The measured 2OG pool value also remained constant during the exponential phase of growth. The data average of 1.4 mM is ∼3-fold higher than that of the NH4+-glycerol-grown cells. Further testing of the filtration and wash strategy on measurements of Glu and Gln pools yielded results similar to those from the no-harvest method.
Fig. 5.

Steady-state pool concentration obtained by the filtration and wash strategy. Cells were grown in NH4+-glycerol, NH4+-glucose, or proline-glycerol medium. Samples were collected at 3 to 5 different cell densities for each culture during its exponential phase of growth. Wash medium was identical to the starting medium with 20 mM either NH4+ or proline. The results were from two sets of independent experiments. Average doubling times were 63 min for the wild type (WT), 68 min for the GDH− mutant, 65 min for the GOGAT− mutant, 48 min for the wild type grown in NH4+-glucose, and 99 min for the wild type grown in proline-glycerol.
KgtP function countering the 2OG leakage.
As shown in Fig. 2A, the 2OG level in the medium was first accumulating and then reached a plateau at ∼1 μM as cells proliferated. So the weak acid was far from equilibration across the membrane, as the plateau concentration was ∼500-fold less than the internal concentration of ∼0.5 mM. 2OG is chemically stable under the growth condition, as we tested its stability for up to 6 h using a cultured medium that is sterilely filtered and with or without addition of extra 2OG standard. Therefore, the data suggest an uptake mechanism to counter the leakage. Otherwise, the external 2OG concentration would have maintained a linear relation with cell density. The external 2OG trend shown in Fig. 2A can be quantitatively explained by a model that balances the leakage with an unregulated function of 2OG uptake (detailed in Fig. S3 in the supplemental material and its legend). E. coli KgtP, a constitutively expressed proton symporter known as a 2OG permease (46, 47), was then considered to be a candidate for this function. To test this, a kgtP deletion mutant was cultured, and both external and internal 2OG concentrations were measured during the exponential phase of growth (Fig. 6 A). Its 2OG leakage profile was distinctly different from that of the wild type, showing a near linear increase along the growth. However, a lack of KgtP did not change either the growth rate or the internal 2OG concentration, whose average of 0.6 mM in the mutant is comparable to that in the wild type shown in Fig. 2B.
Fig. 6.

Continuous and near linear accumulation of external 2OG without a functional KgtP. E. coli (Ec) kgtP mutant strain FG1602 (A) and Salmonella serovar Typhimurium (Se) wild-type strain SK2633 (B) were cultured in NH4Cl-glycerol medium. 2OG concentrations in medium were assayed from immediately after inoculation through the late exponential phase of growth. Internal 2OG concentrations were also measured at different cell densities. The E. coli wild-type external 2OG data in panel A are from Fig. 2A for comparison. (C) kgtP is transcribed in E. coli but silent in Salmonella serovar Typhimurium. The three strains were cultured identically to those described in panels A and B, cells were collected at an OD600 of ∼0.4, and RNA was extracted. cDNA was reverse transcribed and subjected to PCR detection of kgtP expression. Genomic DNA extracted from colonies served as the control template for PCR.
With the above results, we were puzzled by an early brief test on a Salmonella serovar Typhimurium strain whose culture medium gave a much higher 2OG concentration than that of the E. coli wild type. For clarification, the experiment was repeated for a Salmonella serovar Typhimurium wild-type strain (Fig. 6B). Its internal 2OG concentration was steady and about 2-fold greater than that of E. coli, but its leakage level and profile appeared to be much more like those of the E. coli kgtP mutant than the wild type. The apparent contradiction between these two closely related species was investigated. We first inspected their kgtP and adjacent sequences (see the alignment in Fig. S4 in the supplemental material). Although kgtP is annotated in the Salmonella serovar Typhimurium LT2 genome, it appears to have lost its own promoter due to an insertion into ∼50 bp of the equivalent coding region of E. coli kgtP. The insertion contains an open reading frame encoding a putative cytoplasmic protein in the same strand as kgtP. The same is true for each out of 6 genome-sequenced S. enterica serotypes that we inspected, but not for any out of 21 E. coli strains (BioCyc, version 14.5; http://biocyc.org; examples are shown in Fig. S4 in the supplemental material). We then examined transcription of kgtP by reverse transcription-PCR in both species (Fig. 6C). The result showed that kgtP was expressed in E. coli but not in Salmonella serovar Typhimurium, suggesting that Salmonella serovar Typhimurium kgtP is either cryptic or, at least, silent under the culturing condition.
DISCUSSION
In retrospect, measurement of the 2OG pool in E. coli is less problematic than it is in S. enterica. The functional KgtP retains a large portion of the leaked 2OG which would otherwise accumulate in the medium at a high cell density (Fig. 6) and hinder the accuracy of quantification. The effect of KgtP is to change the internal and external concentration ratio. As illustrated in Fig. S2 in the supplemental material, the lower that the proportion of external 2OG is, the more applicable the subtraction strategy becomes. As 2OG may deplete or accumulate rapidly during sampling (Fig. 3), this strategy is preferred when throughput is less of a concern because it is less intrusive during cell handling than the sampling method with washes. Under certain conditions, the subtraction strategy is the only choice if a near perfect or harmless wash cannot be easily formulated—e.g., during the transitional phase of nutrient (nitrogen or carbon) depletion, when the nutrient is rapidly decreasing due to cell consumption, or during the operation of a nutrient-limited continuous culture.
For quantification of cellular metabolites, such a leakage problem is not limited to 2OG: there are many metabolites that cannot be totally contained by a phospholipid bilayer. For example, there are a number of weak acids in central metabolism, such as pyruvate, fumarate, acetate, and lactate, whose leakage has been mentioned (34, 48, 56). The quantification of their internal concentrations would depend on the extent of help provided by their respective uptake mechanisms, similar to what KgtP is to 2OG. Such transporters have been documented in the literature, including a pyruvate transporter (26, 29) and the fumarate/malate/succinate Dct uptake systems (11, 27, 33). Even with a recycle mechanism, however, metabolite leakage presents a challenge for accurate quantification of cellular metabolites. Whether one chooses a no-harvest or whole-broth protocol or any form of harvest method, it is critical to know the excreted level of the metabolite of interest. If a leakage problem was substantial, it is absolutely necessary to take the external amount into account in order to avoid large errors for the internal amount. Our study demonstrated such a challenge in quantification of cellular 2OG, whose leakage problem can be considered only moderate. There are more severe cases, such as cyclic AMP (15).
From the point of cell growth, the leakage may also be a problem. With excess carbon provided in the culture, the cost of carbon leakage and the benefit of uptake may not be obvious: the loss of KgtP function resulted in some carbon losses, but neither the 2OG pool nor the growth rate was affected (Fig. 6A). However, leakage of multiple metabolites may have a cumulative effect for bacteria in their native habitat when available carbon is scarce, and multiple uptake systems for their retention may collectively serve to conserve the carbon flux for bacterial growth.
We chose filtration over centrifugation for cell harvest because our earlier test using centrifugation was unsatisfactory in terms of data variation. There were several possible reasons. Cells within a tightly packed pellet may have less nutrient/oxygen accessibility than those on a wet membrane disc. A potential temperature shift during centrifugation may also affect metabolite pools, as illustrated by the cold treatment (Fig. 4A). A centrifugation process usually took at least 1 min between the times of taking the culture and the acid extraction. In comparison, filtration is a much gentler and faster process. However, in terms of speed, the ∼15-s handling time of the filtration process is still too long compared to the half-life of many central carbon metabolite pools (20). For the 2OG pool, the estimated half-life is 0.5 s. It is then no surprise that a low-glycerol wash can cause an immediate decline of the 2OG pool (Fig. 3A), even though the reduced flux has to pass through many buffer metabolite pools between the glycerol entry and 2OG pool. This illustrates the highly sensitive nature of central metabolite pools; thus, their quantification demands great care during cell sampling. In contrast, the central nitrogen intermediates Gln and Glu have much longer half-lives: about one-half to 1 min. This is mostly due to their larger pool concentrations of 5 mM and 70 mM, respectively, in cells grown with sufficient NH4+ (39). In addition, the temperature effect has to be considered during cell sampling for quantification of metabolites. The commonly exercised cold treatment may not be able to freeze cellular metabolic contents and could lead to an inaccurate conclusion.
The responses of the 2OG pool to both carbon and nitrogen depletions illustrate its dual identity: a central carbon metabolite and a carrier for central nitrogen metabolism. Our results show that 2OG rapidly accumulates upon an NH4+ downshift, while the opposite is observed with a carbon downshift (Fig. 3). The response to a change in NH4+ is more profound: a 5- to 10-fold increase versus a 2-fold decrease. The basis of this accumulation, obviously due to a sudden change of NH4+ assimilation within the central nitrogen metabolism, can be dissected by analyzing the results for mutants. When NH4+ can be assimilated only upon 2OG by the action of GDH in the GOGAT− mutant, a shift of the NH4+ concentration down to the low-mM range is already enough to cause significant 2OG accumulation. By comparison, when NH4+ can be incorporated upon 2OG only through the action of GS/GOGAT in the GDH− mutant, the large increase in the 2OG pool does not occur until the NH4+ concentration is shifted down to the range of ∼0.1 mM. The Kms for NH4+ of GDH and GS are in the range of low mM (36, 43) and 0.1 to 0.2 mM (1, 35), respectively. The agreement of 2OG accumulation in the two mutants and the Kms for NH4+ of the two enzymes indicates that the metabolic fluxes through the two Glu biosynthesis pathways greatly influence cellular 2OG levels. In the wild type with both functional GS/GOGAT and GDH pathways, the 2OG pool changes in both the mM and sub-mM ranges of NH4+ (Fig. 3) and does so more dramatically in the sub-mM (the Km of GS) range. The data also suggest that, although GDH contributes, GS/GOGAT is the dominant pathway for Glu synthesis, even though its action costs more energy. This pathway partitioning has been demonstrated by other means before (19, 58). As for the practical aspects of 2OG pool measurement, the relationship between the Km and the pool disturbance indicates that, if one prefers to perform a harmless wash during sample collection, a near perfectly matched or sufficient NH4+ concentration (far above the Km) has to be present in the wash solution to avoid artifacts.
To interpret the physiological role of a sensory molecule such as 2OG, one needs to understand what it senses and what it regulates. All receptors of 2OG documented so far are exclusively related to nitrogen metabolism. Among those, the quantitative aspect of 2OG regulation on GS function through E. coli GlnB has been well established in vitro, but 2OG is only one of several metabolites affecting GS regulation in enteric bacteria. Studies have shown that Gln plays a more dominant role than 2OG (23, 24, 39). Internal Gln limitation is known to be the signal of external nitrogen deficiency in enteric bacteria (21, 44). From recent observations by our group and others (39, 58) and the mutant analysis after the NH4+ shifts (Fig. 3B and C), it appears that 2OG is also a nitrogen signal in E. coli. Qualitatively, cellular 2OG and Gln levels sense nitrogen availability in opposite directions, and their regulatory effects are antagonistic. Quantitatively, the Gln pool plays a bigger regulatory role in enteric bacteria. In other bacteria, however, the relative importance of the two regulatory metabolites may reverse: 2OG may serve as the only or dominant nitrogen signal when Gln plays no or less of a role (31).
What makes the scheme complex is that 2OG also responds to external carbon availability (Fig. 3A) and hence could be employed as a carbon signal as well. The earliest 2OG pool data related to GS regulation have shown that internal 2OG concentrations correlate with the dilution rates in a glucose-limited chemostat of E. coli (45). As nitrogen assimilation in bacteria costs considerable energy by the action of GS (42), coordinated GS regulation based upon nitrogen and carbon availabilities is suggested through the function of GlnB, and hence, 2OG is considered to be the best candidate for a carbon signal regulating central nitrogen metabolism. With a reliable method established here for quantification of the internal 2OG concentration, its precise role from the carbon side for nitrogen regulation in enteric bacteria can be clarified quantitatively through detailed in vivo studies. Besides 2OG, there may be other sensory metabolites serving the role. For example, carbon availability may in fact reflect on cellular energy status. It has been shown in vitro that ATP/ADP is the third major effector on GS regulation (22), although the physiological implication has yet to be elucidated in vivo.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a research grant (RGP/P0022) from the Human Frontier Science Program.
We thank Timothy Ikeda for developing a prototype of 2OG detection in the laboratory of Sydney Kustu at the University of California at Berkeley, Jilong Wang for assistance in experiments, and Sydney Kustu, Margaret Bauer, and Hiroyuki Okano for critical review of the manuscript. We also thank anonymous reviewers for their comments, especially a suggestion leading to the cold wash experiment.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
Published ahead of print on 5 August 2011.
REFERENCES
- 1. Alibhai M., Villafranca J. J. 1994. Kinetic and mutagenic studies of the role of the active site residues Asp-50 and Glu-327 of Escherichia coli glutamine synthetase. Biochemistry 33:682–686 [DOI] [PubMed] [Google Scholar]
- 2. Baba T., et al. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bennett B. D., et al. 2009. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5:593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boer V. M., Crutchfield C. A., Bradley P. H., Botstein D., Rabinowitz J. D. 2010. Growth-limiting intracellular metabolites in yeast growing under diverse nutrient limitations. Mol. Biol. Cell 21:198–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bolten C. J., Kiefer P., Letisse F., Portais J. C., Wittmann C. 2007. Sampling for metabolome analysis of microorganisms. Anal. Chem. 79:3843–3849 [DOI] [PubMed] [Google Scholar]
- 6. Brown M. S., Segal A., Stadtman E. R. 1971. Modulation of glutamine synthetase adenylylation and deadenylylation is mediated by metabolic transformation of the PII regulatory protein. Proc. Natl. Acad. Sci. U. S. A. 68:2949–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carr P. D., et al. 1996. X-ray structure of the signal transduction protein from Escherichia coli at 1.9 Å. Acta Crystallogr. D Biol. Crystallogr. 52:93–104 [DOI] [PubMed] [Google Scholar]
- 8. Conroy M. J., et al. 2 007. The crystal structure of the Escherichia coli AmtB-GlnK complex reveals how GlnK regulates the ammonia channel. Proc. Natl. Acad. Sci. U. S. A. 104:1213–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Coutts G., Thomas G., Blakey D., Merrick M. 2002. Membrane sequestration of the signal transduction protein GlnK by the ammonium transporter AmtB. EMBO J. 21:536–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Csonka L. N., Ikeda T. P., Fletcher S. A., Kustu S. 1994. The accumulation of glutamate is necessary for optimal growth of Salmonella typhimurium in media of high osmolality but not induction of the proU operon. J. Bacteriol. 176:6324–6333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davies S. J., et al. 1999. Inactivation and regulation of the aerobic C4-dicarboxylate transport (dctA) gene of Escherichia coli. J. Bacteriol. 181:5624–5635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dodsworth J. A., Cady N. C., Leigh J. A. 2005. 2-Oxoglutarate and the PII homologues NifI1 and NifI2 regulate nitrogenase activity in cell extracts of Methanococcus maripaludis. Mol. Microbiol. 56:1527–1538 [DOI] [PubMed] [Google Scholar]
- 13. Durand A., Merrick M. 2006. In vitro analysis of the Escherichia coli AmtB-GlnK complex reveals a stoichiometric interaction and sensitivity to ATP and 2-oxoglutarate. J. Biol. Chem. 281:29558–29567 [DOI] [PubMed] [Google Scholar]
- 14. Ehlers C., Weidenbach K., Veit K., Forchhammer K., Schmitz R. A. 2005. Unique mechanistic features of post-translational regulation of glutamine synthetase activity in Methanosarcina mazei strain Gö1 in response to nitrogen availability. Mol. Microbiol. 55:1841–1854 [DOI] [PubMed] [Google Scholar]
- 15. Epstein W., Rothman-Denes L. B., Hesse J. 1975. Adenosine 3′:5′-cyclic monophosphate as mediator of catabolite repression in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 72:2300–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Flynn K. J. 1990. The determination of nitrogen status in microalgae. Mar. Ecol. Prog. Ser. 61:297–307 [Google Scholar]
- 17. Fong R. N., Kim K. S., Yoshihara C., Inwood W. B., Kustu S. 2007. The W148L substitution in the Escherichia coli ammonium channel AmtB increases flux and indicates that the substrate is an ion. Proc. Natl. Acad. Sci. U. S. A. 104:18706–18711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gruswitz F., O'Connell J., III, Stroud R. M. 2007. Inhibitory complex of the transmembrane ammonia channel, AmtB, and the cytosolic regulatory protein, GlnK, at 1.96 Å. Proc. Natl. Acad. Sci. U. S. A. 104:42–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Helling R. B. 1994. Why does Escherichia coli have two primary pathways for synthesis of glutamate? J. Bacteriol. 176:4664–4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Holms H. 1996. Flux analysis and control of the central metabolic pathways in Escherichia coli. FEMS Microbiol. Rev. 19:85–116 [DOI] [PubMed] [Google Scholar]
- 21. Ikeda T. P., Shauger A. E., Kustu S. 1996. Salmonella typhimurium apparently perceives external nitrogen limitation as internal glutamine limitation. J. Mol. Biol. 259:589–607 [DOI] [PubMed] [Google Scholar]
- 22. Jiang P., Ninfa A. J. 2007. Escherichia coli PII signal transduction protein controlling nitrogen assimilation acts as a sensor of adenylate energy charge in vitro. Biochemistry 46:12979–12996 [DOI] [PubMed] [Google Scholar]
- 23. Jiang P., Peliska J. A., Ninfa A. J. 1998. Reconstitution of the signal-transduction bicyclic cascade responsible for the regulation of Ntr gene transcription in Escherichia coli. Biochemistry 37:12795–12801 [DOI] [PubMed] [Google Scholar]
- 24. Jiang P., Peliska J. A., Ninfa A. J. 1998. The regulation of Escherichia coli glutamine synthetase revisited: role of 2-ketoglutarate in the regulation of glutamine synthetase adenylylation state. Biochemistry 37:12802–12810 [DOI] [PubMed] [Google Scholar]
- 25. Jiang P., et al. 1997. Structure/function analysis of the PII signal transduction protein of Escherichia coli: genetic separation of interactions with protein receptors. J. Bacteriol. 179:4342–4353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jolkver E., et al. 2009. Identification and characterization of a bacterial transport system for the uptake of pyruvate, propionate, and acetate in Corynebacterium glutamicum. J. Bacteriol. 191:940–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kay W. W., Kornberg H. L. 1971. The uptake of C4-dicarboxylic acids by Escherichia coli. Eur. J. Biochem. 18:274–281 [DOI] [PubMed] [Google Scholar]
- 28. Kustu S., Hirschman J., Burton D., Jelesko J., Meeks J. C. 1984. Covalent modification of bacterial glutamine synthetase: physiological significance. Mol. Gen. Genet. 197:309–317 [DOI] [PubMed] [Google Scholar]
- 29. Lang V. J., Leystra-Lantz C., Cook R. A. 1987. Characterization of the specific pyruvate transport system in Escherichia coli K-12. J. Bacteriol. 169:380–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laurent S., et al. 2005. Nonmetabolizable analogue of 2-oxoglutarate elicits heterocyst differentiation under repressive conditions in Anabaena sp. PCC 7120. Proc. Natl. Acad. Sci. U. S. A. 102:9907–9912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leigh J. A., Dodsworth J. A. 2007. Nitrogen regulation in bacteria and archaea. Annu. Rev. Microbiol. 61:349–377 [DOI] [PubMed] [Google Scholar]
- 32. Lie T. J., Wood G. E., Leigh J. A. 2005. Regulation of nif expression in Methanococcus maripaludis: roles of the euryarchaeal repressor NrpR, 2-oxoglutarate, and two operators. J. Biol. Chem. 280:5236–5241 [DOI] [PubMed] [Google Scholar]
- 33. Lo T. C. 1977. The molecular mechanism of dicarboxylic acid transport in Escherichia coli K 12. J. Supramol. Struct. 7:463–480 [DOI] [PubMed] [Google Scholar]
- 34. Lowry O. H., Carter J., Ward J. B., Glaser L. 1971. The effect of carbon and nitrogen sources on the level of metabolic intermediates in Escherichia coli. J. Biol. Chem. 246:6511–6521 [PubMed] [Google Scholar]
- 35. Meek T. D., Villafranca J. J. 1980. Kinetic mechanism of Escherichia coli glutamine synthetase. Biochemistry 19:5513–5519 [DOI] [PubMed] [Google Scholar]
- 36. Miller R. E., Stadtman E. R. 1972. Glutamate synthase from Escherichia coli. An iron-sulfide flavoprotein. J. Biol. Chem. 247:7407–7419 [PubMed] [Google Scholar]
- 37. Muro-Pastor M. I., Reyes J. C., Florencio F. J. 2001. Cyanobacteria perceive nitrogen status by sensing intracellular 2-oxoglutarate levels. J. Biol. Chem. 276:38320–38328 [DOI] [PubMed] [Google Scholar]
- 38. Ninfa A. J., Jiang P. 2005. PII signal transduction proteins: sensors of α-ketoglutarate that regulate nitrogen metabolism. Curr. Opin. Microbiol. 8:168–173 [DOI] [PubMed] [Google Scholar]
- 39. Okano H., Hwa T., Lenz P., Yan D. 2010. Reversible adenylylation of glutamine synthetase is dynamically counterbalanced during steady-state growth of Escherichia coli. J. Mol. Biol. 404:522–536 [DOI] [PubMed] [Google Scholar]
- 40. Oliveira I. C., Coruzzi G. M. 1999. Carbon and amino acids reciprocally modulate the expression of glutamine synthetase in Arabidopsis. Plant Physiol. 121:301–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Osorio A. V., Camarena L., Salazar G., Noll-Louzada M., Bastarrachea F. 1993. Nitrogen regulation in an Escherichia coli strain with a temperature sensitive glutamyl-tRNA synthetase. Mol. Gen. Genet. 239:400–408 [DOI] [PubMed] [Google Scholar]
- 42. Reitzer L. 2003. Nitrogen assimilation and global regulation in Escherichia coli. Annu. Rev. Microbiol. 57:155–176 [DOI] [PubMed] [Google Scholar]
- 43. Sakamoto N., Kotre A. M., Savageau M. A. 1975. Glutamate dehydrogenase from Escherichia coli: purification and properties. J. Bacteriol. 124:775–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schmitz R. A. 2000. Internal glutamine and glutamate pools in Klebsiella pneumoniae grown under different conditions of nitrogen availability. Curr. Microbiol. 41:357–362 [DOI] [PubMed] [Google Scholar]
- 45. Senior P. J. 1975. Regulation of nitrogen metabolism in Escherichia coli and Klebsiella aerogenes: studies with the continuous-culture technique. J. Bacteriol. 123:407–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Seol W., Shatkin A. J. 1992. Escherichia coli α-ketoglutarate permease is a constitutively expressed proton symporter. J. Biol. Chem. 267:6409–6413 [PubMed] [Google Scholar]
- 47. Seol W., Shatkin A. J. 1991. Escherichia coli kgtP encodes an α-ketoglutarate transporter. Proc. Natl. Acad. Sci. U. S. A. 88:3802–3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Siegel W. H., Donohue T., Bernlohr R. W. 1977. Determination of pools of tricarboxylic acid cycle and related acids in bacteria. Appl. Environ. Microbiol. 34:512–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Soupene E., et al. 2003. Physiological studies of Escherichia coli strain MG1655: growth defects and apparent cross-regulation of gene expression. J. Bacteriol. 185:5611–5626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stadtman E. R. 2001. The story of glutamine synthetase regulation. J. Biol. Chem. 276:44357–44364 [DOI] [PubMed] [Google Scholar]
- 51. Tanigawa R., et al. 2002. Transcriptional activation of NtcA-dependent promoters of Synechococcus sp. PCC 7942 by 2-oxoglutarate in vitro. Proc. Natl. Acad. Sci. U. S. A. 99:4251–4255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thorner J. W., Paulus H. 1973. Catalytic and allosteric properties of glycerol kinase from Escherichia coli. J. Biol. Chem. 248:3922–3932 [PubMed] [Google Scholar]
- 53. van Heeswijk W. C., et al. 1996. An alternative PII protein in the regulation of glutamine synthetase in Escherichia coli. Mol. Microbiol. 21:133–146 [DOI] [PubMed] [Google Scholar]
- 54. Vazquez-Bermudez M. F., Herrero A., Flores E. 2002. 2-Oxoglutarate increases the binding affinity of the NtcA (nitrogen control) transcription factor for the Synechococcus glnA promoter. FEBS Lett. 512:71–74 [DOI] [PubMed] [Google Scholar]
- 55. Wang Z. J., Zaitsu K., Ohkura Y. 1988. High-performance liquid chromatographic determination of α-keto acids in human serum and urine using 1,2-diamino-4,5-methylenedioxybenzene as a precolumn fluorescence derivatization reagent. J. Chromatogr. 430:223–231 [DOI] [PubMed] [Google Scholar]
- 56. Wolfe A. J. 2005. The acetate switch. Microbiol. Mol. Biol. Rev. 69:12–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yildiz O., Kalthoff C., Raunser S., Kuhlbrandt W. 2007. Structure of GlnK1 with bound effectors indicates regulatory mechanism for ammonia uptake. EMBO J. 26:589–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yuan J., et al. 2009. Metabolomics-driven quantitative analysis of ammonia assimilation in E. coli. Mol. Syst. Biol. 5:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.