

Abstract
Lipoarabinomannan (LAM) is a structurally heterogeneous amphipathic lipoglycan present in Mycobacterium spp. and other actinomycetes, which constitutes a major component of the cell wall and exhibits a wide spectrum of immunomodulatory effects. Analysis of Mycobacterium smegmatis subcellular fractions and spheroplasts showed that LAM and lipomannan (LM) were primarily found in a cell wall-enriched subcellular fraction and correlated with the presence (or absence) of the mycolic acids in spheroplast preparations, suggesting that LAM and LM are primarily associated with the putative outer membrane of mycobacteria. During the course of these studies significant changes in the LAM/LM content of the cell wall were noted relative to the age of the culture. The LAM content of the M. smegmatis cell wall was dramatically reduced as the bacilli approached stationary phase, whereas LM, mycolic acid, and arabinogalactan content appeared to be unchanged. In addition, cell morphology and acid-fast staining characteristics showed variations with growth phase of the bacteria. In the logarithmic phase, the bacteria were found to be classic rod-shaped acid-fast bacilli, while in the stationary phase M. smegmatis lost the characteristic rod shape and developed a punctate acid-fast staining pattern with carbolfuchsin. The number of viable bacteria was independent of LAM content and phenotype. Taken together, the results presented here suggest that LAM is primarily localized with the mycolic acids in the cell wall and that the cellular concentration of LAM in M. smegmatis is selectively modulated with the growth phase.
INTRODUCTION
The cell envelope (CE) of Mycobacterium spp. is a complex structure that accounts for many of the unique properties displayed by the bacilli, including resistance to host-mediated lysis, low permeability to antibiotics, resistance to desiccation, and acid fastness (9). The CE is thick, with a high lipid content, and disruption, via mutation or exposure to antibiotics, typically results in an increase of CE permeability and a reduction in the virulence of pathogenic spp.
The CE consists of a covalently linked core that is associated with a variety of noncovalently linked lipids, carbohydrates, and proteins (1, 8, 10, 14). The CE core consists of a mycolyl arabinogalactan-peptidoglycan complex (MAPc). In the MAPc peptidoglycan (PG) is linked, via a rhamnose-N-acetylglucosamine phosphate, to a linear d-galactofuran. The d-galactofuran, in turn, is attached to a highly branched d-arabinofuran that has mycolic acids linked, via ester bonds, to the nonreducing termini (1, 25). In 1982 Minnikin (31) proposed that the mycolic acids form an inner leaflet of a putative outer membrane with free lipids forming the outer leaflet. More recently, cryo-electron tomography has provided support for the outer membrane hypothesis (20, 53), but it has been shown that free lipids are distributed over both leaflets of the putative outer membrane (20). Thus, the long proposed compartment analogous to the periplasmic space in Gram-negative bacteria (14) appears to have gained support.
Major components of the noncovalently linked lipids of the cell envelope include the phosphatidylinositol mannose compounds (PIMs), some of which are believed to be precursors of lipomannan (LM) and lipoarabinomannan (LAM) (8, 25). LM and LAM are complex molecules that consist of a polymannosylated phosphatidylinositol core and, in the case of LAM, a large branched arabinan moiety (11, 24). LAM displays species-specific heterogeneity at the nonreducing arabinan termini, which are capped with inositol phosphate residues in Mycobacterium smegmatis or are uncapped or capped with mannose residues in other Mycobacterium spp., including Mycobacterium tuberculosis (13, 17, 27).
LAM is a major immunomodulatory component of the mycobacterial CE implicated in many biological functions; typically, mannose-capped LAM is thought to be anti-inflammatory, whereas inositol phosphate-capped LAM is thought to be proinflammatory. Mannose-capped LAM from M. tuberculosis has been implicated in inhibition of phagosomal maturation, apoptosis, gamma interferon signaling in macrophages, and interleukin-12 secretion of dendritic cells (reviewed in references 10, 12, and 33). LAM mediates entry in to macrophages via the mannose receptor aiding the bacilli in avoiding phagosome-lysosome fusion, oxidative burst and altered calcium and cytokine responses (3, 23, 42, 43, 48). LAM has also been shown to bind to Toll-like receptors and induce signaling events important in the host response in tuberculosis (15, 16). Thus, LAM is generally considered to be a crucial factor in mycobacterial pathogenesis.
Due to its importance in immunogenesis and pathogenesis, LAM has been subjected to intense study; however, the location of LAM in the CE has been poorly defined. Some studies have suggested that LAM was associated with the plasma membrane (21, 22); others have suggested that LAM is anchored in the mycolic acid layer (38) or the recently described outer membrane, with very small amounts associated with the plasma membrane (36). Another recent report indicated that the surface of Mycobacterium bovis BCG is very hydrophobic, a result consistent with the presence of an external layer of mycolic acids but not with a surface of exposed polar head groups of associated lipids intercalated with mycolic acids, suggesting that LAM is not abundant (if present at all) on the surfaces of the bacilli and unlikely to be anchored in the outer membrane (2). Thus, the subcellular localization of LAM has long been subject to debate.
Since earlier work had generated well-characterized spheroplasts from mycobacteria (39–41, 49, 50), it was hypothesized that if the mycolates, which make up the inner leaflet of the putative outer membrane of M. smegmatis, could be removed by this process, it would be possible to determine whether LAM is primarily associated with the plasma membrane or the outer mycolate layer. The results of these studies indicate that LAM and LM are predominantly associated with the putative outer membrane and that the relative concentrations of these molecules in the CE is selectively modulated as cells progress from one growth phase to another.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All chemical reagents were of analytical grade from Sigma unless otherwise specified. M. smegmatis mc2155 was obtained from the American Type Culture Collection, and cells were grown in defined Sauton's medium supplemented with 1% ZnSO4. For spheroplast formation, tryptic soy broth (TSB; Difco) was used.
Spheroplast formation, morphology, and characterization.
M. smegmatis cells were converted to spheroplasts as previously described (49, 50), with minor modification. Briefly, thawed glycerol stock was inoculated into TSB containing 1% glucose and 0.05% Tween 80, followed by incubation at 37°C and 150 rpm until the optical density (OD) reached 0.5. A filter-sterilized solution of 20% glycine was added to 1.2% final concentration, and incubation was continued at 37°C and 120 rpm for 16 to 20 h. The resulting pre-spheroplasts were harvested by centrifugation at 3,500 rpm in a Beckman GS-6R centrifuge and washed with 1× SMM buffer (0.5 M sucrose and 20 mM MgCl2 in 20 mM maleate buffer at pH 6.6). The pellet was resuspended in the original volume of TSB-SMM (TSB containing 0.5 M sucrose, 20 mM MgCl2, and 20 mM maleate buffer at pH 6.6) by gentle pipetting, and then filter-sterilized solutions of lysozyme (50 mg/ml) and glycine (20%) were added to bring the final concentrations to 50 μg/ml and 1.2%, respectively. Spheroplast formation was complete after incubation at 37°C and 100 rpm for 16 to 24 h, and the mixture was filtered through a sterile cell strainer (BD Falcon) to eliminate clumps of cells. The spheroplasts were centrifuged and resuspended in SMM buffer, and changes in morphology were observed by light microscopy. In some cases, the spheroplast preparations were further separated on a self-forming 60% Percoll density gradient by centrifugation at 27,000 × g for 1 h; this process resulted in a low-density population contaminated with intact bacilli and a denser population consisting of pure spheroplasts. Both populations were harvested (see Fig. S1 in the supplemental material) and washed repeatedly with SMM buffer to remove the Percoll. In all preparations the viability was checked by plating cultures on 7H11 plates and using the Bac Titer-Glo microbial cell viability assay from Promega in which cells and spheroplast preparations were serially diluted in the fresh medium and incubated with Bac Titer-Glo reagent at 37°C for 10 min; luminescence was recorded using a Synergy HT plate reader (BioTek).
Microscopy.
Spheroplast preparations were treated with DNase (100 μg/ml) for 30 min before labeling. Cells and spheroplasts were then incubated with fluorescence-labeled vancomycin (BODIPY FL conjugate; Invitrogen). Samples were exposed to vancomycin (1 μg/ml in TSB) for 1 h at 37°C and then washed by repeated centrifugation to remove unbound dye and labeled with DAPI (4′,6′-diamidino-2-phenylindole; Prolong Gold) on a microscope slide by incubation with mount according to the manufacturer's directions. Fluorescent and phase-contrast images were captured on an Olympus IX71 microscope using a Retiga 2000R camera (Qimaging) and Slidebook software (Intelligent Imaging Innovations, Inc.) on a Macintosh G5 computer (Apple Computer).
Extraction of LAM and LM.
A quick extraction procedure was used to obtain LAM and LM from small samples of M. smegmatis and spheroplast preparations (∼10 mg) as described previously (52). Briefly, a mixture of chloroform-methanol-water (10:10:3) was added to the cell pellet and incubated for 30 min at 55°C. The sample was centrifuged, and the organic solvent was removed. Water and phenol saturated with phosphate-buffered saline (1:1) were added to the pellet, and the mixture was incubated at 80°C for 2 h. Chloroform was added, and the sample was centrifuged. The supernatant containing LAM, LM, and PIMs was dialyzed against water overnight using 3,500 molecular-weight-cutoff dialysis tubing (Spectrum Labs). The dialyzed material was analyzed on Novex 10 to 20% Tricine Gels (Invitrogen), stained with periodic acid-Schiff reagent (37). For Western and lectin blotting, LAM and LM were electroblotted from Tricine gels to Protran nitrocellulose membranes (Whatman/Schleicher & Schuell Bioscience), which were then blocked and incubated with monoclonal antibody CS-35 (provided by National Institutes of Health [NIH]/National Institute of Allergy and Infectious Disease [NIAID] contract AI-25469) or concanavalin A (ConA) conjugated to peroxidase (Sigma). For Western blots, membranes were incubated with the secondary antibody (anti-mouse IgG coupled to alkaline phosphatase), and color was developed using the 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium (BCIP/NBT) substrate (Sigma). The lectin blots were visualized using a 4-chloro-1-naphthol/3,3′-diaminobenzidine tetrahydrochloride (CN/DAB) substrate kit according to the manufacturer's instructions (Pierce).
MAMEs.
Mycolic acid methyl esters (MAMEs) were prepared from equal quantities of cells and spheroplast preparations (dry weight). Briefly, the dried pellets were incubated with 15% tetrabutyl ammonium hydroxide at 100°C overnight. The reaction mixture was diluted with 2 ml of water, and methyl esters were prepared by incubating the reaction mixture with iodomethane in the presence of dichloromethane for 2 h at room temperature. The lower organic layer (containing the MAMEs) was washed three times with water. The resultant material was analyzed on silica gel 60 TLC plates (EMD Bioscience) developed four times with hexane-ethyl acetate (95:5). MAMEs were visualized by charring at 110°C for 15 min using a 5% ethanolic molybdophosphoric acid reagent (4).
Monosaccharide composition.
LAM samples were hydrolyzed with 2 M trifluoroacetic acid. The resulting sugars were converted to alditol acetates and analyzed on an Hewlett-Packard gas chromatograph model 5890 fitted with a flame ionization detector and a SP 2380 column (30 m, 0.25-μm film thickness, 0.25-mm inner diameter; Supelco) using a temperature program of 50°C for 1 min, followed by increases at 30°C/min to 170°C and then increases at 4°C/min to 260°C. The data were quantitated by using response factors generated from authentic monosaccharides and scyllo-inositol as an internal standard (29).
Ziehl-Neelsen staining.
Staining was performed according to standard protocol (6). Briefly, bacteria were heat fixed to a glass microscope slide and then stained with carbolfuchsin (Newcomer Supply) by gentle heating until steam was observed for a total of 5 min, washed thoroughly with water, and decolorized with Bacto TB decolorizer. Cell morphology was determined by light microscopy.
DAP analysis.
Cells and spheroplasts were pelleted by centrifugation in a preweighed glass tube. The supernatant was removed, and the samples were dried under vacuum using a Savant (svc100) for 6 h and weighed to determine the dry weight of the sample. Samples were hydrolyzed in 0.2 ml of 6 N HCl (Pierce, Rockford, IL) at 110°C for 18 h. The diaminopimellic acid (DAP) contents of samples obtained from M. smegmatis cells, and spheroplast preparations were determined using an EZ:Fast gas chromatography-mass spectrometry kit according to the instructions supplied by the manufacturer (Phenomenex) and a Varian CP-3800 gas chromatograph fitted with a ZB-5 column (30 m by 0.25 mm by 0.25 μm) connected to a Varian 320 MS-TQ mass spectrometer. Analytical runs were programmed at an initial temperature of 50°C for 2 min and then raised to 310°C at 30°C/min and held at that temperature for 9 min. The gas flow rate was maintained at 1 ml/min, and 1 μl of sample was injected in splitless mode.
RESULTS
LAM and LM content of subcellular fractions.
In an initial attempt to localize the LAM and LM, M. smegmatis cells were sonicated and cell wall, cytosolic, and membrane-enriched fractions were prepared by differential centrifugation (experimental details are provided in the legend for Fig. S2 in the supplemental material). LAM and LM was extracted from each fraction and analyzed by SDS-PAGE and Western blotting (see Fig. S2 in the supplemental material). The majority of LAM and LM was found to be associated with the cell lysate (Fig. S2, lane 1) and the cell wall-enriched fraction (Fig. S2, lane 2), while the membrane fraction (Fig. S2, lane 5) showed small but detectable amounts of material when visualized with an antibody recognizing the arabinan portion of the LAM (CS-35) or a lectin recognizing the terminal mannose residues of LM (ConA). The cytoplasmic components (see Fig. S2, lane 4, in the supplemental material) were essentially devoid of LAM and LM, as expected. These results clearly indicate a major association of LAM and LM with the cell wall-enriched fraction from M. smegmatis.
Preparation and characterization of M. smegmatis spheroplasts.
To further address the issues of LAM and LM localization in the CE, spheroplasts of M. smegmatis were generated by sequential treatment with glycine and lysozyme essentially as previously reported (49). The formation of the spheroplasts was monitored by phase-contrast microscopy (Fig. 1). The spheroplasts were labeled using DAPI (for nucleic acids) and fluorescently tagged vancomycin, which binds to the terminal-d-Ala-d-Ala of the stem peptide found on nascent PG and lipid II. Overlaying the fluorescent images clearly indicated that the cells visualized by bright-field microscopy were also labeled with DAPI and vancomycin, suggesting that these were sealed spheroplasts (Fig. 1). In addition, vancomycin labeling revealed the presence of septa in some of the spheroplasts (Fig. 1B). Other cells in which the septum was not visible showed increased vancomycin associated fluorescence at what appear to be cellular poles.
Fig. 1.

Morphology of spheroplasts as visualized by labeling with DAPI- and BODIPY-labeled vancomycin. M. smegmatis mc2155 cells were treated with glycine and lysozyme as described in Materials and Methods to generate spheroplasts. Cellular morphology was observed by light and fluorescence microscopy after labeling with BODIPY-labeled vancomycin and DAPI. Spheroplasts are shown in panels A to D, and wild-type M. smegmatis cells are shown in panels E to H. (A and E) Bright-field images; (B and F) BODIPY-vancomycin-labeled peptidoglycan and precursor images (green); (C and G) DAPI-labeled nucleic acid images (blue); (D and H) BODIPY-vancomycin-labeled peptidoglycan and DAPI-labeled nucleic acid images overlaid. The arrowhead in panel B indicates a spheroplast with a visible septum.
The viability and intactness of the spheroplast preparation was also assessed by measuring the ATP content of the spheroplasts using the Bac Titer-Glo assay kit (Promega). This procedure showed that the cells contained ATP that could be released by detergent (data not shown), indicating both intactness of the membrane and viability. Subculturing the spheroplast preparation in liquid medium resulted in regeneration of the bacillary morphology (see Fig. S3 in the supplemental material) and pellicle formation in unshaken cultures. Recovery of morphology in liquid medium was gradual, with rod-shaped bacteria appearing after 96 h. When plated on solid medium, colonies that appeared after 3 days of incubation at 37°C were initially smooth and mucoid and slowly developed the irregular rough growth characteristics of typical M. smegmatis colonies (see Fig. S3 in the supplemental material). Conversion to the rough phenotype was generally complete after 12 days. In addition, partial sequencing of 16S rRNA from the spheroplast preparation showed 100% identity with M. smegmatis 16S RNA over 600 bp (data not shown).
Mycolate and DAP content of cells in the spheroplast preparations.
In order to demonstrate that the spheroplast preparations lack the putative outer membrane, the mycolic acid content, following conversion to MAMEs, was analyzed by thin-layer chromatography (TLC). Initial experiments indicated that appreciable amounts of mycolic acids remained associated with the spheroplasts (Fig. 2). Subsequent density gradient centrifugation using a self-forming gradient of 60% Percoll generated two populations of spheroplasts (see Fig. S1 in the supplemental material). The upper, low-density, population was present as a distinct band, while the lower, dense, population was present as a broad, homogeneous zone. Both fractions were collected, washed by gentle centrifugation in SMM buffer to remove the Percoll, and contained spheroplasts as determined by bright-field and fluorescence microscopy. The low-density population also contained bacillus forms and cell clumps, whereas the high-density fraction contained only spheroplasts (Fig. 1A to D). After normalization on the basis of dry weight, virtually all of the mycolates found in the original spheroplast preparation were associated with the low-density population (Fig. 2). MAMEs were undetectable in the dense population when material corresponding to five times more spheroplasts by dry weight than is shown in (Fig. 2) was loaded on the TLC. Thus, the high-density population was used in subsequent experiments.
Fig. 2.
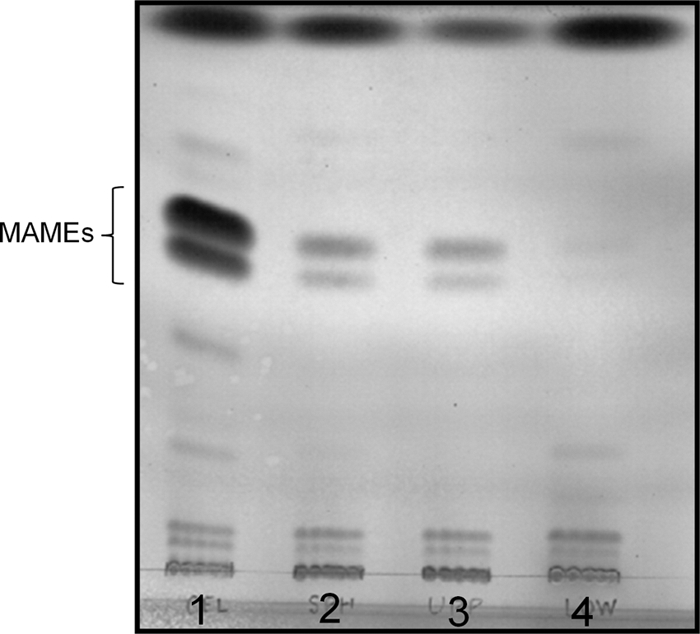
MAME analysis of spheroplast preparations. MAMEs were extracted from equal amounts (by dry weight) of untreated cells or spheroplast preparations and analyzed on silica gel 60 TLC plates developed four times with hexane-ethyl acetate (95:5). MAMEs were visualized by charring. Lane 1, untreated M. smegmatis cells; lane 2, total spheroplasts; lane 3, low-density spheroplast preparation; lane 4, high-density spheroplast preparation.
As a positive control for cell wall removal, the DAP content of the spheroplast preparations relative to untreated cells was also determined. As expected from the vancomycin labeling experiments, the spheroplasts retained measurable amounts of DAP. However, the total DAP, normalized per mg of dry weight, was reduced by 50-fold in the spheroplast preparation relative to the untreated cells (Fig. 3), indicating that the majority of the cell wall PG had been removed.
Fig. 3.

Comparison of the DAP content of M. smegmatis cells and spheroplasts. The analysis was normalized to dry weight; the y axis scale is detector response (area units of the peak identified as DAP) per mg (dry weight) of cells or spheroplasts. The values shown are averages of triplicate measurements, and the error bars indicate the standard deviations.
Analysis of LAM/LM content in M. smegmatis spheroplasts.
After the conditions for the formation of spheroplasts deficient in mycolic acids and PG and, by definition, the outer membrane were established, the LAM and LM content of the spheroplasts was addressed. LAM and LM were analyzed from equal amounts of untreated cells and spheroplast preparation as judged by dry weight. Silver-stained SDS-PAGE gels of LAM/LM extracts from wild-type cells and spheroplasts showed a reduction of both LAM and LM in the spheroplast cells (Fig. 4A) This finding was supported by Western blot analysis of the same samples using the CS-35 antibody and lectin blot analysis using ConA (Fig. 4B and C). In all cases, small amounts of LAM and LM remained detectable in the spheroplast preparations. LAM and LM were restored to untreated levels when the spheroplasts were allowed to regrow into bacillus form.
Fig. 4.

LAM and LM analysis of control cells and spheroplast preparation. LAM and LM were extracted from equivalent amounts (by dry weight) of untreated M. smegmatis cells and spheroplast preparation. The samples were subjected to SDS-PAGE gel analysis visualized by silver staining (A) or by probing electroblots with CS-35 (B) or conjugated ConA (C). Lane 1, untreated cells in mid-log phase; lane 2, cells treated with glycine only; lane 3, cells treated with glycine and lysozyme (spheroplasts); lane 4, spheroplasts subcultured and regrown for 6 days.
Growth-phase-dependent variation in morphology and LAM content.
During the preliminary phases of these studies, it was noted that the LAM content of untreated M. smegmatis cells varied with the age of the culture, prompting an investigation of the potential regulation of this compound from the log phase through the stationary phase. In these experiments, bacteria were grown in Middlebrook 7H9 broth supplemented with oleic acid-albumin-dextrose-catalase (OADC) and 0.05% Tween 80, and aliquots of the culture were taken and plated on Middlebrook 7H11 agar or diluted for OD readings at the time points indicated in Fig. S4 in the supplemental material.
Aliquots from the cultures were also assessed for cell morphology, acid-fast staining (Ziehl-Neelsen), and LAM and LM content. Surprisingly, there were significant changes in the both morphology and staining characteristics of cells as they progressed from log to stationary phase (Fig. 5 A and B). In log-phase growth the bacteria demonstrated the typical rod shape and acid-fast character associated with mycobacteria. However, as the cells progressed into stationary phase, the cells appeared clumpier, lost their rod-like shape, and were less obviously acid-fast, particularly if counterstained with methylene blue (see Fig. S5 in the supplemental material). At higher magnifications (Fig. 5A and B), the phenotypic changes are very pronounced, but it is clear that the cells, although no longer rod-shaped, are acid-fast. The image of the cells in stationary phase (Fig. 5B) shows that there are distinct punctate patterns to the staining that correspond to areas with higher concentrations of fuchsin, since the cells shown in these images were not counterstained. The overall change in cell shape was confirmed using an acid-fast nucleic acid stain (patent pending) developed at Colorado State University (Fig. 5C and D). These results suggested that there may be alterations in the CE as the bacteria enter stationary phase. When subcultured into fresh medium, the cells rapidly regained both the classic rod shape and the acid-fast character (data not shown).
Fig. 5.

Growth-phase-associated changes in the morphology and acid-fast staining of M. smegmatis. M. smegmatis mc2155 cells were grown in 7H9 medium supplemented with OADC and 0.05% Tween 80 at 37°C. (A and B) Cells grown for 24 h (A) or 144 h (B) and subjected to Ziehl-Neelsen acid-fast staining without methylene blue counterstaining and light microscopy. (C and D) Cells grown for 24 h (C) or 144 h (D) and subjected to a proprietary acid-fast nucleic acid stain and fluorescence microscopy.
LAM and LM extracted from equivalent amount of cells from different stages of growth were separated by SDS-PAGE and visualized by silver staining (Fig. 6). The LAM content of the cells had decreased 48 h after inoculation and remained low to 156 h. Western and lectin blots confirmed the loss of LAM in the early stationary phase (data not shown). However, the LM content remained relatively constant through the early to mid stationary phase (96 h) to the late stationary phase (156 h). Subculturing the stationary-phase cells in fresh medium resulted in the rapid reappearance of LAM to log-phase levels (Fig. 6A).
Fig. 6.
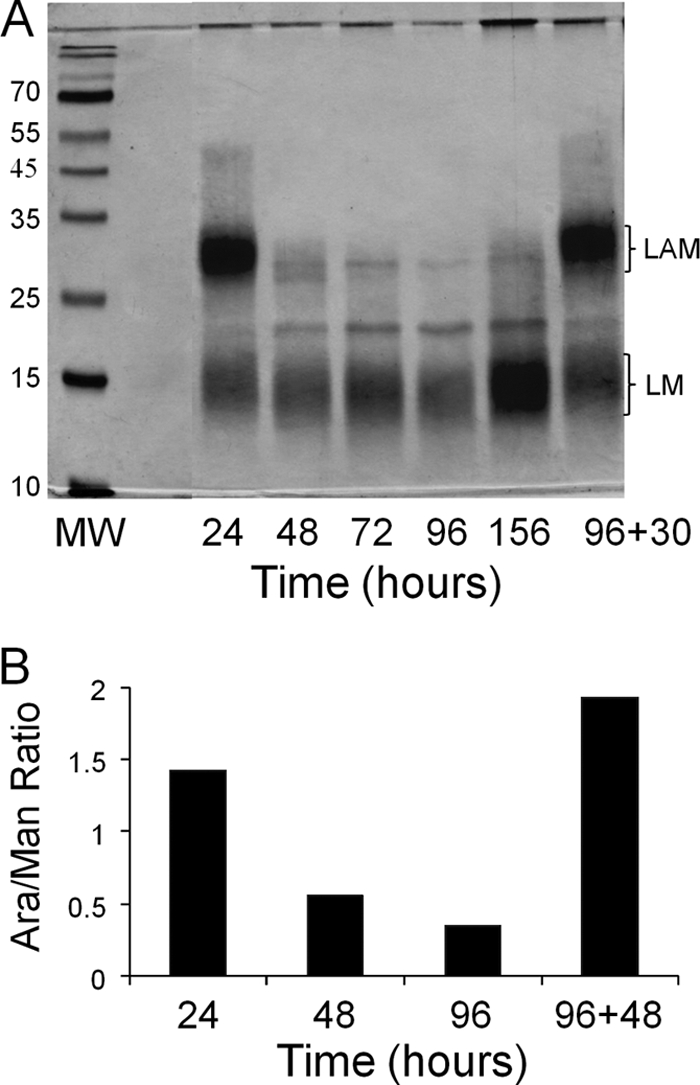
LM and LAM analysis (A) and the Ara/Man ratio of M smegmatis mc2155 (B). M. smegmatis cells were grown in 7H9 medium supplemented with OADC and 0.05% Tween 80. (A) SDS-PAGE analysis of LAM and LM extracted from cells removed from culture at the indicated time points. The material in the lane labeled 96+30 was from an independent aliquot of cells from the 96-h culture that was diluted 1:100 in fresh medium, followed by incubation at 37°C for an additional 30 h. In all cases, the cells were harvested by centrifugation, dried, weighed, and extracted. Extract from equivalent amounts of cell mass was loaded in each lane. (B) Ratio of arabinose to mannose in the LAM and LM extract from cells removed from culture at the indicated time points. The value indicated by the bar labeled 96+48 was from an independent aliquot of cells from the 96-h culture that was diluted 1:100 in fresh medium, followed by incubation at 37°C for an additional 48 h. The analysis, carried out as described in Materials and Methods, is representative of at least three independent experiments.
Based on the quantitative findings, potential qualitative changes in LAM were assessed by determining the ratio of arabinose to mannose in the extracted LAM. The quick LAM/LM extraction (see Materials and Methods) was performed on equivalent amounts of cells taken from culture at 24, 48, and 96 h. The samples were hydrolyzed, reduced, and acetylated. Quantitation using gas chromatography revealed a decrease in the arabinose/mannose (Ara/Man) ratio at 48 and 96 h of culture relative to the value seen at 24 h (Fig. 6B). When the 96-h cultures were subcultured in fresh medium and analyzed after a further 48 h of incubation, the Ara/Man ratio returned to values similar to those seen in log-phase growth (Fig. 6B), confirming the loss of Ara residues associated with LAM relative to mannose presumably associated with LM. However, it is possible that the changes in LAM may be both quantitative and qualitative. No changes were observed in the cellular content of PIMs, mycolic acids, or arabinogalactan with changes in the growth phase in these cultures (data not shown).
DISCUSSION
LAM localization in cell envelope.
Initial subcellular fractionation experiments suggested that the LAM and LM are predominantly associated with a cell wall-enriched subcellular fraction and that a small amount of LAM and LM appears to be associated with the plasma membrane as previously reported (33, 36). Unfortunately, it is not possible to determine, from these experiments, whether the LAM and LM observed in the plasma membrane is due to contamination from the cell wall fraction or whether the relatively small amount of material normally resides in this location.
Therefore, the question of the primary location of the mature LAM and LM was addressed using spheroplast preparations. These preparations were unequivocally shown to have vastly reduced levels of both mycolic acids and DAP and thus are devoid of the putative outer membrane reported in mycobacteria (20, 53). The degree of loss of LAM and LM in the spheroplast preparations in correlation with the loss of the mycolic acids strongly suggests that these molecules are primarily associated with the outer membrane of the CE. The observation that the spheroplasts can regenerate, recovering both LAM and LM, is of considerable importance since it demonstrates that these molecules are restored as the outer membrane is regenerated, supporting the contention that LAM and LM are found primarily in this location.
The fact that detectable amounts of LAM and LM remain in the spheroplast preparations indicates that at least some of the material observed in the membrane-enriched fraction derived from subcellular fractionation of untreated bacilli is not due to contamination. It is highly probable that LAM and LM are synthesized at the inner and outer surfaces of the plasma membrane since these molecules clearly require nucleotide sugars and prenylphosphoryl sugars for synthesis (18, 30); therefore, the material associated with the membrane-enriched subcellular fraction may consist of biosynthetic precursors and/or material that has yet to be transported to the outer membrane.
LAM has generic structural and biological similarities to lipopolysaccharide (LPS) in that both are amphipathic lipids possessing a polysaccharide core and a negatively charged lipid anchor. In addition, the virulence properties of both molecules are determined by species-specific modifications at the nonreducing ends of the polysaccharide. Thus, it is possible that the LAM and LM are associated with the outer leaflet of the outer membrane in analogy to the location of LPS in Gram-negative organisms but the data presented here do not have the resolution to determine whether this is true. However, given the primary localization of LAM and LM in the outer membrane, it does seem likely that LAM and LPS may have similarities in regard to the location and topology of the synthetic machinery and mechanisms of transport to the outer membrane. Since spheroplast preparations and separation of inner and outer membranes were instrumental in understanding the biosynthesis, transport, and localization of LPS in Gram-negative bacteria (19, 34, 34, 35), the observation that LAM and LM concentrations recovered when spheroplasts were regrown may be of particular usefulness. That is, due to their ability to regenerate cell wall components, mycobacteria derived spheroplasts may prove useful in further understanding LAM and LM biogenesis, as well as the biogenesis and transport of other CE components in mycobacteria. Previously, these issues were difficult to address in mycobacteria since separation of inner and outer membrane is unwieldy due to the covalent link between the PG and the inner leaflet of the outer membrane. Thus, the ability to generate bacteria devoid of the outer membrane should afford opportunities to observe the events occurring during its regeneration.
Growth-phase-associated changes in LAM content.
There are ample experimental data showing that Mycobacterium spp. undergo changes in the stationary phase, including changes in colony morphology, generation of “nonculturable” forms, and resistance to osmotic and acid stress (45–47). In addition, alterations in various metabolic and biosynthetic processes, including carbon source utilization, PG cross-linking, and the regulation of mycolic acid synthesis, have been described in the stationary phase (5, 28, 47, 51). However, this appears to be the first publication documenting the variation in cell morphology and staining characteristics shown in Fig. 5. Although these changes are correlated with the loss of LAM, it is possible that other significant changes also occur in the CE, although no changes in the content of PIMs, arabinogalactan, or mycolic acids were noted. Interestingly, the observed changes in morphology and staining characteristics had no apparent effect on the number of CFU in the culture (see Fig. S4 in the supplemental material), and recovery of a “normal” rod shape and acid-fast staining was rapid after subculturing, appearing to occur as soon as the cells re-entered log-phase growth.
To our knowledge, this is also the first study that documents significant changes in the LAM or LM composition in mycobacteria based on growth phase. Previously, an anecdotal observation indicated that there were low amounts of LAM in stationary-phase M. smegmatis, but no data were presented (32). The loss of LAM from stationary-phase cells was unexpected, but regeneration of the LAM on subculturing to induce log-phase growth suggests that these observations are not artifactual or due to culture contamination.
Since the loss of LAM in stationary phase was not accompanied by a loss of PIMs, LM, arabinogalactan, or mycolic acids, the data suggest that the LAM component of the CE is specifically modulated. That is, there does not appear to be a wholesale change in the CE composition, and the presence of the mycolic acids in the stationary phase indicates that there is not a generalized loss of the entire outer membrane. Taken together, these results suggest a well-coordinated, growth-phase-regulated, change in the LAM content. At present, it is unclear whether the observed modulation of LAM synthesis is limited to M. smegmatis or if this is a generic feature of all mycobacteria. Nor has it been determined whether the regulation takes place at the level of synthesis and/or degradation of LAM; although attempts were made to recover and quantitate LAM from the growth medium (data not shown), it was not possible to resolve this issue. However, since LAM is a major immunomodulatory molecule of the pathogenic Mycobacterium spp., further studies focused on understanding regulatory mechanisms underlying these changes in CE content are clearly needed since similar stationary-phase modifications in LPS have been observed in other bacterial species and are reported to have direct effects on the host-pathogen interactions (7, 26, 44). Thus, experiments designed to identify the enzymes, transport mechanisms, potential transcription factors, and their regulation involved in LAM biosynthesis will be of great interest.
Supplementary Material
ACKNOWLEDGMENTS
This research was funded by NIH/NIAID grant AI04915.
We are grateful for the assistance of Stefan Berg.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
Published ahead of print on 12 August 2011.
REFERENCES
- 1. Alderwick L. J., Birch H. L., Mishra A., Eggeling L., Besra G. S. 2007. Structure, function and biosynthesis of the Mycobacterium tuberculosis cell wall: arabinogalactan and lipoarabinomannan assembly with a view to discovering new drug targets. Biochem. Soc. Trans. 35:1325–1328 [DOI] [PubMed] [Google Scholar]
- 2. Alsteens D., et al. 2008. Organization of the mycobacterial cell wall: a nanoscale view. Eur. J. Physiol. 456:117–125 [DOI] [PubMed] [Google Scholar]
- 3. Astarie-Dequeker C., et al. 1999. The mannose receptor mediates uptake of pathogenic and nonpathogenic mycobacteria and bypasses bactericidal responses in human macrophages. Infect. Immun. 67:469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Besra G. S. 1998. Preparation of cell wall fractions from mycobacteria. Methods Mol. Biol. 101:91–107 [DOI] [PubMed] [Google Scholar]
- 5. Betts J. C., Lukey P. T., Robb L. C., McAdam R. A., Duncan K. 2002. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43:717–731 [DOI] [PubMed] [Google Scholar]
- 6. Bishop P. J., Neumann G. 1970. The history of the Ziehl-Neelsen stain. Tubercle 51:196–206 [DOI] [PubMed] [Google Scholar]
- 7. Bravo D., et al. 2008. Growth-phase regulation of lipopolysaccharide O-antigen chain length influences serum resistance in serovars of Salmonella. J. Med. Microbiol. 57:938–946 [DOI] [PubMed] [Google Scholar]
- 8. Brennan P. J., Crick D. C. 2007. The cell-wall core of Mycobacterium tuberculosis in the context of drug discovery. Curr. Top. Med. Chem. 7:475–488 [DOI] [PubMed] [Google Scholar]
- 9. Brennan P. J., Nikaido H. 1995. The envelope of mycobacteria. Annu. Rev. Biochem. 64:29–63 [DOI] [PubMed] [Google Scholar]
- 10. Briken V., Porcelli S. A., Besra G. S., Kremer L. 2004. Mycobacterial lipoarabinomannan and related lipoglycans: from biogenesis to modulation of the immune response. Mol. Microbiol. 53:391–403 [DOI] [PubMed] [Google Scholar]
- 11. Chatterjee D., Hunter S. W., McNeil M., Brennan P. J. 1992. Lipoarabinomannan: multiglycosylated form of the mycobacterial mannosylphosphatidylinositols. J. Biol. Chem. 267:6228–6233 [PubMed] [Google Scholar]
- 12. Chatterjee D., Khoo K. H. 1998. Mycobacterial lipoarabinomannan: an extraordinary lipoheteroglycan with profound physiological effects. Glycobiology 8:113–120 [DOI] [PubMed] [Google Scholar]
- 13. Chatterjee D., Lowell K., Rivoire B., McNeil M. R., Brennan P. J. 1992. Lipoarabinomannan of Mycobacterium tuberculosis: capping with mannosyl residues in some strains. J. Biol. Chem. 267:6234–6239 [PubMed] [Google Scholar]
- 14. Daffe M., Draper P. 1998. The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 39:131–203 [DOI] [PubMed] [Google Scholar]
- 15. Doz E., et al. 2009. Mycobacterial phosphatidylinositol mannosides negatively regulate host Toll-like receptor 4, MyD88-dependent proinflammatory cytokines, and TRIF-dependent costimulatory molecule expression. J. Biol. Chem. 284:23187–23196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Doz E., et al. 2007. Acylation determines the Toll-like receptor (TLR)-dependent positive versus TLR2-, mannose receptor-, and SIGNR1-independent negative regulation of proinflammatory cytokines by mycobacterial lipomannan. J. Biol. Chem. 282:26014–26025 [DOI] [PubMed] [Google Scholar]
- 17. Guerardel Y., et al. 2002. Structural study of lipomannan and lipoarabinomannan from Mycobacterium chelonae: presence of unusual components with α-1,3-mannopyranose side chains. J. Biol. Chem. 277:30635–30648 [DOI] [PubMed] [Google Scholar]
- 18. Gurcha S. S., et al. 2002. Ppm1, a novel polyprenol monophosphomannose synthase from Mycobacterium tuberculosis. Biochem. J. 365:441–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heppel L. A. 1967. Selective release of enzymes from bacteria. Science 156:1451–1455 [DOI] [PubMed] [Google Scholar]
- 20. Hoffmann C., Leis A., Niederweis M., Plitzko J. M., Engelhardt H. 2008. Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc. Natl. Acad. Sci. U. S. A. 105:3963–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hunter S. W., Brennan P. J. 1990. Evidence for the presence of a phosphatidylinositol anchor on the lipoarabinomannan and lipomannan of Mycobacterium tuberculosis. J. Biol. Chem. 265:9272–9279 [PubMed] [Google Scholar]
- 22. Hunter S. W., Gaylord H., Brennan P. J. 1986. Structure and antigenicity of the phosphorylated lipopolysaccharide antigens from the leprosy and tubercle bacilli. J. Biol. Chem. 261:2345–2351 [PubMed] [Google Scholar]
- 23. Kang P. B., et al. 2005. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J. Exp. Med. 202:987–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kaur D., et al. 2006. Biosynthesis of mycobacterial lipoarabinomannan: role of a branching mannosyltransferase. Proc. Natl. Acad. Sci. U. S. A. 103:13664–13669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaur D., Guerin M. E., Skovierova H., Brennan P. J., Jackson M. 2009. Chapter 2: biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv. Appl. Microbiol. 69:23–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khamri W., et al. 2005. Variations in Helicobacter pylori lipopolysaccharide to evade the innate immune component surfactant protein D. Infect. Immun. 73:7677–7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khoo K. H., Dell A., Morris H. R., Brennan P. J., Chatterjee D. 1995. Inositol phosphate capping of the nonreducing termini of lipoarabinomannan from rapidly growing strains of Mycobacterium. J. Biol. Chem. 270:12380–12389 [DOI] [PubMed] [Google Scholar]
- 28. Lavollay M., et al. 2008. The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by l,d-transpeptidation. J. Bacteriol. 190:4360–4366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McNeil M., Chatterjee D., Hunter S. W., Brennan P. J. 1989. Mycobacterial glycolipids: isolation, structures, antigenicity, and synthesis of neoantigens. Methods Enzymol. 179:215–242 [DOI] [PubMed] [Google Scholar]
- 30. Mikusova K., et al. 2005. Decaprenylphosphoryl arabinofuranose, the donor of the d-arabinofuranosyl residues of mycobacterial arabinan, is formed via a two-step epimerization of decaprenylphosphoryl ribose. J. Bacteriol. 187:8020–8025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Minnikin D. E. 1982. Lipids: complex lipids, their chemistry, biosynthesis, and roles, p. 95–184 In Ratledge C., et al. (ed.), The biology of the mycobacteria: physiology, identification, and classification, vol. 1. Academic Press, Inc., New York, NY [Google Scholar]
- 32. Morita Y. S., et al. 2006. PimE is a polyprenol-phosphate-mannose-dependent mannosyltransferase that transfers the fifth mannose of phosphatidylinositol mannoside in mycobacteria. J. Biol. Chem. 281:25143–25155 [DOI] [PubMed] [Google Scholar]
- 33. Nigou J., Gilleron M., Puzo G. 2003. Lipoarabinomannans: from structure to biosynthesis. Biochimie 85:153–166 [DOI] [PubMed] [Google Scholar]
- 34. Osborn M. J., Gander J. E., Parisi E. 1972. Mechanism of assembly of the outer membrane of Salmonella typhimurium: site of synthesis of lipopolysaccharide. J. Biol. Chem. 247:3973–3986 [PubMed] [Google Scholar]
- 35. Osborn M. J., Gander J. E., Parisi E., Carson J. 1972. Mechanism of assembly of the outer membrane of Salmonella typhimurium: isolation and characterization of cytoplasmic and outer membrane. J. Biol. Chem. 247:3962–3972 [PubMed] [Google Scholar]
- 36. Pitarque S., et al. 2008. The immunomodulatory lipoglycans, lipoarabinomannan and lipomannan, are exposed at the mycobacterial cell surface. Tuberculosis 88:560–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Prinzis S., Chatterjee D., Brennan P. J. 1993. Structure and antigenicity of lipoarabinomannan from Mycobacterium bovis BCG. J. Gen. Microbiol. 139:2649–2658 [DOI] [PubMed] [Google Scholar]
- 38. Rastogi N. 1991. Recent observations concerning structure and function relationships in the mycobacterial cell-envelope: elaboration of a model in terms of mycobacterial pathogenicity, virulence, and drug resistance. Res. Microbiol. 142:464–476 [DOI] [PubMed] [Google Scholar]
- 39. Rastogi N., David H. L. 1981. Ultrastructural and chemical studies on wall-deficient forms, spheroplasts, and membrane vesicles from Mycobacterium aurum. J. Gen. Microbiol. 124:71–79 [DOI] [PubMed] [Google Scholar]
- 40. Rastogi N., Rauzier J. Y., Papa F. P., David H. L. 1986. Biochemical and cultural analysis of mycobacterial recombinants obtained by spheroplast fusion. Ann. Inst. Pasteur Microbiol. 137A:135–142 [DOI] [PubMed] [Google Scholar]
- 41. Rastogi N., Venkitasubramanian T. A. 1979. Preparation of protoplasts and whole cell ghosts from Mycobacterium smegmatis. J. Gen. Microbiol. 115:517–521 [DOI] [PubMed] [Google Scholar]
- 42. Schlesinger L. S., Hull S. R., Kaufman T. M. 1994. Binding of the terminal mannosyl units of lipoarabinomannan from a virulent strain of Mycobacterium tuberculosis to human macrophages. J. Immunol. 152:4070–4079 [PubMed] [Google Scholar]
- 43. Schlesinger L. S., Kaufman T. M., Iyer S., Hull S. R., Marchiando L. K. 1996. Differences in mannose receptor-mediated uptake of lipoarabinomannan from virulent and attenuated strains of Mycobacterium tuberculosis by human macrophages. J. Immunol. 157:4568–4575 [PubMed] [Google Scholar]
- 44. Shannon J. G., Howe D., Heinzen R. A. 2005. Virulent Coxiella burnetii does not activate human dendritic cells: role of lipopolysaccharide as a shielding molecule. Proc. Natl. Acad. Sci. U. S. A. 102:8722–8727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shleeva M., Mukamolova G. V., Young M., Williams H. D., Kaprelyants A. S. 2004. Formation of “non-culturable” cells of Mycobacterium smegmatis in stationary phase in response to growth under suboptimal conditions and their Rpf-mediated resuscitation. Microbiology 150:1687–1697 [DOI] [PubMed] [Google Scholar]
- 46. Shleeva M. O., et al. 2002. Formation and resuscitation of “non-culturable” cells of Rhodococcus rhodochrous and Mycobacterium tuberculosis in prolonged stationary phase. Microbiology 148:1581–1591 [DOI] [PubMed] [Google Scholar]
- 47. Smeulders M. J., Keer J., Speight R. A., Williams H. D. 1999. Adaptation of Mycobacterium smegmatis to stationary phase. J. Bacteriol. 181:270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Torrelles J. B., et al. 2008. Identification of mycobacterium tuberculosis clinical isolates with altered phagocytosis by human macrophages due to a truncated lipoarabinomannan. J. Biol. Chem. 283:31417–31428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Udou T., Ogawa M., Mizuguchi Y. 1982. Spheroplast formation of Mycobacterium smegmatis and morphological aspects of their reversion to the bacillary form. J. Bacteriol. 151:1035–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Udou T., Ogawa M., Mizuguchi Y. 1983. An improved method for the preparation of mycobacterial spheroplasts and the mechanism involved in the reversion to bacillary form: electron microscopic and physiological study. Can. J. Microbiol. 29:60–68 [DOI] [PubMed] [Google Scholar]
- 51. Voskuil M. I., Visconti K. C., Schoolnik G. K. 2004. Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis (Edinb.) 84:218–227 [DOI] [PubMed] [Google Scholar]
- 52. Zhang N., et al. 2003. The Emb proteins of mycobacteria direct arabinosylation of lipoarabinomannan and arabinogalactan via an N-terminal recognition region and a C-terminal synthetic region. Mol. Microbiol. 50:69–76 [DOI] [PubMed] [Google Scholar]
- 53. Zuber B., et al. 2008. Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J. Bacteriol. 190:5672–5680 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.