

Abstract
To investigate how different enterohepatic Helicobacter species (EHS) influence Helicobacter pylori gastric pathology, C57BL/6 mice were infected with Helicobacter hepaticus or Helicobacter muridarum, followed by H. pylori infection 2 weeks later. Compared to H. pylori-infected mice, mice infected with H. muridarum and H. pylori (HmHp mice) developed significantly lower histopathologic activity index (HAI) scores (P < 0.0001) at 6 and 11 months postinoculation (MPI). However, mice infected with H. hepaticus and H. pylori (HhHp mice) developed more severe gastric pathology at 6 MPI (P = 0.01), with a HAI at 11 MPI (P = 0.8) similar to that of H. pylori-infected mice. H. muridarum-mediated attenuation of gastritis in coinfected mice was associated with significant downregulation of proinflammatory Th1 (interlukin-1beta [Il-1β], gamma interferon [Ifn-γ], and tumor necrosis factor-alpha [Tnf-α]) cytokines at both time points and Th17 (Il-17A) cytokine mRNA levels at 6 MPI in murine stomachs compared to those of H. pylori-infected mice (P < 0.01). Coinfection with H. hepaticus also suppressed H. pylori-induced elevation of gastric Th1 cytokines Ifn-γ and Tnf-α (P < 0.0001) but increased Th17 cytokine mRNA levels (P = 0.028) at 6 MPI. Furthermore, mRNA levels of Il-17A were positively correlated with the severity of helicobacter-induced gastric pathology (HhHp>H. pylori>HmHp) (at 6 MPI, r2 = 0.92, P < 0.0001; at 11 MPI, r2 = 0.82, P < 0.002). Despite disparate effects on gastritis, colonization levels of gastric H. pylori were increased in HhHp mice (at 6 MPI) and HmHp mice (at both time points) compared to those in mono-H. pylori-infected mice. These data suggest that despite consistent downregulation of Th1 responses, EHS coinfection either attenuated or promoted the severity of H. pylori-induced gastric pathology in C57BL/6 mice. This modulation was related to the variable effects of EHS on gastric interleukin 17 (IL-17) responses to H. pylori infection.
INTRODUCTION
Approximately 50% of the human population is colonized with the gastric pathogen Helicobacter pylori (2, 14). H. pylori persistent colonization initiates chronic active gastritis and in some patients causes peptic ulcer disease, gastric adenocarcinoma, and gastric mucosa-associated lymphoid tissue lymphoma. It has been classified by the World Health Organization as a class I carcinogen (3), with approximately 1 to 2% of the H. pylori-infected population developing gastric tumors. However, the underlying mechanisms governing the clinical outcome of H. pylori infection are poorly understood (6, 14). It is generally accepted that differences in host immune responses, environmental factors, and pathogenicity of H. pylori strains play important roles in disease development. In addition, it has been reported that the presence of endemic parasites may be linked to lower than expected rates of gastric cancer in some African countries and in Colombia, South America, which have especially high prevalence rates of H. pylori infection (4, 11, 31, 50).
The gastrointestinal tract of mammals is colonized by 1012 to 1014 microbes and various parasites which can be mutualistic or pathogenic to human health (21). The interplay among certain organisms can lead to attenuation or promotion of infection-induced pathology. For example, coinfection with Heligmosomoides polygyrus, a natural murine nematode parasite, attenuated gastric atrophy induced by gastric Helicobacter felis in C57BL/6 mice (11). Attenuation of premalignant lesions was associated with reduced expression of proinflammatory Th1 cytokine mRNA levels as well as with increased Th2 cytokine mRNA levels. In contrast, dual infection with H. felis followed by the obligate intracellular protozoan parasite Toxoplasma gondii 20 weeks later led to more severe gastritis associated with increased production of proinflammatory gamma interferon (Ifn-γ) and interleukin 12 (Il-12) in BALB/c mice, which otherwise develop minimal gastritis when infected with H. felis (44). It has also been documented that the interactions between different bacterial species influence infectious intestinal diseases. A recent report noted that prior H. pylori infection attenuated Salmonella enterica serovar Typhimurium-induced colitis in C57BL/6 mice; this protective effect was associated with downregulation of the cecal Th17 response to S. Typhimurium (20). Helicobacter hepaticus, the prototype of enterohepatic Helicobacter species (EHS), is a murine pathogen which causes hepatitis, liver cancer, inflammatory bowel disease, and intestinal carcinoma in susceptible mouse strains (12, 43). Il-10−/− mice (C57BL/6NTac) were protected from H. hepaticus-induced inflammatory bowel disease by administration of polysaccharide A from the human symbiont Bacteroides fragilis. This protection resulted from increased production of IL-10, which suppressed expression of proinflammatory cytokines tumor necrosis factor alpha (TNF-α; Th1) and IL-17A (Th17) (33). Concurrent infection with H. hepaticus delayed recovery and prolonged weight loss of an acute diarrheal disease caused by Citrobacter rodentium in C57BL/6J mice, and this response was associated with upregulation of Il-17A mRNA levels (34). Recently, we showed that coinfection with Lactobacillus reuteri and H. hepaticus induced severe typhlocolitis, whereas monoinfection with either of these organisms in gnotobiotic B6.129P2-IL-10tm1Cgn mice caused no inflammation (51). These lines of evidence indicate that gastrointestinal coinfection with different organisms can modulate host inflammatory responses to pathogenic microbes.
Helicobacter muridarum also has pathogenic potential, since this EHS causes gastritis in experimentally infected BALB/c mice and colitis in T cell-recipient C.B17 SCID mice (24, 26). In addition, H. muridarum elicits proinflammatory TLR2 and NOD1 responses in cultured epithelial cells (5). Recently, we reported that coinfection with another EHS, Helicobacter bilis, significantly decreased the severity of H. pylori-induced gastritis and premalignant lesions in C57BL/6 mice (28). Attenuation of H. pylori-induced gastric pathology correlated with reduced expression of gastric H. pylori-associated proinflammatory mediators in coinfected mice. To investigate whether protection against H. pylori gastritis observed in mice coinfected with H. bilis could be demonstrated with other EHS and also to elucidate the mechanisms responsible for suppression of H. pylori-induced gastric disease, we infected C57BL/6 mice with H. hepaticus or H. muridarum followed by H. pylori and evaluated the progression of gastritis over 11 months post-H. pylori infection.
MATERIALS AND METHODS
Bacterial strains and experimental infections.
Five-week-old female C57BL/6 mice obtained from Taconic Farms (Germantown, NY) were housed in groups of five in polycarbonate microisolator cages on hardwood bedding (PharmaServ, Framingham, MA) under specific-pathogen-free (SPF) conditions (free of Helicobacter spp., Citrobacter rodentium, Salmonella spp., endoparasites, ectoparasites, and exogenous murine viral pathogens) in an Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International)-accredited facility. Mice were maintained at 70 ± 2°F, in 30 to 70% relative humidity, and in a 12-h/12-h light/dark cycle and were fed standard rodent chow (Purina Mills, St. Louis, MO) and water ad libitum. Animal use was approved by the MIT Committee on Animal Care.
Groups of 30 mice were orally gavaged either every other day with 3 doses of 0.2 ml (∼2 × 108 organisms) of H. muridarum strain ST1 (ATCC 49282) or H. hepaticus 3B1 (ATCC 51449) or additionally, followed by 3 doses of H. pylori strain SS1 2 weeks later. The respective infection groups were designated as follows: mono-H. pylori-infected mice, mono-H. muridarum-infected mice, mono-H. hepaticus-infected mice, mice infected with H. muridarum and H. pylori (HmHp mice), and mice infected with H. hepaticus and H. pylori (HhHp mice). At 6 and 11 months postinoculation (MPI) with H. pylori, 15 mice from each group were euthanized with CO2 and necropsied. The sham-dosed control mice, mono-H. pylori-infected mice, mono-H. bilis-infected mice, and mice infected with H. bilis and H. pylori (HbHp mice) described in our previous report were also used as negative and positive controls for select assays, since they were age, gender, and time point matched with the mice used in the present study (28). These two studies were also conducted concurrently under identical husbandry conditions.
Necropsy and histopathology.
At necropsy, stomach samples from the lesser curvature extending from the squamous forestomach through the duodenum were collected and processed as described previously (15, 39). Tissues were graded by a comparative pathologist (S. Muthupalani) blinded to sample identity for inflammation, epithelial defects, atrophy, hyperplasia, pseudopyloric metaplasia, dysplasia, hyalinosis, and mucous metaplasia, as defined elsewhere (15, 39). Gastric lesions were scored on an ascending scale from 0 to 4 using criteria previously described (13, 39). A gastric histologic activity index (HAI) was generated by combining scores for all criteria except hyalinosis and mucous metaplasia, which may develop irrespective of helicobacter infection.
Q-PCR for H. pylori SS1, H. hepaticus, and H. muridarum.
To quantify colonization levels of H. pylori SS1, H. hepaticus, and H. muridarum within the gastric mucosa and H. hepaticus and H. muridarum in cecal tissue, a real-time quantitative PCR (Q-PCR) assay was utilized (15, 32). A standard curve was generated using serial 10-fold dilutions of the respective helicobacter genomic DNA, representing 1 × 106 to 10 genome copies. The copy numbers for H. pylori SS1 and H. hepaticus were calculated based on an average H. pylori genome size of 1.66 Mb of two sequenced isolates and the H. hepaticus 3B1 genome size of 1.8 Mb, respectively (1, 45, 47); the genome size of H. muridarum was estimated to be 1.73 Mb, averaged from the genome sizes of two H. pylori isolates and H. hepaticus 3B1 (1, 47). Primers and probes for quantifying H. pylori and H. hepaticus were previously described (16, 32). A forward primer (5′-AAGAGTGCGCACCCGGGCTAAT-3′) and a reverse primer (5′-CGTTAGCTGCATTACTGCCCTGTC-3′), which hybridize nucleotides 529 to 550 and nucleotides 800 to 823 of the H. muridarum strain ST1 16S rRNA gene (M08205), respectively, were evaluated and selected for measuring colonization levels of H. muridarum. All Q-PCR assays were performed in the 7500 Fast detection system (Applied Biosystems, Carlsbad, CA). Genome copy numbers of H. pylori, H. hepaticus, and H. muridarum were expressed per microgram of murine chromosomal DNA and measured by Q-PCR using a mammalian 18S rRNA gene-based primer and probe mixture (Applied Biosystems) as described previously (17, 49).
Gastric cytokines.
Total RNA from murine stomachs was prepared using TRIzol reagents by following the supplier's instructions (Invitrogen, Carlsbad, CA). The RNA samples were further purified by removing residual DNA using the RNeasy kit (Qiagen, Valencia, CA). cDNA from gastric mRNA (2 μg) was reverse transcribed using the High Capacity cDNA archive kit by following the supplier's instructions (Applied Biosystems, Foster City, CA). Using the 7500 Fast real-time PCR system (Applied Biosystems), mRNA expression of mouse genes involved in innate and adaptive immunity was measured using RT2 Profiler PCR arrays (Super Array Bioscience Corporation, Frederick, MD). For this assay, RNA from 3 mice of each group at 11 MPI was selected for analyses, and selection was based on the HAI scores closest to median scores for each infection group. In addition, gastric RNA from 3 control mice in both the noninfected and mono-H. pylori-infected groups at 11 MPI, described in our previous study (28), were also analyzed. Additionally, mRNA levels of the proinflammatory Th1 cytokines gamma interferon (Ifn-γ), tumor necrosis factor-alpha (Tnf-α), and interlukin-1beta (Il-1β) were measured in gastric tissues of all the mice at 6 MPI, whereas gastric mRNA levels of anti-inflammatory Th2 cytokines Il-10 and Il-13 as well as Foxp3 were examined at both 6 and 11 MPI. Furthermore, transcript levels of proinflammatory gastric Il-17A were compared for all mice from this and prior (the sham control, mono-H. pylori, HbHp, and mono-H. bilis groups) studies at both 6 and 11 MPI (28). All the target genes were normalized to the endogenous control glyceraldehyde-3-phosphate dehydrogenase (Gapdh) mRNA and expressed as fold changes in reference to sham-dosed control mice using the comparative threshold cycle (CT) method (Applied Biosystems User Bulletin No. 2).
Immunohistochemistry for Foxp3+ cells.
As described previously, Foxp3 immunochemistry was performed on paraffin-embedded gastric tissues using Foxp3 antibody (FJK-16S; eBioscience, San Diego, CA) (28). Three mice with HAI scores closest to median scores from each group at 11 MPI were selected for this assay. The number of cells expressing nuclear Foxp3 is reported as the number of Foxp3+ cells per 10 fields (200×) per stomach.
Statistics.
Gastric HAI scores were compared across groups by the Kruskal-Wallis one-way analysis of variance with Dunn's posttest and between groups by the Mann-Whitney U test using Prism software (GraphPad, San Diego, CA). Data on the colonization levels of Helicobacter species and cytokine mRNA levels in the tissues were analyzed using the two-tailed Student t test. P values of <0.05 were considered significant.
RESULTS
Coinfection with H. muridarum but not H. hepaticus attenuated H. pylori-induced gastritis and gastric premalignant lesions.
We previously demonstrated that concurrent infection of H. pylori and H. bilis attenuated H. pylori-induced gastric disease (28). In this study, we tested whether this effect is also attributable to other EHS. C57BL/6 mice infected with H. pylori exhibited moderate gastritis at 6 MPI and severe gastritis with early dysplasia at 11 MPI, whereas there was no infection-associated histopathological alterations of significance observed in sham control mice (28). Lesions in mono-H. pylori mice were characterized by a multifocal to coalescing mucosal and submucosal inflammatory process comprised predominantly of a mixed population of lymphocytes and granulocytes (neutrophils, eosinophils). Inflammatory cell infiltrates were associated with mild to moderate surface epithelial tethering/erosions and glandular ectasia, with occasional crypt abscesses and a moderate degree of oxyntic atrophy, hyperplasia, pseudopyloric metaplasia, and dysplasia (Fig. 1 a and b). Mono-H. pylori-infected mice exhibited mucous metaplasia of the oxyntic mucosa, which contributed to parietal cell atrophy. However, this lesion was not included in the gastric HAI because this is not helicobacter specific (13, 39). As expected, monoinfection with H. muridarum or H. hepaticus did not produce overt gastritis at either time point (Fig. 1c and d, H. muridarum and H. hepaticus, 11 MPI) nor did persistent colonization of the lower bowel with H. muridarum or H. hepaticus elicit lower bowel inflammation (data not shown). Mice colonized with HmHp developed a significantly lower gastric HAI at both 6 and 11 MPI than mice infected with H. pylori alone (n = 15, P < 0.0001 for both time points) (Fig. 1e and f), in a manner similar to that previously observed in HbHp mice at 11 MPI (28) (P = 0.023) (data not shown). Coinfection in HhHp mice did not attenuate gastric pathology at 6 MPI, but rather, this dual infection significantly increased gastric HAI scores compared to mono-H. pylori infection at 6 MPI (Fig. 1B) (P = 0.01). There was no significant difference in the HAI scores between HhHp and mono-H. pylori-infected mice at 11 MPI (Fig. 2) (P = 0.81).
Fig. 1.

Representative hematoxylin and eosin (H&E) images showing the salient histopathological features observed in the stomach at 6 MPI with H. pylori (a), at 11 MPI with H. pylori (b), at 11 MPI with H. muridarum (c), at 11 MPI with H. hepaticus (d), at 6 MPI coinfected with HmHp (e), at 11 MPI coinfected with HmHp (f), at 6 MPI coinfected with HhHp (g), and at 11 MPI coinfected with HhHp (h). Mono-H. pylori-infected (a, b) and HhHp (g, h) mice at both time points showed significant gastric pathology characterized by prominent mucosal and submucosal inflammation, with associated multifocal surface erosions, glandular ectasia, oxyntic atrophy, foveolar and glandular hyperplasia, and early dysplastic changes, as evident by glandular architectural distortion, loss of columnar orientation, and mild cellular atypia. The loss of normal oxyntic glands was frequently a result of metaplastic changes that included both mucous metaplasia and pseudopyloric metaplasia. All lesional parameters were noticeably of greater intensity in HhHp mice at 6 MPI (g) than in H. pylori at 6 MPI (h). H. hepaticus (d) and H. muridarum (c) monoinfection did not induce any significant inflammation or other associated changes, and the mucosal morphology was comparable to that observed in uninfected controls (not shown). (e) At 6 MPI, HmHp mouse stomach showed only mild mucosal inflammation, variable foveolar hyperplasia, mild oxyntic loss, and metaplasia. (f) At 11 MPI, HmHp mouse stomach had slightly increased mild to moderate overall inflammation, mild oxyntic loss, mild hyperplasia, and variable metaplasia but no dysplasia, all of which were appreciably of lesser intensity than those observed in HhHp and mono-H. pylori-infected groups. Bar, 75 μM (c to f), 150 μM (a, b, g, h).
Fig. 2.

Gastric histologic activity index (HAI). For 6 to 11 months, gastric tissues from C57BL/6 mice infected with H. pylori, H. muridarum, or H. hepaticus or from HmHp or HhHp mice (n = 13 to 15 for all groups) were graded for inflammation, epithelial defects, atrophy, hyperplasia, pseudopyloric metaplasia, dysplasia, hyalinosis, and mucous metaplasia. A gastric histologic activity index was generated by combining scores for all criteria except hyalinosis and mucous metaplasia, which may develop irrespective of helicobacter infection. H. muridarum attenuated H. pylori gastritis at 6 and 11 MPI, whereas H. hepaticus enhanced H. pylori gastritis at 6 MPI.
H. pylori-induced upregulation of gastric proinflammatory Th1 cytokine mRNAs was attenuated in mice coinfected with H. muridarum and H. hepaticus.
We previously demonstrated that H. bilis-mediated attenuation of H. pylori-induced gastric pathology in dually infected mice was associated with downregulation of gastric mRNA levels of proinflammatory Th1 cytokines Ifn-γ, Tnf-α, and Il-1β compared to mono-H. pylori-infected mice at 6 and 11 MPI (28). Thus, mRNA expression of these cytokines was examined in all mice at 6 MPI. The levels of gastric Ifn-γ, Tnf-α, and Il-1β mRNAs were significantly decreased in HmHp mice compared to mono-H. pylori-infected (P < 0.0001), HhHp (P < 0.0001) (Fig. 3), and HbHp (P < 0.0003) (data not shown) mice. Even though HhHp mice developed more severe gastric pathology than mono-H. pylori-infected mice, mRNA levels of gastric Ifn-γ and Tnf-α were significantly lower in the HhHp mice than in mono-H. pylori-infected mice (P < 0.0001) (Fig. 3); mRNA levels of Il-1β also trended lower (P = 0.076). Compared to sham controls, the HmHp mice expressed similar baseline mRNA levels for gastric Ifn-γ and Il-1β and lower mRNA levels of Tnf-α (P = 0.015). mRNA expression of gastric Ifn-γ and Tnf-α was significantly decreased in mono-H. muridarum-infected mice (P < 0.0001), and all 3 assayed Th1 cytokines in mono-H. hepaticus-infected mice were also significantly decreased compared to those of the sham controls (P < 0.0003) (Fig. 3).
Fig. 3.
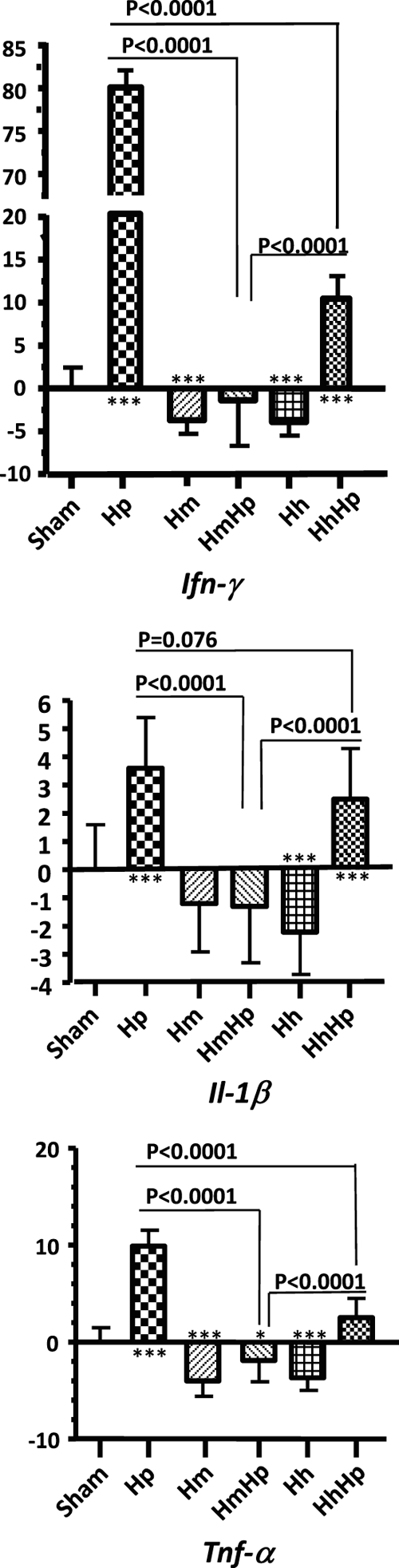
Gastric Th1 cytokine mRNA expression levels. For 6 to 11 months, gastric tissues (n = 13 to 15 per group) from mice infected with H. pylori, H. muridarum, or H. hepaticus or from HmHp or HhHp mice were evaluated by Q-PCR for expression levels of mRNA for proinflammatory cytokines, all normalized to the expression of the housekeeping gene Gapdh. The y axes represent the mean fold changes (± standard deviations) of the mRNA levels in reference to uninfected controls. HmHp or HhHp mice expressed lower levels of proinflammatory mediators Ifn-γ, Il1-β, and Tnf-α at 6 MPI. Numbers on the y axes represent fold changes of gastric mRNA levels compared to those of sham controls. P values compared to those of the sham controls: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To evaluate the expression of innate and adaptive immune response genes to helicobacter infection, we measured mRNA levels of 84 relevant genes in 3 mice from each group using a SuperArray (SABiosciences, Frederick, MD) at 11 MPI (Table 1). The data indicate that mono-H. pylori and HhHp infection upregulated transcription of gastric proinflammatory genes involved in innate and adaptive immunity compared to that of sham control and HmHp infection. Notably, transcript levels for gastric chemokines CXCL1 (mouse homolog of human IL-8) and CXCL2 in mono-H. pylori-infected and HhHp mice were increased by more than 10-fold compared to sham controls and more than 2-fold compared to HmHp mice (Table 1). mRNA levels were elevated for genes encoding all three chains (δ, ε, γ) of CD3, CD4, CD28, and proinflammatory cytokines Ifn-γ, Tnf-α, Il-1β, and Il-21, all of which reflect T cell activation and cell-mediated immunity in response to H. pylori or HhHp infection compared to controls (P < 0.05). However, there were no significant changes in these respective mRNA levels in the gastric tissues of HmHp mice, mono-H. muridarum-infected mice or mono-H. hepaticus-infected mice (P > 0.2). The enhanced expression of Ifn-γ, Tnf-α, and Il-1β mRNA in mono-H. pylori-infected or HhHp mice was consistent with severe gastric inflammation noted previously in H. pylori-infected patients, mice, and gerbils experimentally infected with gastric helicobacters (7, 11, 13, 15, 28, 52).
Table 1.
mRNA levels of innate and adaptive immunity genes in gastric tissue of Helicobacter-infected C57BL/6 mice
| Gene (description)b | mRNA levela |
||||
|---|---|---|---|---|---|
| H. pylori-infected mice | H. muridarum-infected mice | HmHp mice | H. hepaticus-infected mice | HhHp mice | |
| Cxcl5 (chemokine [C-X-C motif] ligand 5) | 147.2 | 7.7 | 139.7 | 45.7 | 595.8 |
| Ifn-γ (gamma interferon) | 56.4 | 1.6 | 2.4 | −1.2 | 33.6 |
| Socs1 (suppressor of cytokine signaling 1) | 17.0 | −2.4 | 1.3 | 1.4 | 11.6 |
| Cxcl1 (chemokine [C-X-C motif] ligand 1) | 11.3 | 2.2 | 4.1 | 1.5 | 22.0 |
| Cd3d (CD3 antigen, δ polypeptide) | 11.1 | 1.1 | 1.4 | −1.4 | 6.8 |
| Cxcl2 (chemokine [C-X-C motif] ligand 2) | 10.8 | 1.7 | 1.8 | 1.5 | 11.6 |
| Cd40lg (CD40 ligand) | 10.4 | 1.6 | 1.7 | −1.3 | 7.9 |
| Cd3γ (CD3 antigen, γ polypeptide) | 9.7 | 1.7 | 1.5 | −1.1 | 8.2 |
| Cd3e (CD3 antigen, ε polypeptide) | 9.5 | 1.2 | 1.0 | −1.5 | 6.9 |
| Cd8a (CD8 antigen, α chain) | 8.6 | −1.3 | −1.2 | −2.3 | 4.4 |
| Tnf-α (tumor necrosis factor alpha) | 8.1 | 1.2 | 1.3 | 1.2 | 10.1 |
| Il-21 (interleukin 21) | 6.8 | −2.7 | −1.0 | −2.1 | 4.4 |
| Il-12b (interleukin 12b) | 6.3 | 2.2 | 1.2 | −1.1 | 3.2 |
| Clec7a (C-type lectin domain family 7, member a) | 5.7 | 1.8 | 1.5 | −1.0 | 5.1 |
| Cd28 (CD28 antigen) | 5.7 | 2.4 | 2.2 | 1.1 | 4.9 |
| Cd2 (CD2 antigen) | 4.5 | 1.5 | 1.3 | −1.1 | 3.8 |
| Ccl20 (chemokine [C-C motif] ligand 20) | 4.4 | 1.4 | 11.3 | −1.0 | 3.6 |
| Icos (inducible T cell costimulator) | 4.3 | 1.9 | 1.3 | 1.2 | 4.2 |
| Cd4 (CD4 antigen) | 4.2 | 1.0 | −1.2 | −1.4 | 2.8 |
| Il-1β (interleukin 1β) | 3.8 | 1.2 | 1.5 | −1.4 | 3.2 |
| Il-7r (interleukin 7 receptor) | 3.7 | 1.9 | 1.0 | −1.2 | 2.7 |
| Il-12r-β2 (interleukin 12 receptor, β2) | 3.6 | 1.4 | −1.2 | 1.1 | 2.4 |
| Ccl1 (chemokine [C-C motif] ligand 1) | 3.5 | 1.6 | −1.0 | −1.7 | 3.9 |
| Tbx21 (T-box 21) | 3.0 | −2.7 | −2.4 | −1.3 | 2.2 |
| Mmp13 (matrix metallopeptidase 13) | 2.7 | 1.2 | 1.2 | 1.1 | 3.5 |
| Il-12r-β1 (interleukin 12 receptor, β1) | 2.6 | −1.7 | −1.9 | −1.3 | 1.9 |
| Icam1 (intercellular adhesion molecule 1) | 2.4 | 1.6 | −1.0 | −1.1 | 2.2 |
| Il-2 (interleukin 2) | 2.4 | 1.1 | −2.3 | −1.5 | −1.2 |
| Ccl2 (chemokine [C-C motif] ligand 2) | 2.2 | 2.6 | 1.0 | −1.0 | 1.6 |
| Il-25 (interleukin 25) | 2.2 | −1.2 | −1.0 | 1.1 | 2.8 |
| Il-23r (interleukin 23 receptor) | 2.1 | 5.0 | 3.7 | 2.1 | 2.5 |
| Csf2 (granulocyte-macrophage colony-stimulating factor 2) | 2.0 | 2.9 | 1.3 | 1.5 | 1.9 |
Fold change of gastric mRNA levels of the respective genes in the infected groups compared to that of sham controls.
Genes listed because mRNA levels increased by ≥2-fold in the stomachs of mono-H. pylori-infected C57BL/6 mice compared to those of sham control mice.
mRNA levels of gastric Il-17A were positively correlated with the increased severity of H. pylori-induced gastric lesions.
It was previously reported that expression of Il-17A by proinflammatory Th17 cells was significantly increased in H. pylori-colonized human gastric mucosa (30). Experimental H. pylori infection in mice and gerbils also upregulated mRNA levels of gastric Il-17A (42, 46). We measured and compared mRNA levels of gastric Il-17A among the infection groups in this study as well as in samples from our previous study (sham control, mono-H. pylori-infected, HbHp, and H. bilis-infected mice) (Fig. 4 A). At both 6 and 11 MPI, all mice infected with H. pylori regardless of coinfection status expressed significantly higher levels of gastric Il-17A mRNA than sham controls (P < 0.0001); there was no significant difference in gastric Il-17A mRNA levels among the sham control, mono-H. muridarum-infected and mono-H. bilis-infected groups (P > 0.2). However, the mono-H. hepaticus-infected mice expressed significantly higher mRNA levels of gastric Il-17A than sham control, mono-H. muridarum-infected, or mono-H. bilis-infected mice at both 6 and 11 MPI (P < 0.001) (Fig. 3A). The mice coinfected with HhHp expressed significantly higher mRNA levels of gastric Il-17A compared to the mono-H. pylori-infected (P < 0.05), HmHp (P < 0.001), or HbHp (P < 0.05) mice at 6 MPI. There was a higher level of gastric Il-17A mRNA in the mono-H. pylori-infected mice than in the HmHp mice (P < 0.01) at 6 MPI. At 11 MPI, levels of gastric Il-17A mRNA in the HhHp mice were significantly higher than those in the HbHp mice (P = 0.028), trended higher compared to those in the HmHp mice (P = 0.059), and were similar to those in the mono-H. pylori-infected mice (P = 0.2). Interestingly, correlation analysis indicated that gastric Il-17A mRNA levels were significantly correlated with HAI scores in all groups (P < 0.0018; r2 > 0.81) (Fig. 4B).
Fig. 4.

Gastric Il-17A mRNA levels and correlation with the severity of H. pylori-induced gastric pathology. (A) For 6 to 11 months, Il-17A mRNA levels in gastric tissues from mice (n = 13 to 15 per group) infected with H. pylori, H. muridarum, or H. hepaticus or from HmHp or HhHp mice were evaluated by Q-PCR. The gastric tissues from mice infected with H. bilis alone or from HbHp mice, as described previously (28), were also included in this assay, since these mice were matched with those used in the present study for age, gender, and infection paradigm and this previously published study was conducted at the same time the current study was performed. Expression levels of gastric Il-17A mRNA were normalized to mRNA levels of the housekeeping gene Gapdh. The y axis represents the mean fold changes (± standard deviations) of the mRNA levels in reference to uninfected controls (C). (B) Linear regression between gastric Il-17A mRNA levels (fold change in reference to the sham controls; x axis) and gastric histologic activity index (HAI; y axis) for all infection and control groups shown in panel A. P values compared to those of the sham controls: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Higher levels of gastric Foxp3 and Il-10 mRNA as well as Foxp3+ cells were associated with more severe gastric pathology.
Foxp3 encodes a transcription factor essential for differential development of anti-inflammatory natural regulatory T cells (10, 22, 40). Increased numbers of CD4+ CD25+ Foxp3+ regulatory T (TREG) cells were present in the gastric tissues of H. pylori-positive patients and mice infected experimentally with H. pylori (19, 36). We previously documented that the mono-H. pylori-infected mice had more Foxp3+ cells and higher mRNA levels of Foxp3 in gastric tissue than HbHp mice at 11 MPI (28). In this study, mono-H. pylori-infected mice contained significantly lower mRNA levels of gastric Foxp3 than HhHp mice (P = 0.0005) at 6 MPI but significantly higher levels than HmHp mice (P < 0.05) at 11 MPI (Fig. 5 A). There was no significant difference in gastric Foxp3 mRNA levels between the mono-H. pylori-infected and HmHp mice at 6 MPI (P = 0.439) and between the mono-H. pylori-infected and HhHp mice at 11 MPI (P = 0.628). The gastric Foxp3 mRNA levels in the HhHp mice were significantly higher than those in the HmHp mice at both time points (P < 0.05). All mice infected with H. pylori contained higher mRNA levels of gastric Foxp3 than the sham controls (P < 0.05) at both time points. These data were positively associated with the severity of H. pylori-induced gastric lesions in the respective groups (Fig. 2). Consistent with the mRNA levels of gastric Foxp3, all H. pylori-infected groups had elevated numbers of gastric Fox3+ cells, particularly in mono-H. pylori-infected and HhHp mice, compared to HmHp mice (Fig. 5B).
Fig. 5.

(A, B) mRNA levels of anti-inflammatory genes (A) and the number of gastric Foxp3+ cells (B). (A) For both 6 and 11 MPI, gastric Foxp3, Il-10, and Il-13 mRNA levels in the stomachs of mice (n = 13 to 15 per group) infected with H. pylori, H. muridarum, or H. hepaticus or of HmHp or HhHp mice were evaluated by Q-PCR and normalized to mRNA levels of the housekeeping gene Gapdh. The y axes represent the mean fold changes (± standard deviations) of the mRNA levels in reference to uninfected controls. (B) Gastric Foxp3+ cells are presented as the total number of Foxp3+ cells per 10 fields (200×) per stomach (n = 3 for each group at 11 MPI). P values compared to those of the sham controls: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Anti-inflammatory Th2 cytokines Il-10 and Il-13 were also measured using Q-PCR. Levels of gastric Il-10 mRNA were significantly higher in mono-H. pylori-infected mice at both time points and in HhHp mice at 11 MPI than in HmHp mice or sham controls (Fig. 5A). There was no significant difference in gastric Il-10 mRNA levels between mono-H. pylori-infected mice and HhHp mice. Higher levels of gastric Il-13 mRNA were detected in mono-H. pylori-infected and HhHp mice than in sham controls at 6 MPI, but no significant difference was noted among any H. pylori-infected groups at 6 MPI (Fig. 5A). At 11 MPI, there were increased levels of gastric Il-13 transcripts in HhHp mice compared to HmHp mice (P < 0.0001).
Interestingly, despite the lack of overt gastric or intestinal pathology in the mono-EHS-infected mice, mRNA levels of gastric Foxp3 in the mono-H. muridarum-infected mice were significantly higher than those in sham controls (P < 0.05) at both time points. In contrast, there was no significant difference in gastric Foxp3 mRNA levels between the mono-H. hepaticus-infected mice and the sham controls at either time point (P > 0.5). In addition, there were higher mRNA levels of gastric Il-10 and more gastric Foxp3+ cells in mono-H. muridarum-infected mice than in mono-H. hepaticus-infected mice at 11 MPI (P < 0.05) Fig. 5A and B).
Colonization of gastric H. pylori was enhanced in mice coinfected with H. hepaticus and H. muridarum.
HmHp mice had significantly higher colonization levels of H. pylori in the stomach than mono-H. pylori-infected mice at 6 and 11 MPI (P < 0.01). Colonization levels of H. pylori were higher by ∼280-fold at 6 MPI (P < 0.05) and by approximately 40-fold at 11 MPI in the HhHp mice than in the mono-H. pylori-infected mice (Fig. 6). Lack of statistical significance for H. pylori colonization levels at 11 MPI (P = 0.14) may be attributed to exceptionally higher numbers of gastric H. pylori in two mice of the mono-H. pylori-infected group. Between the dually infected groups, coinfection with H. muridarum significantly increased H. pylori levels at 6 MPI (P < 0.02) compared to coinfection with H. hepaticus, although there were no significant differences in H. pylori colonization levels at 11 MPI (P = 0.49) (Fig. 6).
Fig. 6.

Quantitation of gastric H. pylori. Copy numbers of the H. pylori SS1 genome were estimated by Q-PCR of gastric samples from mice (n = 13 to 15 per group) infected with H. pylori or dually infected with either H. muridarum and H. pylori or H. hepaticus and H. pylori for 6 to 11 months. P values compared to those of the mono-H. pylori-infected group: #, P < 0.05; ##, P < 0.01.
H. muridarum and H. hepaticus established persistent infection in the lower bowel of C57BL/6 mice.
All mice inoculated with H. muridarum or H. hepaticus became colonized in the cecum (data not shown). The mean colonization levels of H. hepaticus at the cecal-colic junction were at approximately 2 × 107 for all groups (P > 0.31), except for mono-H. hepaticus-infected mice at 11 MPI (2 × 106), in which colonization levels of H. hepaticus were significantly lower than in HhHp mice at either time point (P < 0.0001) and in mono-H. hepaticus-infected mice at 6 MPI (P < 0.0001).
Mean levels of H. muridarum colonization in the cecum were similar in mono-H. muridarum-infected and HmHp mice (∼2 × 106 to 5 × 106). There were significantly lower levels of cecal H. hepaticus in the mono-H. hepaticus-infected mice at 11 MPI than at 6 MPI (P < 0.05). The remaining three groups had similar levels of cecal H. hepaticus (P ≥ 0.2).
Gastric colonization of H. muridarum and H. hepaticus increased over time in coinfected mice.
Although the lower bowel is the primary niche for colonization of H. muridarum and H. hepaticus, both EHS established persistent infection in the stomach (data not shown). The percentage of dually infected mice positive for coinfected EHS in the stomach increased from 6 MPI to 11 MPI, as follows: from 43% (6/14) to 73% (11/15) for H. hepaticus and from 57% to 73% for H. muridarum. In contrast, the percentage of gastric H. muridarum or H. hepaticus in the mono-EHS-infected mice decreased from 6 MPI to 11 MPI, as follows: from 80% to 61% for H. hepaticus and from 60% to 33% for H. muridarum. Mean numbers of gastric H. hepaticus or H. muridarum bacteria were comparable among the groups except for the HmHp group at 11 MPI, in which 6 of 11 gastric H. muridarum-positive mice contained relatively higher levels of gastric H. muridarum.
DISCUSSION
In this study, we demonstrated that H. muridarum significantly attenuated H. pylori-induced gastric pathology in dually infected C57BL/6 mice. In addition, coinfection with H. muridarum had more profound attenuation of H. pylori-associated gastric pathology than that previously reported for mice coinfected with the EHS H. bilis (28). In contrast, coinfection with H. hepaticus did not suppress but rather promoted gastric disease caused by H. pylori at 6 MPI. Despite H. muridarum or H. hepaticus colonization of the murine stomach with H. muridarum or H. hepaticus in a subset of the dually infected or monoinfected mice, levels of gastric EHS colonization did not correlate with the severity of gastric pathology, suggesting that the modulatory effect of EHS coinfection on H. pylori-induced gastric disease resulted from intestinal rather than gastric colonization.
Intriguingly, despite that gastric pathology in HhHp mice at 6 MPI was more severe and at 11 MPI was similar to that in mice infected with H. pylori only, HhHp mice contained lower mRNA levels of gastric Th1 cytokines Tnf-α, Ifn-γ, and Il-1β than mono-H. pylori-infected mice. In contrast, higher mRNA levels of gastric Il-17A in HhHp mice than in mono-H. pylori-infected mice were noted. Previous studies have established a role of a proinflammatory Th17 pathway in the development of H. pylori-induced gastric disease in mouse and gerbil models (9, 42, 46). Our results indicate that the H. hepaticus-associated accentuation of H. pylori-induced gastric pathology was not mediated by upregulation of the Th1 response but instead resulted from a robust Th17 response to HhHp infection. It is likely that prior H. hepaticus infection potentiates a gastric Th17 response to subsequent H. pylori infection. This is supported by significantly higher mRNA levels of gastric Il-17A detected in mono-H. hepaticus-infected mice than those detected in sham control, mono-H. muridarum-infected, or mono-H. bilis-infected mice. Also, suppression of Th1 cytokine mRNAs in HhHp mice compared to that in mono-H. pylori-infected mice, particularly a lower Ifn-γ level, could also contribute in part to the upregulation of gastric Th17 responses, because Ifn-γ has an inhibitory effect on the Th17 pathway (18, 23). However, other immunological factors may have played a pivotal role in enhancing gastric Th17 responses, given that mRNA levels of gastric Ifn-γ were similar in HbHp mice or significantly lower in HmHp mice than in HhHp mice. It is possible that intestinal H. hepaticus-initiated memory Th17 cells migrated from the lower bowel or mesenteric lymph node to the stomach, where they produced a robust Th17 response following H. pylori infection.
Our results indicate that elevated Th17 responses were positively correlated with the severity of H. pylori-induced gastric pathology and also with higher colonization levels of gastric H. pylori in HhHp mice than in mice monoinfected with H. pylori. These findings are consistent with recent data showing that overexpression of IL-17A in mouse stomachs elevated gastric Th17 responses to H. pylori infection and was associated with increased colonization of gastric H. pylori as well as more severe H. pylori-induced gastric pathology in female BALB/c and C57BL/6 mice (41). Although it has been reported that Ifn-γ was essential for clearing H. felis in C57BL/6 mice (40), two recent studies showed that Th17 cells were important in reducing H. pylori colonization in mice. Kao et al. (25) reported that colonization levels of H. pylori were increased when Th17 responses were suppressed by H. pylori-specific dendritic cell-mediated regulatory T (TREG) cells at 2 weeks postinfection (WPI); however, H. pylori colonization was less impacted at 6 WPI, suggesting that the Th17 cell-mediated clearing of H. pylori may be most evident early during infection. Similarly, vaccination of female C57BL/6 mice with H. pylori SS1 cell lysate, followed by challenge with the same H. pylori strain, elicited strong Th17 responses, gastric inflammation, and significantly reduced H. pylori levels compared to those in unimmunized mice by 13 days postinfection (9). Discrepancy on the impact of Th17 responses on H. pylori colonization could partially result from different experimental designs between our study and previous studies, as follows: (i) coinfection with live EHS in this study versus either adoptive transfer of a large number of H. pylori-stimulated dendritic cells (106) (25) or immunization with a cell lysate of H. pylori (9) and (ii) long-term infection duration (6 to 11 MPI) in this study compared to much shorter study periods (∼2 weeks) in other reports (9, 25).
Natural regulatory T (Foxp3+ TREG) cells suppress host inflammatory responses to infectious agents and thereby prevent tissue injury as well as promote physiological homeostasis of host immunity (8). Increased numbers of Foxp3+ cells were located in the inflamed gastric tissues of H. pylori-positive patients and H. pylori-infected mice compared to uninfected controls; depletion of TREG cells by treatment with monoclonal C61 antibody in H. pylori-infected mice led to enhanced expression of gastric proinflammatory cytokines as well as more severe gastritis (19, 36). CD4+ CD25+ TREG cells from H. pylori-positive patients have higher potency in the suppression of memory T cell responses (Ifn-γ production and proliferation) to in vitro stimulation with dendritic cells pulsed with H. pylori SS1 membrane proteins than TREG cells from H. pylori-negative patients (29, 37). In Rag2−/− mice lacking T and B lymphocytes, we previously reported that H. pylori-induced gastritis was suppressed by adoptive transfer of TREG cells harvested from IL-10-competent C57BL/6 donor mice but not from Il-10−/− mice, demonstrating that IL-10-dependent TREG cells play a crucial role in suppressing H. pylori-induced gastric disease (27). Consistent with these previous findings, our data show that mRNA levels of gastric Fopx3 and Il-10 as well as the number of gastric Foxp3+ cells were significantly higher in the mono-H. pylori-infected or HhHp mice than in sham controls or HmHp mice. Furthermore, HbHp mice with attenuated gastritis had fewer Foxp3+ cells and lower levels of gastric Foxp3 mRNA than mice infected with H. pylori alone (28). We propose that coinfection with H. bilis or H. muridarum sensitizes TREG cells, which are then more efficient in suppressing H. pylori-induced proinflammatory responses than nonsensitized TREG cells. This hypothesis is supported by a previous finding that splenocytes transferred from H. pylori-infected C57BL/6 mice, compared to naïve splenocytes, were more efficient in attenuating H. pylori-induced gastritis and premalignant lesions in SCID mice (35). In addition, we previously demonstrated that sensitized TREG cells from H. hepaticus-infected C57BL/6 mice were more potent in inhibiting intestinal inflammation in ApcMin/+ mice (38). In the current study, higher mRNA levels of gastric Foxp3 and Il-10 as well as larger numbers of gastric Foxp3+ cells were also noted in mono-H. muridarum-infected mice than in mono-H. hepaticus-infected mice or sham controls. We hypothesize that TREG cells are sensitized by specific antigens shared between H. muridarum or H. bilis and H. pylori, probably by a process coined “heterologous immunity,” in which protective immunity is elicited against subsequent infection with a different agent through memory T cells sensitized by shared antigens from a previous infection (48). Further investigations are needed using adoptive transfer of TREG cells to identify which EHS antigens are required to sensitize TREG cells during coinfection with an EHS and H. pylori.
Taken together, we propose a conceptual model showing that attenuation of H. pylori-induced gastric disease by EHS coinfection is EHS dependent (Fig. 7). In this model, the interaction between bacteria colonizing the lower bowel can either attenuate or promote H. pylori-induced gastritis. Additional studies using this model with identification of specific bacterial antigens, mechanisms involving antigen presentation, sensitized T cell trafficking, and disruption of key proinflammatory pathways will help delineate how the interaction among pathogenic or commensal microbes in the gastrointestinal tract may affect human disease.
Fig. 7.

Proposed working model underlying EHS-dependent effects on H. pylori-induced gastric pathology in C57BL/6 mice. H. muridarum/H. bilis-sensitized TREG (sTREG) cells with anti-inflammatory properties migrate to the stomach, where they dampen host proinflammatory responses to subsequent H. pylori infection by decreasing expression of proinflammatory cytokines, including Ifn-γ, Tnf-α, and Il-1β, as well as Il-17A. This downregulation, despite higher colonization levels of gastric H. pylori, attenuates H. pylori-induced gastritis. In contrast, H. hepaticus may be less potent in sensitizing TREG cells and more potent in stimulating Th17 cells (HhTh17), which then also migrate to the stomach upon H. pylori infection. H. hepaticus sTREG cells may moderately downregulate proinflammatory Th1 cytokines, such as Ifn-γ, which play an important role in helicobacter clearance (40), thereby permitting higher colonization levels of H. pylori in HhHp mice than in mice infected with H. pylori only. H. hepaticus Th17 cells in addition to relatively lower levels of Ifn-γ would markedly increase signals of the Th17 pathway and thereby lead to a more severe H. pylori-induced gastric pathology in HhHp mice than in mono-H. pylori-infected mice. Our data show that alteration of these H. pylori-induced proinflammatory cytokines by coinfection with an EHS was more evident at 6 MPI than at 11 MPI, indicating that the interplay among the Th1, TREG, and Th17 pathways occurred early in infection, potentially prior to development of overt gastritis. iPI, EHS-induced proinflammatory responses; PI, proinflammatory responses; AI, anti-inflammatory responses. Upward and downward arrows depict up- and downregulation of associated pathways compared to mono-H. pylori infection, respectively.
ACKNOWLEDGMENTS
We thank Elaine Robbins for technical support in the preparation of the figures.
This work was supported by the following NIH grants given to J.G.F.: R01CA67529, R01AI51404, T32RR07036, P30 ES02109, and P01CA028842.
Footnotes
Published ahead of print on 25 July 2011.
REFERENCES
- 1. Alm R. A., et al. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176–180 [DOI] [PubMed] [Google Scholar]
- 2. Amieva M. R., El-Omar E. M. 2008. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology 134:306–323 [DOI] [PubMed] [Google Scholar]
- 3. Anonymous. 1994. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. IARC Monogr. Eval. Carcinog. Risks Hum. 61:1–241 [PMC free article] [PubMed] [Google Scholar]
- 4. Bravo L. E., van Doom L. J., Realpe J. L., Correa P. 2002. Virulence-associated genotypes of Helicobacter pylori: do they explain the African enigma? Am. J. Gastroenterol. 97:2839–2842 [DOI] [PubMed] [Google Scholar]
- 5. Chaouche-Drider N., et al. 2009. A commensal Helicobacter sp. of the rodent intestinal flora activates TLR2 and NOD1 responses in epithelial cells. PLoS One 4:e5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheung T. K., Wong B. C. 2008. Treatment of Helicobacter pylori and prevention of gastric cancer. J. Dig. Dis. 9:8–13 [DOI] [PubMed] [Google Scholar]
- 7. Crabtree J. E., et al. 2004. Gastric mucosal cytokine and epithelial cell responses to Helicobacter pylori infection in Mongolian gerbils. J. Pathol. 202:197–207 [DOI] [PubMed] [Google Scholar]
- 8. Curotto de Lafaille M. A., Lafaille J. J. 2009. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity 30:626–635 [DOI] [PubMed] [Google Scholar]
- 9. DeLyria E. S., Redline R. W., Blanchard T. G. 2009. Vaccination of mice against H. pylori induces a strong Th-17 response and immunity that is neutrophil dependent. Gastroenterology 136:247–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fontenot J. D., Gavin M. A., Rudensky A. Y. 2003. Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat. Immunol. 4:330–336 [DOI] [PubMed] [Google Scholar]
- 11. Fox J. G., et al. 2000. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat. Med. 6:536–542 [DOI] [PubMed] [Google Scholar]
- 12. Fox J. G., Ge Z., Whary M. T., Erdman S. E., Horwitz B. H. 2011. Helicobacter hepaticus infection in mice: models for understanding lower bowel inflammation and cancer. Mucosal Immunol. 4:22–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fox J. G., et al. 2007. Accelerated progression of gastritis to dysplasia in the pyloric antrum of TFF2−/− C57BL6 x Sv129 Helicobacter pylori-infected mice. Am. J. Pathol. 171:1520–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fox J. G., Wang T. C. 2007. Inflammation, atrophy, and gastric cancer. J. Clin. Invest. 117:60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fox J. G., et al. 2003. Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology 124:1879–1890 [DOI] [PubMed] [Google Scholar]
- 16. Ge Z., White D. A., Whary M. T., Fox J. G. 2001. Fluorogenic PCR-based quantitative detection of a murine pathogen, Helicobacter hepaticus. J. Clin. Microbiol. 39:2598–2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haggerty T. D., Perry S., Sanchez L., Perez-Perez G., Parsonnet J. 2005. Significance of transiently positive enzyme-linked immunosobent assay results in detection Helicobacter pylori in stool samples from children. J. Clin. Microbiol. 43:2220–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harrington L. E., et al. 2005. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6:1123–1132 [DOI] [PubMed] [Google Scholar]
- 19. Harris P. R., et al. 2008. Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology 134:491–499 [DOI] [PubMed] [Google Scholar]
- 20. Higgins P. D., Johnson L. A., Luther J., Zhang M., Kao J. Y. 2011. Prior Helicobacter pylori infection ameliorates Salmonella typhimurium-induced colitis: mucosal crosstalk between stomach and distal intestine. Inflamm. Bowel Dis. 17:1398–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hooper L. V., Gordon J. I. 2001. Commensal host-bacterial relationships in the gut. Science 292:1115–1118 [DOI] [PubMed] [Google Scholar]
- 22. Hori S., Nomura T., Sakaguchi S. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science 299:1057–1061 [DOI] [PubMed] [Google Scholar]
- 23. Jiang H., Chess L. 2004. An integrated view of suppressor T cell subsets in immunoregulation. J. Clin. Invest. 114:1198–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang H. Q., Kushnir N., Thurnheer M. C., Bos N. A., Cebra J. J. 2002. Monoassociation of SCID mice with Helicobacter muridarum, but not four other enterics, provokes IBD upon receipt of T cells. Gastroenterology 122:1346–1354 [DOI] [PubMed] [Google Scholar]
- 25. Kao J., et al. 2010. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology 138:1046–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee A., et al. 1993. Long term infection of the gastric mucosa with Helicobacter species does induce atrophic gastritis in an animal model of Helicobacter pylori infection. Zentralbl. Bakteriol. 280:38–50 [DOI] [PubMed] [Google Scholar]
- 27. Lee C. W., et al. 2007. Wild-type and interleukin-10-deficient regulatory T cells reduce effector T-cell-mediated gastroduodenitis in Rag2−/− mice, but only wild-type regulatory T cells suppress Helicobacter pylori gastritis. Infect. Immun. 75:2699–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lemke L. B., et al. 2009. Concurrent Helicobacter bilis infection in C57BL/6 mice attenuates proinflammatory H. pylori-induced gastric pathology. Infect. Immun. 77:2147–2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lundgren A., Suri-Payer E., Enarsson K., Svennerholm A. M., Lundin B. S. 2003. Helicobacter pylori-specific CD4+ CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect. Immun. 71:1755–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luzza F., et al. 2000. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J. Immunol. 165:5332–5337 [DOI] [PubMed] [Google Scholar]
- 31. Maizels R. M., Yazdanbakhsh M. 2003. Immune regulation by helminth parasites: cellular and molecular mechanisms. Nat. Rev. Immunol. 3:733–744 [DOI] [PubMed] [Google Scholar]
- 32. Maurer K. J., et al. 2006. Helicobacter pylori and cholesterol gallstone formation in C57L/J. mice: a prospective study. Am. J. Physiol. Gastrointest. Liver Physiol. 290:G175–G182 [DOI] [PubMed] [Google Scholar]
- 33. Mazmanian S. K., Round J. L., Kasper D. L. 2008. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453:620–625 [DOI] [PubMed] [Google Scholar]
- 34. McBee M. E., Zheng P. Z., Rogers A. B., Fox J. G., Schauer D. B. 2008. Modulation of acute diarrheal illness by persistent bacterial infection. Infect. Immun. 76:4851–4858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peterson R. A., Hoepf T., Eaton K. A. 2003. Adoptive transfer of splenocytes in SCID mice implicates CD4+ T cells in apoptosis and epithelial proliferation associated with Helicobacter pylori-induced gastritis. Comp. Med. 53:498–509 [PubMed] [Google Scholar]
- 36. Rad R., et al. 2006. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology 131:525–537 [DOI] [PubMed] [Google Scholar]
- 37. Raghavan S., Suri-Payer E., Holmgren J. 2004. Antigen-specific in vitro suppression of murine Helicobacter pylori-reactive immunopathological T cells by CD4CD25 regulatory T cells. Scand. J. Immunol. 60:82–88 [DOI] [PubMed] [Google Scholar]
- 38. Rao V. P., Poutahidis T. G. Z., Nambiar P. R. H. B. H., Fox J. G., Erdman S. E. 2006. Proinflammatory CD4+ CD45RB(hi) lymphocytes promote mammary and intestinal carcinogenesis in ApcMin/+ mice. Cancer Res. 66:57–61 [DOI] [PubMed] [Google Scholar]
- 39. Rogers A. B., et al. 2005. Helicobacter pylori but not high salt induces gastric intraepithelial neoplasia in B6129 mice. Cancer Res. 65:10709–10715 [DOI] [PubMed] [Google Scholar]
- 40. Sayi A., et al. 2009. The CD4+ T cell-mediated IFN-gamma response to Helicobacter infection is essential for clearance and determines gastric cancer risk. J. Immunol. 182:7085–7101 [DOI] [PubMed] [Google Scholar]
- 41. Shi Y., et al. 2010. Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J. Immunol. 184:5121–5129 [DOI] [PubMed] [Google Scholar]
- 42. Shiomi S., et al. 2008. IL-17 is involved in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Helicobacter 13:518–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Solnick J. V., Schauer D. B. 2001. Emergence of diverse Helicobacter species in the pathogenesis of gastric and enterohepatic diseases. Clin. Microbiol. Rev. 14:59–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stoicov C., et al. 2004. Coinfection modulates inflammatory responses and clinical outcome of Helicobacter felis and Toxoplasma gondii infections. J. Immunol. 173:3329–3336 [DOI] [PubMed] [Google Scholar]
- 45. Suerbaum S., et al. 2003. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc. Natl. Acad. Sci. U. S. A. 100:7901–7906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sugimoto M., Ohno T., Graham D. Y., Yamaoka Y. 2009. Gastric mucosal interleukin-17 and -18 mRNA expression in Helicobacter pylori-induced Mongolian gerbils. Cancer Sci. 100:2152–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tomb J. F., et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547 [DOI] [PubMed] [Google Scholar]
- 48. Welsh R. M., Fujinami R. S. 2007. Pathogenic epitopes, heterologous immunity and vaccine design. Nat. Rev. Microbiol. 5:555–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Whary M. T., et al. 2001. Long-term colonization levels of Helicobacter hepaticus in the cecum of hepatitis-prone A/JCr mice are significantly lower than those in hepatitis-resistant C57BL/6 mice. Comp. Med. 51:413–417 [PubMed] [Google Scholar]
- 50. Whary M. T., et al. 2005. Intestinal helminthiasis in Colombian children promotes a Th2 response to Helicobacter pylori: possible implications for gastric carcinogenesis. Cancer Epidemiol. Biomarkers Prev. 14:1464–1469 [DOI] [PubMed] [Google Scholar]
- 51. Whary M. T., et al. 2011. Lactobacillus reuteri promotes Helicobacter hepaticus-associated typhlocolitis in gnotobiotic B6.129P2-IL-10(tm1Cgn) (IL-10−/−) mice. Immunology 133:165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wiedemann T., et al. 2009. Helicobacter pylori cag-pathogenicity island-dependent early immunological response triggers later precancerous gastric changes in Mongolian gerbils. PLoS One 4:e4754. [DOI] [PMC free article] [PubMed] [Google Scholar]