

Abstract
We have previously reported that compromised interleukin 17A (IL-17A) production in the lungs increased susceptibility to infection with the invasive fungal pathogen Aspergillus fumigatus. Here we have shown that culturing lung cells from A. fumigatus-challenged mice ex vivo demonstrated Dectin-1-dependent IL-17A production. In this system, neutralization of IL-23 but not IL-6, IL-1β, or IL-18 resulted in attenuated IL-17A production. Il23 mRNA expression was found to be lower in lung cells from A. fumigatus-challenged Dectin-1-deficient mice, whereas bone marrow-derived dendritic cells from Dectin-1-deficient mice failed to produce IL-23 in response to A. fumigatus in vitro. Addition of recombinant IL-23 augmented IL-17A production by wild-type (WT) and Dectin-1-deficient lung cells, although the addition of IL-6 or IL-1β did not augment the effect of IL-23. Intracellular cytokine staining of lung cells revealed lower levels of CD11b+ IL-17A+ and Ly-6G+ IL-17A+ cells in A. fumigatus-challenged Dectin-1-deficient mice. Ly-6G+ neutrophils purified from the lungs of A. fumigatus-challenged Dectin-1-deficient mice displayed lower Il17a mRNA expression but surprisingly had intact Rorc and Rora mRNA expression. We further demonstrated that Ly-6G+ neutrophils required the presence of myeloid cells for IL-17A production. Finally, upon in vitro stimulation with A. fumigatus, thioglycolate-elicited peritoneal neutrophils were positive for intracellular IL-17A expression and produced IL-17A in a Dectin-1- and IL-23-dependent manner. In summary, Dectin-1-dependent IL-17A production in the lungs during invasive fungal infection is mediated in part by CD11b+ Ly-6G+ neutrophils in an IL-23-dependent manner.
INTRODUCTION
Aspergillus fumigatus, the etiological agent of invasive pulmonary aspergillosis (IPA), is a ubiquitous mold that causes severe, invasive life-threatening infections in patients that are severely immunocompromised. Disease acquisition includes such risk factors as neutropenia, impaired neutrophil function, and myeloablative-immunosuppressive therapies associated with hematopoietic stem-cell transplantation (16). Despite available antifungal therapy, the prognosis of IPA remains poor and mortality ranges from 30 to 90% (3) (10). This is thought to be due in part to the relatively small arsenal of effective antifungal drugs, some of which, specifically amphotericin B, cause severe nephrotoxicity and are associated with low response rates of between 10% and 40% (28). IPA has risen dramatically over the last several decades due to an increase in immunosuppressed patients, and by the early 1990s, 60% of invasive fungal infections diagnosed at autopsy were IPA (22). It must also be stated that IPA is not only associated with stem-cell transplantation but also presents in whole-organ transplantation, primarily that of the lung and heart, with mortality rates of 68% to 78% (35).
Interleukin 17 (IL-17), first discovered more than 15 years ago (46) and now called IL-17A, is a proinflammatory cytokine that upregulates a number of cytokines and chemokines, leading to the recruitment of neutrophils to sites of inflammation. In terms of infection, IL-17A has been demonstrated to have a protective role against multiple microorganisms, predominantly extracellular bacteria (9). However, IL-17A is also the classic example of a “double-edged sword” in that it clearly functions as an immunopathogenic mediator of inflammation in such diseases as inflammatory bowel disease, lupus, and psoriasis (17). IL-17A gained prominence when it was discovered to be produced by CD4 T cells, a lineage now termed T helper IL-17, or Th17, cells (18). Recent studies have begun to uncover non-CD4 T cells as important sources of IL-17A. Among T cell lineages, CD8 T cells (15), γδ T cells (31), and invariant natural killer T (iNKT) (25) cells can produce IL-17A. Lymphoid tissue inducer-like cells, which are CD4+ CD3− NK1.1− CD11b− Gr1− CD11c− B220− cells, have been shown to produce IL-17A (38), as have CD11b+ F4/80+ alveolar macrophages (36) and structural cells, such as Paneth cells in the gut (37). However, the cytokines and transcription factor(s) that drive IL-17A production by nonlymphoid cells have not been thoroughly investigated.
A role for Dectin-1 in the generation of Th17/IL-17A-mediated responses has recently been identified (19). The Dectin-1/Syk/CARD9 signaling axis promotes dendritic cell (DC) activation and the secretion of proinflammatory mediators, including IL-23 (19). Dectin-1-mediated activation of dendritic cells has been shown to bias T helper cell differentiation to a Th17 phenotype (19). Moreover, infection with Candidaalbicans resulted in the natural generation of Th17 cells that did not develop in CARD9-deficient mice (19). A follow-up study showed that DCs activated via Dectin-1 produced IL-23 and converted Foxp3+ regulatory T cells (Tregs) to IL-17A-producing cells (27). A more recent study reported that DC-expressed phospholipase C-γ2 was the dominant signaling intermediate for DC-mediated Th1 and Th17 differentiation, again with a focus of IL-23 production (39). With respect to fungus-associated IL-17A responses, a recent study has shown that purified mannan from C. albicans is a potent inducer of IL-17A responses via the macrophage mannose receptor (MR), although blockage of MR, Dectin-1, and Toll-like receptor 2 (TLR2) separately had an inhibitory effect on peripheral blood mononuclear cell (PBMC) IL-17A production (41). Intriguingly, stimulation of PBMCs from three human subjects identified as being deficient in Dectin-1 has shown attenuated IL-17A production in response to C. albicans yeast (41) and more recently A. fumigatus (6), indicating a role for Dectin-1 in IL-17A production by T cells in humans.
We have previously reported that mice deficient in the beta glucan receptor Dectin-1 were inherently susceptible to lung infection with A. fumigatus as a result of multiple defects in innate immune mechanisms that control infection (43). Among these, we reported that IL-17A production in the lung within the first 24 to 48 h after A. fumigatus exposure was dependent on Dectin-1 and was critical for clearance of A. fumigatus from the lung, since neutralization of IL-17A in wild-type (WT) mice resulted in a >10-fold increase in the fungal burden. In this work, we sought to characterize mechanisms driving IL-17A production in a Dectin-1-dependent manner during lung infection with A. fumigatus.
MATERIALS AND METHODS
Mice.
C57BL/6NTac mice, 6 to 8 weeks old, were purchased from Taconic Farms Incorporated (Germantown, NY). Dectin-1-deficient mice were generated on the 129/SvEv background as described previously (40), backcrossed 10 generations to the C57BL/6 background, and bred at Taconic. All mice were maintained in a specific-pathogen-free environment in microisolator cages within an American Association for Laboratory Animal Science-certified animal facility in the Lyons Harrison Research Building at the University of Alabama at Birmingham. Animal studies were reviewed and approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee (IACUC).
Preparation of A. fumigatus and in vivo challenge.
A. fumigatus isolate 13073 (ATCC, Manassas, VA) was maintained on potato dextrose agar for 5 to 7 days at 37°C. Conidia were harvested by washing the culture flask with 50 ml of sterile phosphate-buffered saline supplemented with 0.1% Tween 20. The conidia were then passed through a sterile 40-μm nylon membrane to remove hyphal fragments and enumerated on a hemacytometer. Mice were lightly anesthetized with isoflurane and administered (5 to 7) × 107 A. fumigatus conidia in a volume of 50 μl intratracheally.
Lung cell isolation and culture, cytokine neutralizations, and stimulations.
Mice were anesthetized with intraperitoneal ketamine/xylazine and sacrificed by exsanguination 18 h postinfection. Both lungs were collected and minced in Iscove's modified Dulbecco's medium (IMDM) (Sigma, St. Louis, MO) supplemented with 1% penicillin-streptomycin-glutamine (Pen-Strep-Glut) (Mediatech, Herndon, VA), 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA), and 0.4 mg/ml polymyxin B (Thermo Fisher), followed by incubation for 60 min with tissue-culture-grade type IV collagenase (1 mg/ml; Sigma, St. Louis, MO) in a 37°C orbital shaker at 100 rpm. The cell suspension was filtered through sterile 70-μm and 40-μm nylon filters, and red blood cells were lysed with ACK lysing buffer (Lonza, Walkersville, MD) to create lung cell preparations. For lung cell cultures, cells were enumerated on a hemacytometer and plated at 1 × 106 cells in a volume of 0.2 ml. Supernatants were collected after 24 h, clarified by centrifugation, and stored at −80°C. IL-6, IL-1β, and IL-17A levels were quantified by Bio-Plex or enzyme-linked immunosorbent assay (ELISA) as described previously (43). In specific experiments, neutralizing antibodies were added to lung cells to assess the effects of cytokine neutralization on IL-17A production. For this, anti-mouse IL-1β, IL-6, IL-18, or IL-23 (all neutralizing antibodies were purchased from R&D Systems) were added to lung cell cultures at a final concentration of 2 to 5 μg/ml for 24 h. Rat (IL-6 and IL-18) or goat (IL-1β and IL-23) isotype antibodies were added to lung cell cultures as a control. Supernatants were collected after 24 h and clarified by centrifugation, and IL-17A levels were quantified by ELISA (R&D Systems). In specific experiments, recombinant murine IL-23, IL-1β, or IL-6 (all from R&D Systems), alone or in combination, was added to lung digest cells at 1 or 10 ng/ml for 24 h. Supernatants were collected after 24 h and clarified by centrifugation, and IL-17A levels were quantified by ELISA (R&D Systems).
Lung cell surface marker flow cytometry and intracellular analysis of IL-17A production.
Lung cells were prepared as described above. Cells were washed, and Fc receptors were blocked with Mouse BD Fc Block (BD Biosciences, San Diego, CA) at 4°C for 20 min. Thereafter, cells were stained with a single-color Live/Dead fixable dead cell stain (Invitrogen), followed by labeling with specific immune cell surface markers. The following staining parameters were employed: macrophages were identified as CD11blo/neg F4/80lo CD11c+, eosinophils as CD11b+ Siglec F+ Ly-6Clo/neg, neutrophils as CD11b+ Ly-6G+, dendritic cells as CD11b+ CD11c+ F4/80med, natural killer cells as CD11b+ DX5+, monocytes as CD11b+ Ly-6C+ Ly6G−, B cells as CD19+, and T cells as CD3+ (all antibodies purchased from eBiosciences and BD Biosciences). Of note, Ly-6G+ cells were identified using the 1A8 clone. Samples were acquired using a four-laser, 20-parameter analytic BD LSR II flow cytometer, and data were analyzed using the FlowJo software program (Tree Star, Ashland, OR). For intracellular IL-17A staining, lung cells were isolated from A. fumigatus-exposed mice and cultured overnight, the last 8 to 10 h in the presence of GolgiStop inhibitor(BD Biosciences, San Diego, CA). Cells were Fc blocked (as above), stained with a live/dead stain (as above), CD11b, or Ly-6G, and then fixed and permeabilized with BD Biosciences Cytofix/Cytoperm buffers followed by staining with rat anti-mouse IL-17A-Alexa 647 (clone eBio17B7; eBioscience). Unstained cells served as a control for background fluorescence and gating. Samples were acquired using a BD LSR II cytometer (BD Biosciences), and data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Lung CD11b+, CD11c+, and Ly-6G+ cell purification and culture.
Lung cells were prepared as described above. Thereafter, cells were incubated with anti-murine CD11b, anti-murine CD11c+, or murine anti-Ly-6G MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) and magnetically isolated per the manufacturer's instructions. Assessment of cells by flow cytometry routinely demonstrated >95% purity (data not shown). Each cell population was cultured individually or in a ratio of 60% Ly-6G+, 30% CD11b+, and 10% CD11c+ cells with A. fumigatus at a 1:1 cell-to-conidium ratio. Supernatants were collected after 24 h and clarified by centrifugation, and IL-17A levels were quantified by ELISA (R&D Systems).
Il17a, Rorc, Rora, Ahr, and Irf4 analysis.
Total RNA was isolated from lung cells or purified Ly-6G+ cells immediately after isolation by a single-step method using TRIzol LS reagent (Invitrogen) as per the manufacturer's instructions. Thereafter, RNA was transcribed to cDNA (iScript cDNA synthesis kit; Bio-Rad), and real-time PCR for Il17a (Mm00439618_m1; Applied Biosystems), Rorc (Mm00441139_m1; Applied Biosystems), Rora (Mm00443103_m1; Applied Biosystems), Ahr (Mm01291777_m1; Applied Biosystems), and Irf4 (Mm00516431_m1; Applied Biosystems) was carried out (iQ Supermix; Bio-Rad). mRNA levels were normalized to Gapdh mRNA levels (primers/probe from Applied Biosystems) using the 2−(ΔΔCT) method.
Thioglycolate peritoneal neutrophil isolation, culture, and intracellular IL-17A analysis.
To isolate neutrophils, C57BL/6 and Dectin-1-deficient mice were administered 1.5 ml of 3% sterile thioglycolate intraperitoneally for 4 h as previously described (2, 8). Thereafter, animals were sacrificed, and peritoneal lavage was performed using 10 ml of tissue culture medium (Sigma, St. Louis, MO). The lavage fluid was centrifuged at 600 × g for 10 min, and the subsequent cell pellet was enumerated using a hemacytometer. For IL-17A production, cells (1 × 106 in a volume of 100 μl) were stimulated for 24 h with A. fumigatus conidia (1 × 106 conidia in a volume of 100 μl). IL-17A was measured in clarified supernatants by ELISA. For intracellular IL-17A staining, cells were isolated and stimulated as above for 12 h in the presence of brefeldin A (BioLegend). Thereafter, cells were stained with rat anti-mouse IL-17A-Alexa 647 (clone eBio17B7; eBioscience) and Ly-6G as detailed above.
Statistics.
Data were analyzed using the GraphPad Prism statistical software program, version 5.0. Comparisons between groups when data were normally distributed were made with Student's t test. Significance was accepted at a P value of <0.05.
RESULTS
Whole-lung versus lung cell IL-17A production from A. fumigatus-challenged Dectin-1-deficient mice.
In our previous work, mice deficient in Dectin-1, originally developed on the 129/SvEv background (40), had a 2.5-fold reduction in IL-17A levels in lungs after A. fumigatus challenge (43). We have now backcrossed 129/Dectin-1-deficient mice for 10 generations to the C57BL/6 background, and initial studies confirmed that BL/6 Dectin-1-deficient mice were equally susceptible to lung infection with A. fumigatus (8- to 10-fold-higher A. fumigatus lung burden at 48 to 72 h postchallenge; unpublished data). In addition, IL-17A production in the lungs after A. fumigatus challenge was also dependent on Dectin-1 expression, since BL/6 Dectin-1-deficient mice demonstrated 5-fold-lower IL-17A levels in the lungs 48 h after exposure (Fig. 1 A). We next collected lungs from C57BL/6 (WT) and Dectin-1-deficient (KO) mice after A. fumigatus challenge and subjected them to enzymatic digestion to determine whether single-cell suspensions could replicate the differences in IL-17A levels observed in whole-lung homogenates. Initial experiments isolated lung cells at 48 h postchallenge, followed by an additional 24 h of culture. However, this design resulted in fungal overgrowth, particularly in the Dectin-1-deficient lung cell cultures, resulting in both exacerbated and erratic IL-17A production (data not shown). Therefore, in an alternative experimental design, we isolated lung cells at an earlier time point, 18 h postchallenge. Real-time PCR assessment of Il17a mRNA levels in 18-h lung cells from A. fumigatus-exposed Dectin-1-deficient mice revealed a 4-fold reduction in Il17a mRNA (Fig. 1B). Upon ex vivo culturing overnight of lung cells isolated at 18 h postchallenge (in the absence of additional stimulation), lung cells from Dectin-1-deficient mice produced only a third of the IL-17A levels produced by WT lung cells (Fig. 1C). We next examined the expression of multiple immune cell surface markers on lung cells isolated 18 h postinfection. Cumulative data in Fig. 1D indicated that the major constituent of lung cells isolated from WT mice, approximately 68% of all cells, was CD11b+ Ly-6G+ cells, i.e., neutrophils. CD11b+ Ly-6G+ cells were also the major cell constituent in lung cells from Dectin-1-deficient mice but constituted only approximately 53% of all cells (Fig. 1D). The remaining myeloid cells (CD11b+ Ly-6Glo/neg) were comprised of monocytes, eosinophils, dendritic cells, and natural killer (NK) cells. Total eosinophils, which were defined as CD11b+, Ly-6Clo/neg, and Siglec F+ (33), were surprisingly increased in Dectin-1=deficient mice (Fig. 1D). CD3+ T cells and CD19+ B cells were also present in equal numbers in WT and Dectin-1 KO mice. Thus, CD11b+ Ly-6G+ cells are the predominant cell type in lung cells from A. fumigatus-exposed mice. Moreover, Dectin-1-deficient mice on the C57BL/6 background have attenuated IL-17A production in the lungs after A. fumigatus challenge, and ex vivo culturing of enzymatically isolated lung cells replicates in vivo Dectin-1-dependent IL-17A production.
Fig. 1.

Whole-lung versus lung cell IL-17A production from A. fumigatus-challenged Dectin-1-deficient mice. (A) C57BL/6 wild-type (WT) and Dectin-1-deficient (KO) mice were challenged intratracheally with (5 to 7) × 107 A. fumigatus conidia, and 48 h after exposure, IL-17A levels in lung homogenates were quantified by ELISA. Data are expressed as mean pg/ml + SEM. Cumulative data from three independent studies (n=5 mice/group for each study) are illustrated. “**” represents a P value of <0.01 (unpaired two-tailed Student's t test). (B) C57BL/6 wild-type (WT) and Dectin-1-deficient (KO) mice were challenged intratracheally with (5 to 7) × 107 A. fumigatus conidia, and 18 h after exposure, lungs were collected and enzymatically digested. Immediately after single-cell suspensions were collected, total RNA was isolated from 1 × 106 cells and transcribed to cDNA, and quantitative real-time PCR was performed for Il17a. Gene expression was normalized to Gapdh expression, and fold changes between WT (set at 1) and KO mice were determined using the 2−ΔΔCT method. Cumulative data from five independent studies are shown. “***” represents a P value of <0.001 (paired two-tailed Student's t test). (C) Lung cells were isolated as for panel B, and 1 × 106 cells were cultured for 24 h in a volume of 0.2 ml. IL-17A levels were quantified in clarified coculture supernatants by ELISA. Cumulative data from five independent studies are shown. Data are expressed as mean pg/ml + SEM. “**” represents a P value of <0.01 (unpaired two-tailed Student's t test). (D) Lung cells were isolated as described for panel B, Fc blocked, stained with a live/dead staining kit, and thereafter stained with fluorochrome-conjugated antibodies against various immune cell surface markers (see Materials and Methods). Cumulative data from three independent studies are shown. Data are expressed as absolute numbers of live cells in the lung digest cell preparation. “*” and “**” represent P values of <0.05 and 0.01, respectively (unpaired two-tailed Student's t test). PMNs, polymorphonuclear leukocytes; Eos, eosinophils; Monos, monocytes; Macs, macrophages.
IL-17A production by lung cells after A. fumigatus challenge partially requires IL-23.
Ex vivo culturing of lung cells from A. fumigatus-challenged mice replicated in vivo observations (Fig. 1), providing us an experimental in vitro system to determine which cytokines were involved in Dectin-1-dependent IL-17A production. To date, multiple cytokines have been identified as being critical for inducing IL-17A production by CD4+ T cells (Th17), including transforming growth factor β (TGF-β) and IL-6 (reviewed in reference 17). IL-23 and IL-1β are thought to maintain IL-17A production and enhance the expansion of Th17 cells (17). Similar roles have been identified for IL-23 and IL-1β in IL-17A production by γδ T cells (23) and LTi cells (38). Finally, IL-18 has been shown to synergize with IL-23 to induce STAT4-mediated IL-17A production in Th1-polarized, IL-23-primed CD4+ T cells in vitro (24). We therefore determined whether these Th17/IL-17A-associated cytokines were involved in IL-17A production by lung cells. Initial analysis indicated that both IL-6 and IL-1β were produced by lung cells from A. fumigatus-challenged mice in a Dectin-1-dependent manner (for IL-6, WT, 2,481 ± 415 pg/ml, versus KO, 1,484 ± 238; P < 0.05; for IL-1β, 977 ± 143 pg/ml, versus KO, 541 ± 114; P < 0.05). Despite their dependency on Dectin-1 for optimal production in the lungs, neutralization of IL-6 and IL-1β had no effect on IL-17A production by lung cells (data not shown). In contrast, neutralization of IL-23 resulted in a 50% decrease in IL-17A production (Fig. 2 A), although neutralization of IL-18 had no effect (Fig. 2A). A previous report has shown that human monocyte-derived DCs stimulated with A. fumigatus hyphae, but not conidia, produced IL-23 (7), whereas another study has shown that Dectin-1-deficient mice challenged with a nonlethal dose of A. fumigatus surprisingly had increased Il23 mRNA in the lungs (30). In the current study, analysis of IL-23 in lung cells revealed undetectable levels in culture supernatants in both WT and Dectin-1-deficient mice (data not shown); however, real-time PCR assessment of Il23 mRNA showed a significant reduction in lung cells from Dectin-1-deficient mice (Fig. 2B). Furthermore, bone marrow-derived DCs (BMDCs) from WT mice stimulated with A. fumigatus overnight demonstrated low but detectable IL-23 production, which was absent in BMDCs from Dectin-1-deficient mice (Fig. 2C). We next determined whether recombinant IL-23 had the capacity to induce IL-17A production in WT and Dectin-1-deficient lung cells. Results shown in Fig. 2D indicated that the addition of IL-23 induced a dose-dependent increase in IL-17A production in both WT and Dectin-1-deficient lung cells. The induction of IL-17A was lower in lung cells from Dectin-1-deficient mice, which we hypothesize is possibly a result of lower neutrophil numbers (as detailed in Fig. 1D). The addition of IL-6 or IL-1β, alone or in combination with IL-23, did not augment IL-17A production above the level induced by IL-23 (data not shown). Thus, IL-17A production by lung cells from A. fumigatus-challenged mice is partially dependent on Dectin-1-mediated IL-23, putatively from DCs, but IL-17A production after A. fumigatus challenge does not require IL-18, IL-1β, or IL-6. Furthermore, IL-23 can promote the induction of IL-17A, even in the setting of Dectin-1 deficiency.
Fig. 2.

IL-17A production by lung cells after A. fumigatus challenge partially requires IL-23. (A) Lung cells were isolated as described in the legend to Fig. 1B, and 1 × 106 cells were cultured for 24 h in a volume of 0.2 ml. Neutralizing antibodies against IL-23 and IL-18 were added at a final concentration of 2 to 5 μg/ml at the beginning of the culture (α-18 and α-23). Rat (IL-18) or goat (IL-23) isotype antibodies (Iso 18 and Iso 23) were included as a control. IL-17A levels were quantified in clarified coculture supernatants by ELISA. Cumulative data from five independent studies are shown. Data are expressed as mean pg/ml + SEM. “**” and “***” represent P values of <0.01 and 0.001, respectively (unpaired two-tailed Student's t test). (B) Immediately after lung cells were collected, total RNA was isolated from 1 × 106 cells and transcribed to cDNA, and quantitative real-time PCR was performed for Il23. Gene expression was normalized to that of Gapdh, and fold changes between WT (set at 1) and KO mice were determined using the 2−ΔΔCT method. Cumulative data from five independent studies are shown. “***” represents a P value of <0.001 (paired two-tailed Student's t test). (C) Bone marrow-derived dendritic cells (BMDCs) were isolated and cultured at 1 × 106 cells in a volume of 100 μl. A. fumigatus conidia (A.f) were added at a 1:1 ratio to BMDCs in a volume of 100 μl. Supernatants were collected after 24 h and clarified by centrifugation. IL-23 levels were quantified by ELISA. Cumulative data from three independent studies are shown. “*” represents a P value of <0.05 (unpaired two-tailed Student's t test). Unstim, unstimulated. (D) Lung cells were isolated from WT and KO mice as described for Fig. 1B, and 1 × 106 cells were cultured for 24 h in a volume of 0.2 ml. Recombinant murine IL-23 was added at 1 and 10 ng/ml at the beginning of the culture. Controls included lung cells cultured in the absence of IL-23. IL-17A levels were quantified in clarified coculture supernatants by ELISA. Cumulative data from three independent studies are shown. Data are expressed as mean pg/ml + SEM. “*,” “**,” and “***” represent P values of 0.05, 0.01, and 0.001, respectively (unpaired two-tailed Student's t test).
IL-17A is not produced by T cells in a Dectin-1-dependent manner after A. fumigatus challenge.
As discussed previously, CD4 T cells, as well as other cell types, may be a cellular source of IL-17A (36–38). Due to the rapid production of IL-17A in the lungs after A. fumigatus challenge, we questioned whether CD3+ cells were the source of Dectin-1-dependent IL-17A early after A. fumigatus challenge. Our initial hypothesis was that due to its rapid production after challenge, IL-17A was not likely to be significantly derived from a T cell source. Indeed, analysis of intracellular IL-17A expression by CD3+ cells from phorbol myristate acetate-ionomycin (PMA/I)-stimulated lung cells did not show significant induction of IL-17A nor a dependency on Dectin-1 (Fig. 3). Intracellular IL-17A expression by CD4+ cells from PMA/I-stimulated lung cells also demonstrated similar results (data not shown). Thus, CD3+ CD4+ T cells and CD3+ γδ T cells do not appear to be a significant source of Dectin-1-dependent IL-17A production early after A. fumigatus challenge.
Fig. 3.
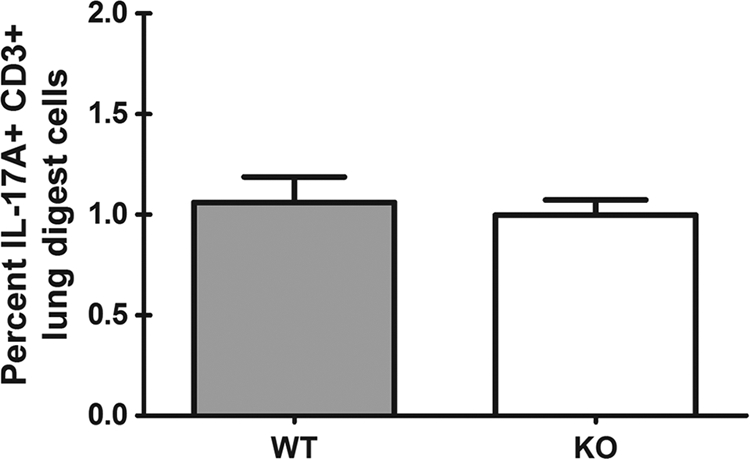
IL-17A is not produced by T cells in a Dectin-1-dependent manner after A. fumigatus challenge. Lung cells were isolated as described for Fig. 1B and stimulated with PMA/I for 5 h in the presence of GolgiStop inhibitor. Cells were Fc blocked, stained with a live/dead staining kit, and thereafter stained with an anti-CD3 antibody, followed by fixation/permeabilization, and stained for intracellular IL-17A. Cumulative data from two independent studies are shown. Data are expressed as the percentages of CD3+ cells that are IL-17A+.
Lung CD11b+ Ly-6G+ cells are a source of Dectin-1-dependent IL-17A after A. fumigatus challenge.
We next determined whether the major constituent in lung cells, CD11b+ Ly-6G+ cells, was a source of Dectin-1-dependent IL-17A in the lungs after A. fumigatus challenge. For this, we employed the in vitro culture system developed for Fig. 1C. Lung cells were collected 18 h after A. fumigatus challenge and cultured overnight in the absence of additional stimulation. To capture intracellular IL-17A, the last 8 to 10 h of this culture was in the presence of a protein transport inhibitor. Employing the global myeloid cell marker CD11b and the granulocyte/neutrophil-associated marker Ly-6G, representative flow cytometric results illustrate a CD11b+ IL-17A+ population (Fig. 4 A) and a Ly-6G+ IL-17A+ population (Fig. 4B) in lung cells from A. fumigatus-challenged WT (left) versus Dectin-1-deficient (right) mice. Cumulative data show that CD11b+ IL-17A+ lung cells (Fig. 4C) and Ly-6G+ IL-17A+ lung cells (Fig. 4D) are reduced by half and two-thirds, respectively, in Dectin-1-deficient mice. We also gated on CD11b+ Ly-6G+ cells and determined the percentage that were IL-17A+, which indicated a 40% reduction in Dectin-1-deficient mice (Fig. 4E). Thus, CD11b+ Ly-6G+ neutrophils from the lungs of A. fumigatus-exposed Dectin-1-deficient mice have a reduction in intracellular IL-17A expression.
Fig. 4.

Lung CD11b+ Ly-6G+ cells are a source of Dectin-1-dependent IL-17A after A. fumigatus challenge. (A) Lung cells were isolated from WT and KO mice as described for Fig. 1B, and 1 × 106 cells were cultured for 24 h in a volume of 0.1 ml (second half of the culture in the presence of GolgiStop inhibitor). Cells were Fc blocked, stained with a live/dead staining kit, and thereafter stained for CD11b, followed by fixation/permeabilization, and stained for intracellular IL-17A. Representative flow cytometric plots after gating on live cells followed by gating on CD11b+ IL-17A+ cells are shown. Double-positive populations (CD11b+ IL-17A+; square gates) were based on unstained controls. (B) Lung cells were isolated and cultured as described for Fig. 1B. Cells were Fc blocked, stained with a live/dead staining kit, and thereafter stained for Ly-6G, followed by fixation/permeabilization, and stained for intracellular IL-17A. Representative flow cytometric plots after gating on live cells followed by gating on Ly-6G+ IL-17A+ cells are shown. Double-positive populations (Ly-6G+ IL-17A+; square gates) were based on unstained controls. (C) Cumulative flow cytometric data from panel A from three independent studies with lung cells cultured and analyzed in triplicate. Data are expressed as the percentages of CD11b+ IL-17A+ cells. “***” represents a P value of <0.001 (unpaired two-tailed Student's t test). (D) Cumulative flow cytometric data from panel B from three independent studies with lung cells cultured and analyzed in triplicate. Data are expressed as the percentage of Ly-6G+ IL-17A+ cells. “**” represents a P value of <0.01 (unpaired two-tailed Student's t test). (E) Cumulative flow cytometric data from three independent studies with lung cells cultured and analyzed in triplicate. Data are expressed as the percentages of CD11b+ Ly-6G+ lung cells that are IL-17A+. “***” represents a P value of <0.001 (unpaired two-tailed Student's t test).
Lung Ly-6G+ cells require the presence of myeloid cells for IL-17A production.
We next questioned whether lung Ly-6G+ lung cells produced IL-17A in vitro. In initial studies, we bead purified Ly-6G+ cells from the lungs 18 h after infection and immediately isolated total RNA. Assessment of Il17a mRNA levels by real-time PCR indicated that purified Ly-6G+ cells from Dectin-1-deficient mice had a >2-fold reduction in Il17a mRNA levels compared to those for WT mice (Fig. 5 A). However, overnight culture of purified Ly-6G+ cells from either WT or Dectin-1-deficient mice unexpectedly resulted in little to no IL-17A production in vitro (data not shown). Similar results were obtained when CD11b+ Ly-6G+ cells were sorted from WT and Dectin-1-deficient lung cells by flow cytometry and cultured overnight (data not shown). This observation led us to hypothesize that neutrophils were unable to produce IL-17A without the presence of other cells, particularly those that may serve as a source of IL-23. To address this, we bead purified Ly-6G+, CD11b+, and CD11c+ cells from 18-h lung cells and cultured them at a ratio consisting of approximately 60% Ly-6G+ lung cells, 30% CD11b+ lung cells (some of which are neutrophils), and 10% CD11c+ cells in an effort to replicate the percentages observed in Fig. 1D. Results showed that IL-17A was undetectable in purified Ly-6G+, CD11b+, and CD11c+ cells cultured individually in the presence of A. fumigatus (Fig. 5B). Interestingly, the addition of CD11b+ or CD11c+ cells to Ly-6G+ cells failed to induce IL-17A production (Fig. 5B). However, upon combination of all three populations, we consistently detected IL-17A, albeit at low levels (Fig. 5B). Thus, neutrophils require the presence of additional myeloid cells for optimal IL-17A production.
Fig. 5.

Lung Ly-6G+ cells require the presence of CD11b+ and CD11c+ cells for IL-17A production. (A) Lung cells were isolated from WT and KO mice as described for Fig. 1B, and Ly-6G+ cells were purified via magnetic bead isolation. Immediately thereafter, total RNA was isolated from 1 × 106 cells and transcribed to cDNA, and quantitative real-time PCR was carried out for Il17a. Gene expression was normalized to that of Gapdh, and fold changes between WT (set at 1) and KO mice were determined using the 2−ΔΔCT method. Cumulative data from three independent studies are shown. “*” represents a P value of <0.05 (paired two-tailed Student's t test). (B) Lung cells were isolated from WT mice as described for Fig. 1B, and Ly-6G+, CD11b+, and CD11c+ cells were purified by magnetic bead selection followed by culturing with A. fumigatus for 24 h individually or in various combinations. IL-17A levels were quantified in clarified coculture supernatants by ELISA. Cumulative data from three independent studies, with cultures performed in duplicate or triplicate in each, are represented. Data are expressed as mean pg/ml + SEM. “*” represents a P value of <0.05 (paired two-tailed Student's t test).
Dectin-1 is not required for Rorc, Rora, Ahr, or Irf4 mRNA expression in lung cells after A. fumigatus challenge.
The transcription factors retinoid-related orphan receptor gamma and alpha, encoded by Rorc and Rora, are essential for IL-17A production by CD4 T cells (14, 45). To date, Rorc has been observed to be a marker of all lymphoid-like cells that produce IL-17A. The aryl hydrocarbon receptor (Ahr) (42) (23) and interferon regulatory factor 4 (Irf4) (5) are additional transcription factors characteristic of IL-17A-producing cells. Whether lung cells producing IL-17A also express these transcription factors and whether they are dependent on Dectin-1 for expression during A. fumigatus lung infection have not been explored. Unexpectedly, despite having impaired Il17a mRNA expression (Fig. 1B), lung cells from A. fumigatus-challenged Dectin-1-deficient mice did not demonstrate an impairment in Rorc and Rora mRNA expression but rather had significant increases in both transcription factors (Fig. 6 A). In contrast, Ahr and Irf4 mRNA levels were not modulated by Dectin-1 deficiency (Fig. 6A). Similar to unfractionated lung cells (Fig. 6B), purified CD11b+ Ly-6G+ cells from Dectin-1-deficient mice also demonstrated an increase in Rorc (Fig. 6B) and Rora (data not shown) mRNA levels. Thus, impaired IL-17A production in Dectin-1-deficient mice does not correlate with diminished Rorc, Rora, Ahr, or Irf4 mRNA expression.
Fig. 6.

Dectin-1 is not required for Rorc, Rora, Ahr, or Irf4 mRNA expression in lung cells after A. fumigatus challenge. (A) Lung cells were isolated from WT and KO mice as described for Fig. 1B. Immediately after single-cell suspensions were collected, total RNA was isolated from 1 × 106 cells and transcribed to cDNA, and quantitative real-time PCR was performed for Rorc, Rora, Ahr, and Irf4. Gene expression was normalized to that of Gapdh, and fold changes between WT (set at 1) and KO mice were determined using the 2−ΔΔCT method. Cumulative data from 10 independent studies are shown. “**” represents a P value of <0.01 (paired two-tailed Student's t test). (B) Lung cells were isolated from WT and KO mice as described for Fig. 1B, and Ly-6G+ cells were purified via magnetic bead isolation. Immediately thereafter, total RNA was isolated from 1 × 106 cells and transcribed to cDNA, and quantitative real-time PCR was performed for Rorc. Gene expression was normalized to that of Gapdh, and fold changes between WT (set at 1) and KO mice were determined using the 2−ΔΔCT method. Cumulative data from six independent studies are represented. “*” represents a P value of <0.05 (paired two-tailed Student's t test).
Thioglycolate-elicited peritoneal neutrophils produce IL-17A in a Dectin-1- and IL-23-dependent manner in response to A. fumigatus in vitro.
Data thus far have indicated that in response to A. fumigatus infection in vivo, neutrophils appear to be a source of Dectin-1-dependent IL-17A. To further support this observation, we questioned whether naive neutrophils had the capacity to produce IL-17A in a Dectin-1-dependent manner in response to A. fumigatus in vitro. We therefore isolated peritoneal neutrophils via thioglycolate elicitation (8) (2) and assessed IL-17A production after overnight stimulation with A. fumigatus. Results shown in Fig. 7 indicated similar levels of neutrophil recruitment (Fig. 7A) and cellular composition (Fig. 7B) in WT (left) and Dectin-1 KO (right) mice in response to thioglycolate. Upon overnight culture with A. fumigatus, peritoneal neutrophils from Dectin-1-deficient mice produced only a third of the IL-17A produced by WT neutrophils (Fig. 7C). In support of the observation with lung cells, we observed that IL-23 was also required for optimal IL-17A production by thioglycolate-elicited peritoneal neutrophils since IL-23 neutralization reduced IL-17A production by more than half (Fig. 7D). Furthermore, similar to the case with lung Ly-6G+ cells (Fig. 4B), intracellular IL-17A was detected in WT thioglycolate-elicited Ly-6G+ cells stimulated with A. fumigatus in vitro and was at a nearly 8-fold lower level in Dectin-1-deficient thioglycolate-elicited Ly-6G+ cells (Fig. 7E). Thus, IL-23 and Dectin-1 are required for optimal peritoneal neutrophil IL-17A production in response to A. fumigatus.
Fig. 7.

Thioglycolate-elicited peritoneal neutrophils produce Dectin-1- and IL-23-dependent IL-17A in response to A. fumigatus in vitro. (A) Thioglycolate was injected intraperitoneally (3%; 1.5 ml) into naive WT and Dectin-1-deficient mice. After 4 h, mice were lavaged with 10 ml of prewarmed tissue culture medium, and cells were isolated via centrifugation. Neutrophil recruitment was assessed by Wright staining of cytospun peritoneal lavage cell preparations. Representative Wright stains of WT (left; 22/27 are neutrophils [81%]) and Dectin-1-deficient (right; 27/33 are neutrophils [77%]) mice are shown. (B) Cumulative cell differential data as determined by Wright staining of cytospun peritoneal lavage cell preparations. Cumulative data from three independent studies are shown. (C) Thioglycolate-elicited peritoneal neutrophils (1 × 106) were cultured in a volume of 100 μl. A. fumigatus conidia were added at a 1:1 ratio to a volume of 100 μl. Supernatants were collected after 24 h and clarified by centrifugation. IL-17A levels were quantified by ELISA. Cumulative data from four independent studies are shown. “*” represents a P value of <0.05 (unpaired two-tailed Student's t test). (D) Thioglycolate-elicited peritoneal neutrophils (1 × 106) from WT mice were cultured with A. fumigatus in the presence of neutralizing antibodies against IL-23. Goat isotype antibodies were included as a control. IL-17A levels were quantified in clarified coculture supernatants by ELISA after 24 h. Cumulative data from five independent studies with 1 to 2 mice per group analyzed in duplicate are shown. Data are expressed as mean pg/ml + SEM. “**” represents a P value of <0.01 (unpaired two-tailed Student's t test). (E) Representative flow cytometric data of intracellular IL-17A staining of thioglycolate-elicited Ly-6G+ cells from WT and Dectin-1-deficient mice after coculture with A. fumigatus in vitro.
DISCUSSION
We have previously reported that mice deficient in the beta-glucan receptor Dectin-1 (129/SvEv background) are susceptible to lung infection with A. fumigatus (43). A correlate of susceptibility was an impaired proinflammatory cytokine and chemokine response in the lungs, and among these, IL-17A was found to be a critical mediator of host defense and clearance of the organism from the lung (43). Here we report that an essential cell type required for clearing A. fumigatus from the lungs, CD11b+ Ly-6G+ neutrophils (4), appears to also be a cellular source of Dectin-1-dependent IL-17A. With the recent identification of human Dectin-1 polymorphisms (12) and the fact that the highest cellular expression of Dectin-1 in humans is on neutrophils (44), our observations further illuminate the role of both Dectin-1 and neutrophils in defense against A. fumigatus.
We initiated our study by demonstrating that immune cells collected from enzymatic lung digestion 18 h after A. fumigatus exposure were capable of producing IL-17A in vitro, which was highly dependent on Dectin-1. We chose to isolate lung cells 18 h postinfection, followed by 24 h of culture, since we felt that this best represented events occurring in the lungs within the first 48 h of infection. This provided us with an experimental system to pursue cytokines that were required for lung IL-17A production and potentially which cell type(s) was a source of early, Dectin-1-dependent IL-17A production after lung A. fumigatus challenge. The most surprising findings employing this system were that neutrophils were a Dectin-1-dependent cellular source of IL-17A and that IL-23 but not IL-1β, IL-6, or IL-18 was required for optimal IL-17A production. TGF-β is an additional cytokine critical for IL-17A production (21); however, TGF-β was not produced in the lungs in a Dectin-1-dependent manner after A. fumigatus exposure (nor in lung cell cultures; unpublished data) and thus was a low-priority candidate for driving IL-17A production. In contrast, IL-23 was a high-priority candidate in that we have previously reported that IL-23 is produced at low levels in the lungs in a Dectin-1-dependent manner after A. fumigatus exposure (43). In turn, we have demonstrated here that lung cells from Dectin-1-deficient mice had reduced Il23 mRNA expression and BMDCs from Dectin-1-deficient mice failed to produce IL-23 upon stimulation with A. fumigatus, suggesting the strong possibility that IL-23 was a needed factor for IL-17A production. Subsequently, IL-17A production by WT lung cells was reduced by half when IL-23 was neutralized, indicating that IL-23 was required for optimal IL-17A production by lung cells. However, since neutralization of IL-23 in the lung cell cultures resulted in only a 50% reduction in IL-17A production, we hypothesize that an additional cytokine(s) may also be playing a role in Dectin-1-dependent IL-17A production by lung cells/CD11b+ Ly-6G+ cells. Studies are under way to identify other cytokines differentially expressed in lung cell cultures from WT versus Dectin-1-deficient mice and to determine whether they play a role in IL-17A production.
The most well-described cellular source of IL-17A is CD4+ T cells (17). In addition, γδ+ T cells are a known source of IL-17A in several models, including A. fumigatus infection (32). In fact, anti-CD3-stimulated αβ+ and γδ+ T cells isolated from p47phox-deficient mice demonstrated heightened IL-17A production in vitro (32). However, a caveat to this observation was the 3-fold-higher A. fumigatus lung burden in these mice at the time points examined (32), which may play a role in amplifying IL-17A production by these cells types, especially in p47phox-deficient mice, which are known to hyperrespond to A. fumigatus (26). Nevertheless, published data indicated that a focus on T cell sources of IL-17A during A. fumigatus infection was warranted. However, we did not observe a significant presence of CD4+ T cells or γδ+ T cells (less than 1% in WT and Dectin-1-deficient mice; data not shown) in lung cells from either WT or Dectin-1-deficient mice at the time point we employed in our studies (18 h postinfection). In turn, stimulating lung cells with PMA/I, which is extensively utilized for both CD4+ T cell and γδ+ T cell intracellular IL-17A production (21) (23), did not result in a significant induction of intracellular Dectin-1-dependent IL-17A in CD3+ cells.
To focus our efforts on identifying a Dectin-1-dependent cell source(s) of IL-17A in the lungs during A. fumigatus infection, we determined the cellular composition of lung cells isolated from WT and Dectin-1-deficient mice. These results indicated that CD11b+ Ly-6G+ cells, commonly defined as neutrophils, constituted the majority of cells in the lungs within the first 18 h after A. fumigatus exposure, which therefore positioned this cell type as a candidate for the source of innate Dectin-1-dependent IL-17A. We then applied intracellular cytokine staining to determine which cell population(s) produced IL-17A in a Dectin-1-dependent manner. We discovered that intracellular IL-17A detection in myeloid cells was most consistently observed when the cells were cultured overnight with the addition of a protein transport inhibitor in the second half of the culture. Employing this method allowed us to elucidate that CD11b+ lung cells were positive for intracellular IL-17A and were reduced by 45% in Dectin-1-deficient mice. We subsequently demonstrated that Ly-6G+ lung cells also expressed intracellular IL-17A and were also at a significantly lower level in Dectin-1-deficient mice. Finally, intracellular IL-17A was observed in WT thioglycolate-elicited peritoneal neutrophils stimulated with A. fumigatus, and these cells secreted IL-17A in a Dectin-1-dependent manner. Collectively, our data support a new role, i.e., IL-17A production by CD11b+ Ly-6G+ neutrophils, for the essential cell type required for elimination of A. fumigatus from the lungs. In other experiments, we asked two additional questions. (i) What is the effect of neutrophil depletion on IL-17A levels in the lungs? (ii) can adoptive transfer of Dectin-1+ neutrophils augment IL-17A production in Dectin-1-deficient mice? In neutrophil depletion experiments, we were unable to determine an inoculum that did not result in a significantly higher lung fungal burden, which consequently resulted in higher IL-17A levels (both in lung homogenates and in lung cell cultures; data not shown). These results lead us to hypothesize that other cell types have the capacity to produce IL-17A when the A. fumigatus lung burden becomes exacerbated. We hypothesize that by this point in the infection, IL-17A-mediated defenses are likely immunopathogenic and along with a higher fungal burden may contribute to mortality. Previous studies in CDG mice support this contention (32). In neutrophil transfer experiments, we were unable to observe an effect on IL-17A production in Dectin-1-deficient mice that received Dectin-1+ neutrophils (data not shown). This outcome could be dependent on many factors, such as the number and timing of neutrophils being transferred. However, we hypothesize that the most likely explanation is that the lung environment of Dectin-1-deficient mice, i.e., impaired production of IL-1α, IL-1β, and tumor necrosis factor alpha (TNF-α) among others, is not conducive to activation/survival of the transferred Dectin-1+ neutrophils. Moreover, since Dectin-1-deficient mice have compromised IL-23 production, it is quite likely that transferred Dectin-1+ neutrophils would not receive the proper signals required for IL-17A production. One possible limitation of our study is that additional A. fumigatus-induced IL-17A-producing cells are present in the lungs and remain to be identified. However, our goal for this study was to not identify every cell type producing IL-17A but rather to identify which cell(s) produced IL-17A specifically in a Dectin-1-dependent manner. To this end, we have determined that CD11b+ Ly-6G+ neutrophils are a source of Dectin-1-dependent IL-17A. Clearly, a goal in future studies is to identify an additional IL-17A-producing cell population(s) in our model. However, we feel that more sensitive methods will need to be employed (an IL-17A reporter mouse, for example), which we anticipate will be a more efficient method than intracellular cytokine staining for identifying these IL-17A+ cells.
In regard to neutrophil IL-17A production, a recent report has shown that intranasal lipopolysaccharide (LPS) administration led to recruitment of CD4+ cells, CD8+ cells, and neutrophils, all of which were positive for Il17a mRNA expression by nonquantitative reverse transcriptase PCR (RT-PCR) (11). In a model of vasculitis induced by myeloperoxidase (MPO)-specific antineutrophil cytoplasmic autoantibodies (ANCA), IL-17A+ neutrophils were identified by intracellular flow cytometry in casein-elicited peritoneal exudate cells when stimulated with ANCA in the presence of a C. albicans cell wall antigen (13). Upon purification from peritoneal exudate cells, neutrophils produced IL-17A upon ANCA stimulation, albeit at extremely low levels (3 pg/ml versus 0.5 pg/ml in unstimulated cells) (13). A more recent study employing a model of kidney ischemia-reperfusion injury identified IL-17A-producing cells collected from kidney digests as being Gr-1+ using an IL-17A secretion assay (20). Similar to our study, this study identified IL-17A-producing neutrophils in an organ digest cell preparation (kidney). Adoptive transfer of CD11b+ Gr-1+ neutrophils isolated from Il17a−/− bone marrow resulted in attenuated kidney injury, suggesting that IL-17A produced by neutrophils was mediating kidney injury (20). Although adoptive transfer of purified bone marrow Gr-1+ neutrophils mediated IL-17A-dependent pathology in vivo, production of IL-17A by purified neutrophils in vitro was not directly tested in this study (20). We further hypothesize that IL-17A production by neutrophils requires additional “help,” both in vivo and in the presence of other cells in vitro. Indeed, Ly-6G+ cells purified from the lung were unable to produce IL-17A in vitro unless other cells, such as CD11b+ and CD11c+ cells, were present. Similarly, although neutrophils were more enriched in thioglycolate-elicited peritoneal lavages, small numbers of other cell types were present. Moreover, the addition of recombinant IL-23 to lung cells from both WT and Dectin-1-deficient mice resulted in significantly increased IL-17A production; however, IL-23 supplementation failed to induce IL-17A production in purified Ly-6G+ cells, again demonstrating the requirement for the presence of additional myeloid cells. Thus, we cannot exclude the possibility that cell contact between CD11b+ Ly-6G+ neutrophils and other cells in the lung digest (or present in thioglycolate-elicited peritoneal cells) in the presence of IL-23 and possibly other secreted mediators are required for optimal Dectin-1-dependent IL-17A production. Indeed, integrins such as CD11b/CD18 (1) and surface receptors such as TREM-1 (29) are critical for neutrophil activation and inflammatory responsiveness and thus may be playing a role in the interactions of CD11b+ Ly-6G+ cells and other lung cells in driving IL-17A production.
In summary, we have identified CD11b+ Ly-6G+ cells, i.e., neutrophils, as one cell type capable of producing Dectin-1-dependent IL-17A during invasive fungal infection with A. fumigatus. We can partially attribute lower IL-17A levels in the lungs of A. fumigatus-challenged Dectin-1-deficient mice to several observations: (i) impaired recruitment of CD11b+ Ly-6G+ neutrophils, i.e., a lower number of IL-17A-producing cells in the lungs, (ii) lower IL-23 production in lung cells/dendritic cells, i.e., inadequate levels of factors required for optimal IL-17A induction, and (iii) lower Il17a mRNA in purified Ly-6G+ neutrophils and lower IL-17A production by A. fumigatus-stimulated thioglycolate-elicited peritoneal neutrophils, i.e., an inherent defect in neutrophil IL-17A production. Our studies also suggest that soluble mediators in addition to IL-23 may also be playing a role in Dectin-1-dependent IL-17A production by CD11b+ Ly-6G+ neutrophils. In addition, we are the first to report that Rorc, Rora, Ahr, and Irf4 mRNA expression during A. fumigatus lung infection is not dependent on Dectin-1, further suggesting that there may be additional Dectin-1-dependent mechanisms that mediate lung IL-17A production during A. fumigatus infection. In conclusion, our data provide additional insight into the complex role of Dectin-1 in antifungal immunity and the generation of IL-17A responses.
ACKNOWLEDGMENTS
We thank Lee Quinton, Boston University, and Hubert Tse, University of Alabama at Birmingham, for helpful discussions and critical reading of the manuscript.
This work was supported by Public Health Service grants AI068917, HL080317, and HL096702.
Footnotes
Published ahead of print on 1 August 2011.
REFERENCES
- 1. Abram C. L., Lowell C. A. 2009. The ins and outs of leukocyte integrin signaling. Annu. Rev. Immunol. 27:339–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Averill M. M., et al. 2011. S100A9 differentially modifies phenotypic states of neutrophils, macrophages, and dendritic cells. Circulation 123:1216–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baddley J. W., Stroud T. P., Salzman D., Pappas P. G. 2001. Invasive mold infections in allogeneic bone marrow transplant recipients. Clin. Infect. Dis. 32:1319–1324 [DOI] [PubMed] [Google Scholar]
- 4. Barnes P. D., Marr K. A. 2007. Risks, diagnosis and outcomes of invasive fungal infections in haematopoietic stem cell transplant recipients. Br. J. Haematol. 139:519–531 [DOI] [PubMed] [Google Scholar]
- 5. Brustle A., et al. 2007. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 8:958–966 [DOI] [PubMed] [Google Scholar]
- 6. Chai L. Y., et al. 2010. Aspergillus fumigatus conidial melanin modulates host cytokine response. Immunobiology 215:915–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chamilos G., et al. 2010. Generation of IL-23 producing dendritic cells (DCs) by airborne fungi regulates fungal pathogenicity via the induction of T(H)-17 responses. PLoS One 5:e12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheung Y. Y., et al. 2007. Impaired neutrophil activity and increased susceptibility to bacterial infection in mice lacking glucose-6-phosphatase-β. J. Clin. Invest. 113:784–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Curtis M. M., Way S. S. 2009. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology 126:177–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Denning D. W. 1998. Invasive aspergillosis. Clin. Infect. Dis. 26:781–803 [DOI] [PubMed] [Google Scholar]
- 11. Ferretti S., Bonneau O., Dubois G. R., Jones C. E., Trifilieff A. 2003. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J. Immunol. 170:2106–2112 [DOI] [PubMed] [Google Scholar]
- 12. Ferwerda B., et al. 2009. Human dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med. 361:1760–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoshino A., et al. 2008. MPO-ANCA induces IL-17 production by activated neutrophils in vitro via classical complement pathway-dependent manner. J. Autoimmun. 31:79–89 [DOI] [PubMed] [Google Scholar]
- 14. Ivanov I. I., et al. 2006. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126:1121–1133 [DOI] [PubMed] [Google Scholar]
- 15. Kondo T., Takata H., Matsuki F., Takigucki M. 2009. Cutting edge: Phenotypic characterization and differentiation of human CD8+ T cells producing IL-17. J. Immunol. 182:1794–1798 [DOI] [PubMed] [Google Scholar]
- 16. Kontoyiannis D. P., Bodey G. P. 2002. Invasive aspergillosis in 2002: an update. Eur. J. Clin. Microbiol. Infect. Dis. 21:161–172 [DOI] [PubMed] [Google Scholar]
- 17. Korn T., Bettelli E., Oukka M., Kuchroo V. K. 2009. IL-17 and Th17 cells. Annu. Rev. Immunol. 27:485–517 [DOI] [PubMed] [Google Scholar]
- 18. Langrish C. L., et al. 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201:233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. LeibundGut-Landmann S., et al. 2007. Syk- and Card9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 8:630–638 [DOI] [PubMed] [Google Scholar]
- 20. Li L., et al. 2010. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J. Clin. Invest. 120:331–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mangan P. R., et al. 2006. Transforming growth factor induces development of the TH17 lineage. Nature 441:231–234 [DOI] [PubMed] [Google Scholar]
- 22. Marr K. A., Patterson T., Denning D. 2002. Aspergillosis pathogenesis, clinical manifestations and therapy. Infect. Dis. Clin. North Am. 16:875–894 [DOI] [PubMed] [Google Scholar]
- 23. Martin B., Hirota K., Cua D. J., Stockinger B., Veldhoen M. 2009. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity 31:321–330 [DOI] [PubMed] [Google Scholar]
- 24. Mathur A. N., et al. 2007. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J. Immunol. 178:4901–4907 [DOI] [PubMed] [Google Scholar]
- 25. Michel M. L., et al. 2007. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J. Exp. Med. 204:995–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morgenstern D. E., Gifford M. A., Li L. L., Doerschuk C. M., Dinauer M. C. 1997. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J. Exp. Med. 185:207–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Osorio F., et al. 2008. DC activated via dectin-1 convert Treg into IL-17 producers. Eur. J. Immunol. 38:3274–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patterson T. F., et al. 2000. Invasive aspergillosis. Disease spectrum, treatment practices, and outcomes. I3 Aspergillus Study Group. Medicine 79:250–260 [DOI] [PubMed] [Google Scholar]
- 29. Radsak M. P., Salih H. R., Rammensee H. G., Schild H. 2004. Triggering receptor expressed on myeloid cells-1 in neutrophil inflammatory responses: differential regulation of activation and survival. J. Immunol. 172:4956–4963 [DOI] [PubMed] [Google Scholar]
- 30. Rivera A., et al. 2011. Dectin-1 diversifies Aspergillus fumigatus-specific T cell responses by inhibiting T helper type 1 CD4 T cell differentiation. J. Exp. Med. 208:369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roark C. L., Simonian P. L., Fontenot A. P., Born W. K., O'Brien R. L. 2008. gammadelta T cells: an important source of IL-17. Curr. Opin. Immunol. 20:353–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Romani L., et al. 2008. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature 451:211–215 [DOI] [PubMed] [Google Scholar]
- 33. Rose C. E., et al. 2010. Murine lung eosinophil activation and chemokine production in allergic airway inflammation. Cell Mol. Immunol. 7:361–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reference deleted.
- 35. Singh N. 2003. Fungal infections in the recipients of solid organ transplantation. Infect. Dis. Clin. North Am. 17:113–134 [DOI] [PubMed] [Google Scholar]
- 36. Song C., et al. 2008. IL-17-producing alveolar macrophages mediate allergic lung inflammation related to asthma. J. Immunol. 181:6117–6124 [DOI] [PubMed] [Google Scholar]
- 37. Sugimoto K., et al. 2008. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118:534–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takatori H., et al. 2009. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 206:35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tassi I., et al. 2009. Requirement of phospholipase C-gamma2 (PLCgamma2) for Dectin-1-induced antigen presentation and induction of TH1/TH17 polarization. Eur. J. Immunol. 39:1369–1378 [DOI] [PubMed] [Google Scholar]
- 40. Taylor P. R., et al. 2007. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat. Immunol. 8:31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van de Veerdonk F. L., et al. 2009. The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe 5:329–340 [DOI] [PubMed] [Google Scholar]
- 42. Veldhoen M., et al. 2008. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453:106–109 [DOI] [PubMed] [Google Scholar]
- 43. Werner J., et al. 2009. Requisite role for the Dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J. Immunol. 182:4938–4946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Willment J. A., et al. 2005. The human beta-glucan receptor is widely expressed and functionally equivalent to murine Dectin-1 on primary cells. Eur. J. Immunol. 35:1539–1547 [DOI] [PubMed] [Google Scholar]
- 45. Yang X. O., et al. 2008. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28:29–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yao Z., et al. 1995. Human IL-17: a novel cytokine derived from T cells. J. Immunol. 155:5483–5486 [PubMed] [Google Scholar]