

Abstract
The transmembrane protein Tim-3 has been shown to negatively regulate T-cell-dependent immune responses and was recently demonstrated to be associated with the phenomenon of immune exhaustion, which can occur as a consequence of chronic viral infection. Unlike other negative regulators of T-cell function (e.g., PD-1), Tim-3 does not contain any obvious inhibitory signaling motifs. We have found that ectopic expression of Tim-3 in T cells leads to enhancement of T-cell receptor (TCR)-dependent signaling pathways, which was observed at the level of transcriptional reporters and endogenous cytokine production. We have exploited this observation to dissect what elements within the cytoplasmic tail of Tim-3 are required for coupling to downstream signaling pathways. Here we have demonstrated that two of the more membrane-proximal cytoplasmic tail tyrosines are required for Tim-3 signaling to T-cell activation pathways in a redundant fashion. Furthermore, we show that Tim-3 can directly bind to the Src family tyrosine kinase Fyn and the p85 phosphatidylinositol 3-kinase (PI3K) adaptor. Thus, at least under conditions of short-term stimulation, Tim-3 can augment T-cell activation, although this effect can be blocked by the inclusion of an agonistic antibody to Tim-3. These findings should help further the study of Tim-3 function in other physiological settings, such as those that lead to immune exhaustion.
INTRODUCTION
During the expansion phase of an immune response to acute infection, newly activated antigen-specific T cells expand rapidly and acquire effector functions. This is then followed by a period of contraction, where all but 5 to 10% of these effector T cells succumb to apoptosis. The remaining T cells constitute the memory pool—multifunctional T cells that persist in the host in an antigen-independent manner with the ability to respond quickly upon reexposure to viral antigen. However, during chronic viral infection, effector CD8+ T cells generated during the expansion phase fail to develop into memory CD8+ T cells. Instead, these effector CD8+ T cells appear to become exhausted (2, 33).
T-cell exhaustion is characterized as the progressive and stepwise loss of the ability to secrete interleukin 2 (IL-2), tumor necrosis factor alpha (TNF-α), and gamma interferon (IFN-γ) in response to antigenic stimulation, culminating (in the most extreme cases) in apoptosis (33). This system of clonal deletion has been documented under conditions of persistent antigen stimulation, such as high-grade chronic viral infections in both mouse and human and, most recently, in patients with advanced melanoma. Exhausted CD8+ T cells have a distinct molecular signature that resembles that of effector T cells more than that of memory T cells (34). Of the several hundred genes differentially upregulated in exhausted CD8+ T cells, some are inhibitory receptors, such as CTLA-4, LAG-3, and PD-1. Several studies have confirmed that PD-1 is upregulated on exhausted CD4+ and CD8+ T cells (4, 6, 7, 10). However, blocking the interaction between PD-1 and its ligand does not always completely restore the effector functions of exhausted T cells, which suggests the involvement of other receptors. Recently it was shown that blocking Tim-3/Tim-3L interactions, along with those of PD-1/PD-1L, has an additive and sometimes synergistic effect on the invigoration of exhausted T cells, either in the setting of a chronic viral infection (16, 17) or in a solid tumor (15, 27).
Tim-3 is a type I glycoprotein receptor whose expression often parallels that of PD-1 under conditions of chronic inflammation. Prior to its implication in T-cell exhaustion, Tim-3 was shown to be important in the induction of tolerance and suppression of effector T-cell function (20, 30). Recent studies show that Tim-3 may promote the suppression of T-cell function by expanding myeloid-derived suppressor cells (MDSC). This activity appears to be mediated by an interaction between galectin-9 (one of the known Tim-3 ligands) expressed by MDSC and Tim-3 on CD4+ T cells (9). The cooperation between Tim-3 and PD-1 in maintaining T-cell exhaustion indicates that these two receptors either employ the same signaling pathway (quantitative effect) or distinct signaling pathways (qualitative effect) when ligated. Despite the wealth of literature on the in vivo effects of these receptors, relatively little is known of their signaling mechanisms, although PD-1 has been more extensively studied in this regard (25, 26). The PD-1 cytoplasmic tail contains ITIM and ITSM motifs. Ligation of either CD3 alone or with CD28 and PD-1 leads to the recruitment of the tyrosine phosphatase SHP-2, predominantly to the ITSM motif of PD-1. The Tim-3 cytoplasmic tail has six well-conserved tyrosines, although there are no obvious inhibitory signaling motifs. One of these tyrosines has been shown to be phosphorylated in HEK293 cells stimulated with pervanadate (32), but the role of the cytoplasmic tail tyrosines in the downstream signaling of Tim-3 in T cells has not yet been determined.
In this study, we explored the possible function of the tyrosines in the cytoplasmic tail of Tim-3, using a system that we employed in the past to show that Tim-1 is a costimulatory molecule while Tim-2 is inhibitory, observations that have since been validated by other groups. Interestingly, we found that expression of Tim-3 augments activation of T-cell receptor (TCR)- and CD28-dependent signaling pathways that lead to increased NFAT/AP-1 and NF-κB-dependent transcription, as well as downstream signaling leading to IL-2 production. This activity requires the cytoplasmic tail of Tim-3 and is regulated by phosphorylation of one or more tyrosines.
MATERIALS AND METHODS
Antibodies and reagents.
Antibody to the Jurkat TCR was purified from the C305.2 hybridoma, which was obtained from ATCC (Manassas, VA). Antibodies to human and mouse CD28 (10F3 and 37.51, respectively) and mouse CD3 (2C11) were from Caltag/Invitrogen (Carlsbad, CA). Polyclonal antibody to murine Tim-3 was from R&D Systems (Minneapolis, MN). Anti-Flag antibody M2, phenyl phosphate, and peroxide were from Sigma (St. Louis, MO). Doxycycline, phorbol myristate acetate (PMA), ionomycin, sodium orthovanadate, aprotinin, leupeptin, pepstatin, and 4-(2-aminoethyl)benzene sulfonyl fluoride (AEBSF) were from Calbiochem/EMD Biosciences (San Diego, CA). Antiphosphotyrosine antibody 4G10 and anti-Fyn were from Millipore (Billerica, MA). Horseradish peroxidase (HRP)-conjugated antimouse and protein A were from Pierce and GE Healthcare (Piscataway, NJ), respectively. [γ-32P]ATP was from Perkin Elmer (Waltham, MA). Peptide N-glycosidase F (PNGase F) was from New England BioLabs (Beverly, MA). Directly conjugated fluorescent antibody to phospho-S6 was obtained from Cell Signaling Technology (Danvers, MA). Phospho-specific antibodies to phospholipase C-γ1 (PLC-γ1) (Y783) and phospho-extracellular signal-regulated kinase (ERK) (T202/Y204) were from BD Biosciences (San Diego, CA). Near-full-length Fyn was expressed in Sf9 insect cells with an N-terminal His tag as described previously for the homologous Src family members Lyn, Hck, and c-Src (31). Recombinant Fyn was purified from 1 liter of infected Sf9 cells using a combination of ion-exchange and immobilized metal affinity chromatography. The purity and concentration of the final kinase preparation were confirmed by SDS-PAGE.
DNA constructs.
Luciferase reporter constructs have been described previously (14, 19). Wild-type murine Tim-3 cDNA was obtained by reverse transcription-PCR (RT-PCR), starting with whole-spleen RNA made from C57BL/6 mice. Briefly, first-strand cDNA was generated using the ImProm-II reverse transcription system from Promega (Madison, WI). Full-length Tim-3 (without the signal sequence) was then amplified with specific primers and cloned into a mammalian expression vector containing a signal sequence followed by a Flag tag, as described previously for Tim-1 (14). An IMAGE clone containing the full-length human TIM-3 (hTIM-3) cDNA was obtained from Open Biosystems. The open reading frame (ORF) was amplified by PCR and cloned into the expression vector pCDEF3. Truncation mutants were generated by PCR with Expand high-fidelity polymerase from Roche (Indianapolis, IN). Point mutants of Tim-3 were generated with the QuikChangeII site-directed mutagenesis kit from Stratagene (San Diego, CA). Tetracycline (Tet)-inducible Tim-3 was generated by subcloning full-length Tim-3 into the pBI-EGFP vector from Clontech/BD Biosciences (San Jose, CA). The CD2-Tim3 chimera was generated by fusing the ecto- and transmembrane (TM) domains of human CD2 with the cytoplasmic tail of murine Tim-3. All constructs were verified by automated sequencing. Primers for PCR and sequencing were obtained from Operon (Huntsville, AL).
Transfections and luciferase assays.
Jurkat, D10, and 293T cells were cultured and transiently transfected as previously described (14, 19, 22). Luciferase assays were conducted as described previously (18, 22). To generate Jurkat T cells stably expressing Tim-3, cells were transfected with full-length, Flag-tagged, murine Tim-3. Two days later, cells were cultured in medium containing 2 mg/ml G418 and aliquoted into 96-well plates for eventual selection of single colonies. For selection of cells with Tet-inducible Tim-3 expression, Jurkat T cells were transfected with the pBI-EGFP-Tim3 construct and an otherwise empty mammalian expression vector (pCDEF3) containing the G418 resistance cassette.
SDS-PAGE/Western blotting.
Cells were stimulated in microcentrifuge tubes as appropriate at 37°C. Cells were pelleted at 13,200 rpm for 30 s. Pellets were lysed with ice-cold 1% NP-40 lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl) supplemented with AEBSF, aprotinin, leupeptin, pepstatin, sodium fluoride, sodium orthovanadate, and beta-glycerophosphate. Lysates were then incubated on ice for 10 min before being cleared by centrifugation. Sample buffer (2×; Bio-Rad, Hercules, CA) was added to the cleared lysates before loading onto 10% SDS-PAGE gels. Proteins were transferred onto polyvinylidene difluoride (PVDF) membranes, probed with 4G10 antibody, and detected with anti-mouse HRP. Blots were imaged on a Kodak ImageStation 4000R system. Images were exported in JPEG format and assembled in the Canvas software program.
Kinase assays.
Flag-tagged Tim-3 was purified from transfected 293T cells with anti-Flag beads, followed by extensive washing and elution with Flag peptide. Purified Tim-3 was mixed with recombinant Fyn in kinase buffer (5 mM morpholinepropanesulfonic acid [MOPS], pH 7.2, 2.5 mM glycerol 2-phosphate, 4 mM MgCl2, 5 mM MnCl2, 1 mM EGTA, 0.4 mM EDTA, 0.25 mm dithiothreitol [DTT], 20 μM [32P]ATP ATP [200 μCi/ml]). Reactions were carried out at 30°C and terminated by the addition of 2× SDS sample buffer. Samples were separated by SDS-PAGE and blotted to PVDF. Membranes were initially exposed to X-ray film and then rehydrated, blocked, and probed for Flag-tagged Tim-3.
SH2 domain arrays.
Membranes with recombinant SH2 domains were obtained from Panomics (Santa Clara, CA) and processed according to the manufacturer's instructions. The biotinylated Tim-3 peptide used to probe the array was generated at the University of Pittsburgh Peptide Synthesis Core facility and was used at 0.33 μg/ml (approximately 0.2 μM). Where indicated, competition was performed in the presence of 200 mM phenyl phosphate.
Flow cytometry and ELISA.
Tet-inducible Tim-3 Jurkat T cells were induced for 2 days with doxycycline and then stimulated as appropriate for measurement of intracellular phospho-S6, phospho-ERK, or secreted IL-2. For intracellular Phospho-flow analysis, cells were fixed in methanol immediately after stimulation for the appropriate times and then processed for staining with antibody specific for phospho-S6 or phospho-ERK. All flow cytometry was carried out on a BD LSR II instrument. Data were exported and analyzed in the FlowJo software program (Treestar, Ashland, OR). For IL-2 ELISA, cells were stimulated for 24 h with anti-TCR/CD28 antibodies or PMA-ionomycin. Cell-free supernatants were analyzed by ELISA for human IL-2, using OptEIA matched antibodies (BD Bioscience).
Retroviral infections.
Purified murine CD4+ T cells were stimulated under Th1 conditions and infected with recombinant MSCV retroviruses expressing the indicated constructs, essentially as previously reported (3). Infected T cells were then restimulated, again under Th1 conditions, followed by intracellular cytokine staining 2 days later for IFN-γ.
RESULTS
The Tim-3 cytoplasmic tail couples to signaling pathways associated with T-cell activation.
We previously reported that ligation of Tim-3 can augment the activation of dendritic cells (DCs) (1), in contrast to its previously described ability to induce apoptosis of Th1 T cells. Paradoxically, we also found that Tim-3 ligation was capable of inducing NF-κB activation in both DCs and T cells (1). We were intrigued by the fact that the Tim-3 cytoplasmic tail contains six conserved tyrosine residues (Fig. 1 A), which might couple to intracellular signaling pathways that regulate T-cell activation (either positively or negatively). We therefore expressed murine Tim-3 in T-cell lines to study the signals responsible for such effects, similar to previous work from our group on Tim-1 and Tim-2 (14, 19). As shown in Fig. 1B (left panel), expression of full-length Tim-3 in Jurkat T cells (which lack endogenous Tim-3; data not shown) enhanced NFAT/AP-1 reporter activation, both in the basal state (i.e., with no further stimulation) and in conjunction with TCR/CD28 cross-linking. To determine whether signaling to NFAT/AP-1 requires the cytoplasmic tail of Tim-3, we generated two truncation mutations. Truncation 1 (T1) contains all but the three most C-terminal tyrosines, while truncation 2 (T2) lacks all but one tyrosine, which is predicted to reside very close to the membrane (Fig. 1A). The T1 construct functioned at least as well as wild-type (WT) Tim-3 to augment NFAT/AP-1 reporter activity (Fig. 1B). Conversely, the shorter T2 truncation construct lost all ability to enhance NFAT/AP-1 activity. All Flag-tagged Tim-3 constructs were expressed at equivalent levels (Fig. 1B, right panel). Consistent with our previous demonstration of Tim-3-induced IκB degradation in a T-cell clone (1), Tim-3 expression in Jurkat T cells induced activation of an NF-κB transcriptional reporter, again in a manner requiring the more N-terminal tyrosine residues (data not shown). We also found that human TIM-3 could enhance induction of NFAT/AP-1 (Fig. 1C) and NF-κB (data not shown), thus demonstrating that these effects of Tim-3 are conserved. In addition, the human TIM-3 construct was not epitope tagged, ruling out any secondary effects of the Flag tag contained in the mouse construct used above.
Fig. 1.

Ectopic Tim-3 expression augments activation of NFAT/AP-1 and NF-κB. (A) Alignment of the C-terminal regions of murine and human Tim-3. The boundary of the TM/cytoplasmic tail is indicated, as are truncations discussed below. Relevant tyrosines are indicated with asterisks. (B) Effect of ectopic wild-type or truncated (Trunc.) Tim-3 expression on NFAT/AP-1 induction in Jurkat T cells (left panel). Luciferase activity is presented as absolute light units. Equivalent transfection efficiency was confirmed by expression of a cotransfected GFP (not shown). Equal expression of the constructs was confirmed by flow cytometry (right panel). (C) Enhancement of NFAT/AP-1 induction by human TIM-3. Jurkat T cells were transfected with Flag-tagged mTim-3 or untagged hTIM-3. NFAT/AP-1 luciferase activity was determined after a 6-h stimulation (left panel). Human TIM-3 expression was confirmed by flow cytometry (right panel). Luciferase results in panel C are the percentage of the maximal stimulation obtained with PMA-ionomycin (PMA/Iono). Results shown in panels B and C are the average ± SD for triplicate samples from a single experiment, representative of at least three that were performed in each case.
Requirements for downstream signaling of Tim-3.
To investigate the role of individual tyrosine residues in Tim-3 signaling, we focused on two tyrosines retained in the T1 but not T2 Tim-3 construct (Fig. 1A). Therefore, we mutated tyrosines at positions 256 and 263 to phenylalanine, both individually and together. As with the truncation constructs shown in Fig. 1, these mutations had no detectable effect on Tim-3 expression (data not shown). By abolishing the ability of these tyrosines to be phosphorylated, we hoped to abolish binding of downstream mediators that may interact directly and indirectly with the Tim-3 cytoplasmic tail when it is phosphorylated. Interestingly, we observed that full-length Tim-3 carrying the Y256/263F double mutation loses most but not all of its ability to enhance NFAT/AP-1 activation by anti-TCR/CD28 stimulation (Fig. 2 A). However, the Y256/263F double mutation on the T1 background led to a much more dramatic loss of NFAT/AP-1 activation (Fig. 2B). Mutating either Y256 or Y263 individually to phenylalanine did not affect the ability of Tim-3 to enhance NFAT/AP-1 activation (data not shown). We also observed a similar decrease in activity of the Y256/263F mutant, with more of an effect on the T1 background, in cells transfected with an NF-κB luciferase reporter (Fig. 2C), suggesting that common receptor-proximal signaling proteins couple Tim-3 to both NFAT and NF-κB induction. Importantly, the same effects observed in Jurkat T cells with each Tim-3 construct described above were also obtained in a nontransformed T cell line—the Th2 clone D10.G41—using the NFAT/AP-1 luciferase reporter as a readout (Fig. 2D). Thus, Y256 and Y263 may be able to compensate for each other's loss. Furthermore, while the dominant activity of the more C-terminal residues appears to be inhibitory in nature, a somewhat paradoxical positive role is revealed when Y256 and Y263 are mutated.
Fig. 2.

Requirements for transcriptional activation by Tim-3. Jurkat T cells (A to C) were transfected with an NFAT/AP-1 (A and B) or NF-κB (C) reporter plus the indicated Tim-3 constructs. D10 T cells (D) were also transfected with an NFAT/AP-1 reporter and the indicated Tim-3 constructs. Jurkat T-cell lines lacking expression of either ZAP-70 (E) or SLP-76 (F) were transfected with NFAT/AP-1-luciferase and the indicated constructs. The next day, cells were left unstimulated (white bars) or stimulated with anti-TCR/CD28 monoclonal antibodies (MAbs) (black bars). Luciferase activity is presented as the percentage of maximal stimulation obtained with PMA-ionomycin. Luciferase activity is presented as the percentage of the maximal response obtained with PMA plus ionomycin. Results shown are the average ± SD of data for triplicate samples from a single experiment, representative of at least three that were performed in each case.
Next, we focused further on the TCR signaling pathway to better define the requirements for Tim-3 signaling to NFAT and NF-κB. We took advantage of the existence of mutant Jurkat T-cell lines lacking expression of ZAP-70 and SLP-76, a critical tyrosine kinase and adaptor, respectively. As shown in Fig. 2E, expression of wild-type Tim-3 in ZAP-70-deficient Jurkat T cells (35) led to a modest, although reproducible, increase in basal NFAT/AP-1 reporter activity (white bars). This level of reporter activity was not enhanced further with TCR/CD28 stimulation (black bars), consistent with the strict requirement for ZAP-70 in the TCR signaling pathway. In ZAP-70 mutant cells transiently reconstituted with wild-type ZAP-70, we observed higher basal activity. We also explored the function of Tim-3 in a Jurkat mutant line (J.14) lacking expression of the adaptor protein SLP-76 (37), which is critical for nucleation of signaling complexes downstream of the TCR/CD3 complex. Thus, expression of Tim-3 in J.14 cells led to a small increase in NFAT/AP-1 reporter activity that was not enhanced by TCR/CD28 stimulation (Fig. 2F). Similar results were seen in both cell lines with an NF-κB reporter (data not shown). These results indicate that Tim3-mediated T-cell activation shares signaling pathways employed by TCR/CD3.
The results shown in Fig. 1 and 2 demonstrate that tyrosines in the cytoplasmic tail of Tim-3 are involved in its ability to augment TCR/CD28 signaling. However, the function of the ectodomain of Tim-3 remained more cryptic. In order to examine the roles of the ecto- and cytoplasmic domains of Tim-3 in more detail, we constructed a series of chimeric receptors containing various domains from murine Tim-3 (Fig. 3 A). All of these constructs were efficiently expressed at the cell surface of transfected T cells (data not shown). First, we deleted either the IgV domain alone or most of the ectodomain from full-length Tim-3 (with an extracellular Flag tag). As shown in Fig. 3B, these deletion constructs still retained the costimulatory effect of Tim-3 expression observed in the presence of TCR/CD28 ligation (dark-gray bars). While the activity of full-length (FL) Tim-3 was not affected by anti-Flag cross-linking, activity of the Del.IgV construct was slightly enhanced. However, deletion of both the IgV and mucin domains (Del.Ecto) did not allow for a cross-linking-mediated increase in Tim-3 activity. These somewhat paradoxical results suggest that the IgV domain actually inhibits dimerization and/or association of Tim-3 with another receptor that helps to mediate its activity in a mucin domain-dependent fashion. Finally, we examined whether the cytoplasmic tail of Tim-3 is sufficient to mediate any downstream signaling in the context of a heterologous ectodomain, for which we constructed a CD2-Tim3 chimera. As shown in Fig. 3C, expression of this chimera in Jurkat T cells led to a dose-dependent costimulatory effect on NFAT/AP-1 induction, in conjunction with anti-TCR/CD28 stimulation. However, the activity of CD2-Tim3 was not modulated by cross-linking the chimera with anti-CD2 antibody, and similar results were observed in a murine T cell line, D10 (data not shown). Thus, the results in Fig. 1 to 3 demonstrate that the cytoplasmic tail is necessary and sufficient for Tim-3 signaling to NFAT/AP-1, although we cannot rule out modulating effects of the ectodomain in Tim-3 function.
Fig. 3.

The cytoplasmic domain of Tim-3 is sufficient to costimulate TCR/CD28-induced NFAT/AP-1. Jurkat T cells were transfected with an NFAT/AP-1 reporter and the indicated constructs and then stimulated as indicated before determination of luciferase activity. (A) Schematic of Tim-3 constructs used in these experiments. (B) Effect of ectodomain deletions on Tim-3 costimulation of NFAT/AP-1. (C) Costimulation of NFAT/AP-1 activation by a CD2-Tim3 chimera.
Inducible tyrosine phosphorylation of Tim-3.
We next proceeded to determine more directly whether the tyrosine residues in Tim-3 examined above were available for phosphorylation. We transfected the constructs discussed in Fig. 1 and 2 into cells and treated the cells with pervanadate, a protein tyrosine phosphatase inhibitor and a potent inducer of tyrosine phosphorylation. Flag-tagged Tim-3 constructs were immunoprecipitated (IP'd) from cell lysates and then probed with antibody to phosphotyrosine. As shown in Fig. 4 A, wild-type Tim-3 was efficiently phosphorylated, with a partial loss of phosphorylation of the T1 construct (compare antiphosphotyrosine signals [upper panel] to total Tim-3 [lower panel]). Note that the majority of the Tim-3 protein is contained in a rather large smear (indicated by the solid arrow), which appears to be the result of extensive glycosylation. This was confirmed by treatment of IPs with PNGase F, which resulted in a single band at the molecular weight predicted for Tim-3 (data not shown), equivalent in size to the minor fraction of Tim-3 denoted by the dashed arrow in Fig. 4A and subsequent figures. However, the T2 construct was incapable of being phosphorylated even by pervanadate, consistent with its one remaining tyrosine being closely juxtaposed to the predicted transmembrane domain (see Fig. 1A). We next examined tyrosine phosphorylation of the Tim-3 constructs with individual tyrosine point mutations, all on the T1 construct background. Mutation of both Y256 and Y263 (“2YF”) abolished the phosphorylation of Tim-3 by pervanadate, with the mutation of Y256 having a partial effect (Fig. 4B). In contrast, we observed no detectable effect of the Y263F mutation on overall levels of tyrosine phosphorylation, suggesting either that this site is not recognized efficiently by the antiphosphotyrosine antibody or that it is phosphorylated at low stoichiometry. Finally, we examined the tyrosine phosphorylation of Tim-3 in the context of a T cell. To improve the signal in these experiments, IPs were treated with PNGase F prior to SDS-PAGE, resulting in a more compact Tim-3 band. Thus, induction of Tim-3 expression on these cells with doxycycline consistently revealed basal tyrosine phosphorylation of Tim-3, which was rather modestly increased after TCR/CD28 stimulation (Fig. 4C). The specificity of the phosphotyrosine signal was confirmed by preincubating cells with the Src kinase inhibitor PP2, which completely reversed both the basal and inducible tyrosine phosphorylation of Tim-3 (data not shown). These findings are consistent with the reporter assays shown in Fig. 1 and 2 and provide further support for the model that phosphorylation of the Tim-3 cytoplasmic tail is important for its function.
Fig. 4.
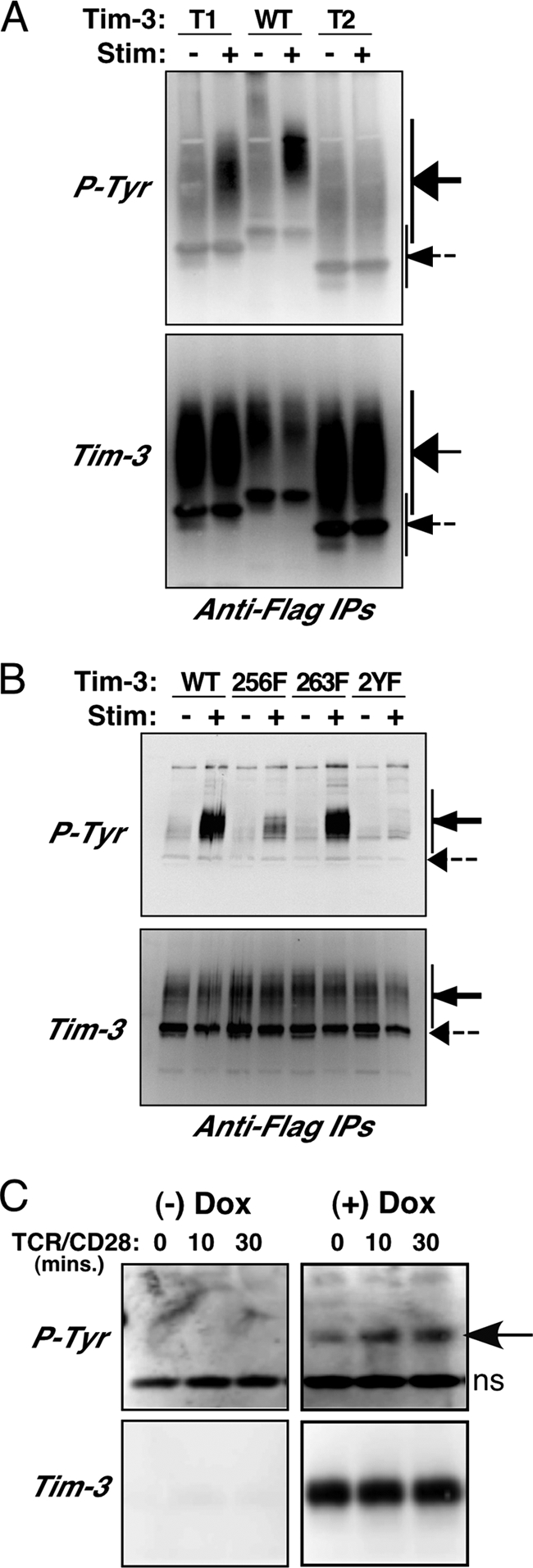
Availability of Tim-3 cytoplasmic tyrosine residues for phosphorylation. (A and B) 293T cells were transfected with the indicated Flag-tagged Tim-3 constructs and then split and left unstimulated or treated with pervanadate. Lysates were prepared and subjected to IP with an anti-Flag antibody, followed by SDS-PAGE and Western blotting. Blots were first probed for Tim-3 (lower panel) and then stripped and reprobed with an antiphosphotyrosine antibody (upper panel). (C) Parental Jurkat T cells or Jurkat T cells stably transfected with Flag-tagged Tim-3 were stimulated as indicated and prepared for IP and Western blotting as described above. After IP, samples were treated with PNGase F prior to SDS-PAGE.
The results discussed above suggest that phosphorylation of tyrosines 256 and/or 263 in Tim-3 couples this protein to TCR signaling pathways. To begin to map this pathway in more detail, we performed a solid-phase screen for SH2 domains that might interact with phosphorylated Tim-3. The various SH2 domains contained on the commercially available membrane are shown in Fig. 5 A. This membrane was probed with a biotinylated and phosphorylated peptide corresponding to the region around Y256 and Y263 in the cytoplasmic tail of Tim-3 (Fig. 5A). As shown in Fig. 5B, this peptide interacted strongly with several SH2 domains, including those from the Src family kinase Fyn and the phosphatidylinositol 3-kinase (PI3K) adaptor p85. Weaker interactions were noted with SH2 domains from PLC-γ1, RasGAP1, and two additional Src-family members, Hck and c-Yes. We also performed this assay in the presence of excess phenylphosphate, which was used to compete off phospho-specific interactions (Fig. 5C). While most of the interactions were effectively eliminated by phenylphosphate, the interaction with the Fyn SH2 domain was not, suggesting that this interaction either is of very high affinity or is independent of phosphorylation. We sought to confirm this binding by performing an IP experiment with full-length Tim-3 and endogenous Fyn. Thus, control or Flag-Tim3-transfected Jurkat T cells were stimulated and then subjected to IP with an anti-Flag antibody. As shown in Fig. 5D, IP of Flag-Tim-3 resulted in co-IP of endogenous Fyn expressed in these cells, an interaction that did not appear to vary significantly with stimulation of the cells. Conversely, p85 PI3K recruitment to Tim-3 required tyrosine phosphorylation of Tim-3, since the binding was abolished in pervanadate-stimulated cells when the Y256/263F form of Tim-3 was used (Fig. 5E). The results shown in Fig. 5 suggest that expression of Tim-3 on T cells results in constitutive recruitment of the Fyn tyrosine kinase to the cell surface via its interaction with Tim-3, while recruitment of p85 PI3K is dependent on inducible phosphorylation of Tim-3.
Fig. 5.

Identification of SH2 domain-containing proteins capable of interacting with the cytoplasmic tail of Tim-3. (A) Detail of the phospho-peptide derived from the Tim-3 sequence (top) used to screen a solid-phase array containing recombinant SH2 domains from the indicated proteins (bottom). (B) Representative results of screening the array with the Tim-3 phospho-peptide. (C) Representative results of a screen performed with the same array and peptide, except in the presence of 200 mM phenyl phosphate. (D) Confirmation of activation-independent interaction of Fyn with Tim-3. Control or Tim3-expressing Jurkat T cells were stimulated (Stim) for the indicated times with anti-TCR antibody (TCR) or pervanadate (PV), lysed, and subjected to IP with anti-Flag antibody. Western blots were probed for endogenous Fyn (upper panel) or the transfected Tim-3 (lower panel). (E) Confirmation of p85 PI3K binding to Tim-3. Jurkat T cells were transfected with the indicated plasmids and stimulated with pervanadate. Anti-Flag IPs were performed and analyzed by blotting for p85, followed by anti-Flag.
Although a previous report proposed that tyrosine 256 in the cytoplasmic tail of Tim-3 could be phosphorylated by the Tec family kinase Itk (32), our analysis of the sequence around tyrosines 256 and 263 with the ScanSite algorithm (23, 24) revealed that it was likely that a Src family kinase could phosphorylate one or both of these sites (data not shown). Thus, we expressed wild-type Tim-3 either alone or together with the Src family tyrosine kinase Lck or Fyn in 293 cells and then stimulated the cells with pervanadate. As shown in Fig. 6 A, when Tim-3 was expressed alone, stimulation with pervanadate induced significant, although somewhat transient, tyrosine phosphorylation. However, when Lck was coexpressed with Tim-3, stimulation led to more robust and sustained phosphorylation (Fig. 6A). Similar results were obtained when the Fyn tyrosine kinase was coexpressed with Tim-3, although in this case the basal level of Tim-3 phosphorylation was consistently higher than that when Lck was expressed with Tim-3. These results demonstrate that both Lck and Fyn can mediate phosphorylation of Tim-3 and that Fyn may do so more efficiently. The level of phosphorylation observed after stimulation of cells transfected with Tim-3 alone is most likely the result of endogenous tyrosine kinases expressed in the 293 cells. In an attempt to determine whether Src kinases (particularly Fyn) might directly phosphorylate Tim-3, we performed in vitro kinase assays with WT Tim-3 or a tyrosine mutant (“5YF,” in which all five tyrosines discussed above were mutated to phenylalanine). Thus, the purified Tim-3 protein was incubated with purified Fyn kinase and 32P-labeled ATP. As shown in Fig. 6B, while WT Tim-3 was phosphorylated by Fyn, the 5YF construct was not. Similar results were obtained with the Y256/263F mutant form of the Tim-3 T1 construct (data not shown). These results demonstrate that Fyn can directly phosphorylate tyrosines in the cytoplasmic tail of Tim-3, although the relative efficiency and stoichiometry of this putative phosphorylation are still unclear.
Fig. 6.

Role of Src kinases in Tim-3 phosphorylation and function. (A) 293T cells were transfected with full-length Tim-3, alone or with Lck or Fyn. Cells were stimulated with pervanadate for the indicated times. Tim-3 was IP'd from lysates, and the presence of Tim-3 was first confirmed by Western blotting (lower panel), followed by stripping and reprobing with an antiphosphotyrosine (P-Tyr) antibody (upper panel). (B) In vitro kinase assay showing direct phosphorylation of Tim-3 by Fyn (upper panel). Purified wild-type Tim-3 or a version lacking all cytoplasmic tyrosines (except for the membrane proximal tyrosine) was subjected to an in vitro kinase assay with purified active Fyn kinase. Levels of purified Tim-3 were confirmed by Western blotting (lower pnael). (C) Lck-deficient JCaM.1 cells were transfected with an NFAT/AP-1 reporter and the indicated plasmids. Cells were stimulated the next day as indicated. Luciferase activity is presented as the percentage of the maximal response obtained with PMA and ionomycin. Tim-3 expression was verified by flow cytometry (not shown).
To understand how Src family tyrosine kinase interaction with and phosphorylation of Tim-3 might contribute to downstream signaling, we focused further on Lck and Fyn. J.CaM.1 is an Lck-deficient Jurkat T-cell line (29) with detectable endogenous levels of Fyn (data not shown). To determine if Tim-3 requires Lck and/or Fyn to enhance TCR/CD28 signaling, we transfected J.CaM.1 cells with wild-type Tim-3 and either Lck or Fyn and then assayed for modulation of NFAT/AP-1 activity in the presence or absence of signals from TCR/CD28. Transfection of J.CaM.1 cells with Tim-3 alone led to a small but reproducible increase in NFAT/AP-1 reporter activity (Fig. 6C, second set of bars). This was surprising given the lack of any detectable increase after TCR/CD28 stimulation of these cells. Coexpression of Lck enhanced the ability of Tim-3 to activate NFAT/AP-1, either alone or together with anti-TCR/CD28, to levels similar to what we observed in parental Jurkat T cells (Fig. 1 and 2). J.CaM.1 cells cotransfected with Fyn and Tim-3 also demonstrated enhanced NFAT/AP-1 activity (Fig. 6C, last set of bars), although to a lesser extent than in the presence of Lck. Similar results were obtained in J.CaM.1 cells transfected with an NF-κB reporter (data not shown). Thus, while Lck expression is required for optimal Tim-3 (and any TCR) function, the residual Fyn kinase activity in J.CaM.1 cells appears to allow some level of Tim-3 function, most likely due to the direct and constitutive interaction between the two proteins.
Intersection of Tim-3 signaling with TCR/CD3 signaling pathways.
Next, we sought to connect our observations of increased NFAT/AP-1 and NF-κB reporter activity in the presence of Tim-3 to endogenous TCR signaling and activation pathways. To facilitate these experiments, we established a Jurkat T-cell line with a stably integrated, inducible Tim-3 construct. This line, like the parental Jurkat cells, is uniformly negative for Tim-3 expression until induction by doxycycline, after which the cells are Tim-3+ (and GFP+, due to a coexpressed reporter) by flow cytometry (data not shown). First, we examined the inducible tyrosine phosphorylation of signaling intermediates important for TCR signaling. The substrate that yielded the most consistent difference in tyrosine phosphorylation in the presence of Tim-3 expression was PLC-γ1 (at Y783), as shown in Fig. 7 A. Enhanced phosphorylation of PLC-γ1 is consistent with the binding of Fyn to Tim-3. Furthermore, consistent with enhanced activation of PLC-γ1, we also noted a Tim3-dependent increase in phosphorylation of the ERK mitogen-activated protein (MAP) kinase (Fig. 7B), which is a downstream target of PLCγ1-induced diacylglycerol production.
Fig. 7.

Augmented TCR signaling and cytokine production by Tim-3. (A) Tet-on-Tim3 or parental Jurkat T cells expressing only the reverse tetracycline transactivator (rtTA) were cultured for 2 days without or with doxycycline (Dox). Cells were stimulated as indicated for analysis of phospho-PLC-γ1 (Y783) by Western blotting, followed by probing for beta-actin as a loading control (left). Average results (± SD) from five separate experiments are shown (*, P < 0.05; **, P < 0.01) (right). (B and C) Tet-on-Tim3 Jurkat T cells were cultured for 2 days without or with Dox. Cells were then stimulated as indicated and processed for intracellular staining of phospho-ERK (average of results from three experiments ± SD; ***, P < 0.001) (B) or phospho-S6 (C). In addition, Dox-treated cells were subdivided into GFPlo (nonexpressing) and GFPhi (Tim3 expressing). (D) Tet-on-Tim3 Jurkat T cells were cultured with the indicated concentrations of Dox. Analysis by flow cytometry of Tim-3 expression (x axis) and a coexpressed GFP reporter (y axis) is shown (left). Cells were stimulated for 24 h with soluble anti-TCR/CD28 antibodies; cell-free supernatants were harvested, and secreted IL-2 was measured by ELISA (right).
Given our finding that a peptide derived from Tim-3 could interact with an SH2 domain of the p85 adaptor subunit of PI3K (Fig. 5), we next examined the effect of Tim-3 on a downstream target of the PI3K pathway: phosphorylation of the S6 ribosomal subunit. As shown in Fig. 7C (upper panels), stimulation of noninduced Tim-3 Jurkat T cells with an optimal dose of anti-TCR antibody resulted in some increase in phosphorylation of S6. This phosphorylation was reproducibly greater in the cells cultured in doxycycline, and therefore expressing Tim-3, and was further confirmed by gating on GFP+ cells after induction. The effect of Tim-3 on S6 phosphorylation was even more dramatic in the presence of a suboptimal dose of anti-TCR antibody (Fig. 7C, lower panels). Tim-3 expression did not detectably alter the phosphorylation of S6 when proximal TCR signaling was bypassed by PMA-ionomycin stimulation (data not shown). Next, we determined the effect of Tim-3 expression on IL-2 production. For this experiment, we cultured our inducible Tim-3 Jurkat clone in increasing concentrations of doxycycline in order to obtain graded expression of Tim-3. As shown in Fig. 7D, we were able to obtain cells with various amount of Tim-3 surface expression, as determined by flow cytometry for Tim-3 and a coexpressed GFP. We then stimulated these cells with a suboptimal amount of soluble anti-TCR/CD28 antibodies and collected supernatant for measurement of IL-2 secretion. Cells were also stimulated with PMA and ionomycin to provide an internal control (data not shown). As an additional control, we also stimulated the parental Jurkat clone, which expresses only the reverse tetracycline transactivator (rtTA) and not Tim-3. Thus, treatment of Jurkat with this suboptimal stimulus of soluble anti-TCR/CD28 resulted in no detectable IL-2 production except in the cases where Tim-3 expression was induced (Fig. 7D, right panel). Furthermore, the response correlated with the level of Tim-3 expression. Thus, expression of Tim-3 can enhance T-cell activation, as determined by the production of endogenous IL-2.
Finally, we sought to determine if Tim-3 expression could enhance the activation of primary T cells. Murine CD4+ T cells were isolated from lymph nodes, stimulated in vitro under Th1 conditions, and then infected with retroviruses encoding wild-type Tim-3 or the T1 or T2 truncation and restimulated for determination of cytokine production and proliferation. As shown in Fig. 8A, a much greater proportion of T cells expressing wild-type Tim-3 were capable of making IFN-γ upon restimulation than was the case for cells infected with GFP control virus. Consistent with data discussed above, the T1 construct was at least as efficient as wild-type Tim-3 at enhancing IFN-γ production, while the T2 construct was deficient in such activity. Similar results were obtained when secreted IFN-γ was measured in the supernatant of restimulated T cells infected with the same retroviral constructs (Fig. 8B). Given that Tim-3 has been most commonly described as a negative regulator of T-cell responses, we determined whether cross-linking Tim-3 with a previously described agonistic antibody would modulate the effects noted above. Thus, T cells were infected as described above and restimulated in the presence of a control antibody or an agonistic Tim-3 antibody (5D12). As shown in Fig. 8C, inclusion of this antibody in the restimulation significantly inhibited the stimulatory activity of Tim-3. Thus, while expression of Tim-3 on T cells can initially augment T-cell activation, cross-linking of the protein, at least under these conditions, leads to rapid inhibition of T-cell activation.
Fig. 8.

Costimulation of acute T-cell cytokine production by ectopic Tim-3 expression. Naive murine CD4+ T cells were stimulated with anti-CD3/CD28 antibodies under Th1 conditions and then infected with retrovirus encoding the indicated constructs and restimulated with the indicated concentrations of soluble anti-CD3 antibody, again under Th1 conditions. Two days later, cells were processed for intracellular cytokine staining and flow cytometry. (A) Representative flow cytometry of intracellular IFN-γ and retroviral coexpressed GFP. (B and C) T cells were restimulated with the indicated concentrations of anti-CD3, and supernatant was collected for determination of IFN-γ concentrations by ELISA.
DISCUSSION
In this study, we have demonstrated for the first time that Tim-3 can enhance signaling pathways that lead to T-cell activation, at least under the conditions of acute stimulation examined here. In T-cell lines stimulated through TCR/CD3 and CD28, Tim-3 enhances the secretion of IL-2 and the induction of transcription factors important for T-cell activation, i.e., NFAT, AP-1, and NF-κB. Importantly, we have also demonstrated that Tim-3 can enhance cytokine production and proliferation by primary T cells, again in conjunction with stimulation through CD3 and CD28. These activities of Tim-3 occurred without the addition of exogenous ligand, and structure/function studies suggested that cell surface expression of Tim-3 may be sufficient for its ability to augment T-cell activation. Conversely, we also found that at least one antibody to Tim-3 could prevent its enhancement of T-cell activation, so the extracellular domain of Tim-3 can indeed modulate its function under the right conditions, which is consistent with previous functional studies using this antibody (1).
It will be of interest in the future to determine how different Tim-3 antibodies and natural ligands can affect the signaling pathways delineated here. In addition to galectin-9 (38), Tim-3 has also been shown to interact with the apoptotic cell marker phosphatidylserine (PS) (11, 21) and possibly another, as yet unknown ligand (8). The stimulatory effects of acute Tim-3 expression that we have observed do not appear to require interaction with PS, since they occur in the Tet-inducible cell line, which contains minimal numbers (<1%) of apoptotic cells in the steady state (data not shown). In addition, we observed enhanced NFAT/AP-1 activity even after expression of Tim-3 lacking most of the ectodomain. However, these findings do not rule out a role for the ectodomain of Tim-3 in modulating its function, as discussed above.
We have also shown that five tyrosine residues within the Tim-3 cytoplasmic tail regulate T-cell signaling by Tim-3 in a complex fashion, with two tyrosines (residues 256 and 263 in mouse Tim-3 [mTim-3]) being particularly important. Although these tyrosines appear to be the most critical for Tim-3 function, their mutation to phenylalanine has the most dramatic effects when the more distal three tyrosines are also removed by truncation. While this finding suggests a role for phosphorylation of the distal tyrosines, it is still formally possible that the distal part of the Tim-3 cytoplasmic tail amplifies signaling via a mechanism that is independent of tyrosine phosphorylation. However, there are no obvious signaling motifs in this part of the protein. A previous report showed that Y265 in human TIM-3 (corresponding to Y256 in mouse Tim-3) is phosphorylated by the Tec family kinase Itk (32). While our data do not rule out a role for Itk in phosphorylation of Tim-3, our results demonstrate that both Lck and Fyn are capable of mediating Tim-3 phosphorylation and NFAT activation, with Fyn possibly being the more efficient of the two. This phosphorylation may then promote the recruitment of one or more SH2 domain-containing proteins to Tim-3, including the PI3K p85 adaptor proteins and possibly others (see the model in Fig. 9). Secondary recruitment of proteins like p85 could explain why the phosphorylation-independent binding of Fyn to Tim-3 is not sufficient to mediate downstream signaling to NFAT/AP-1 and NF-κB.
Fig. 9.
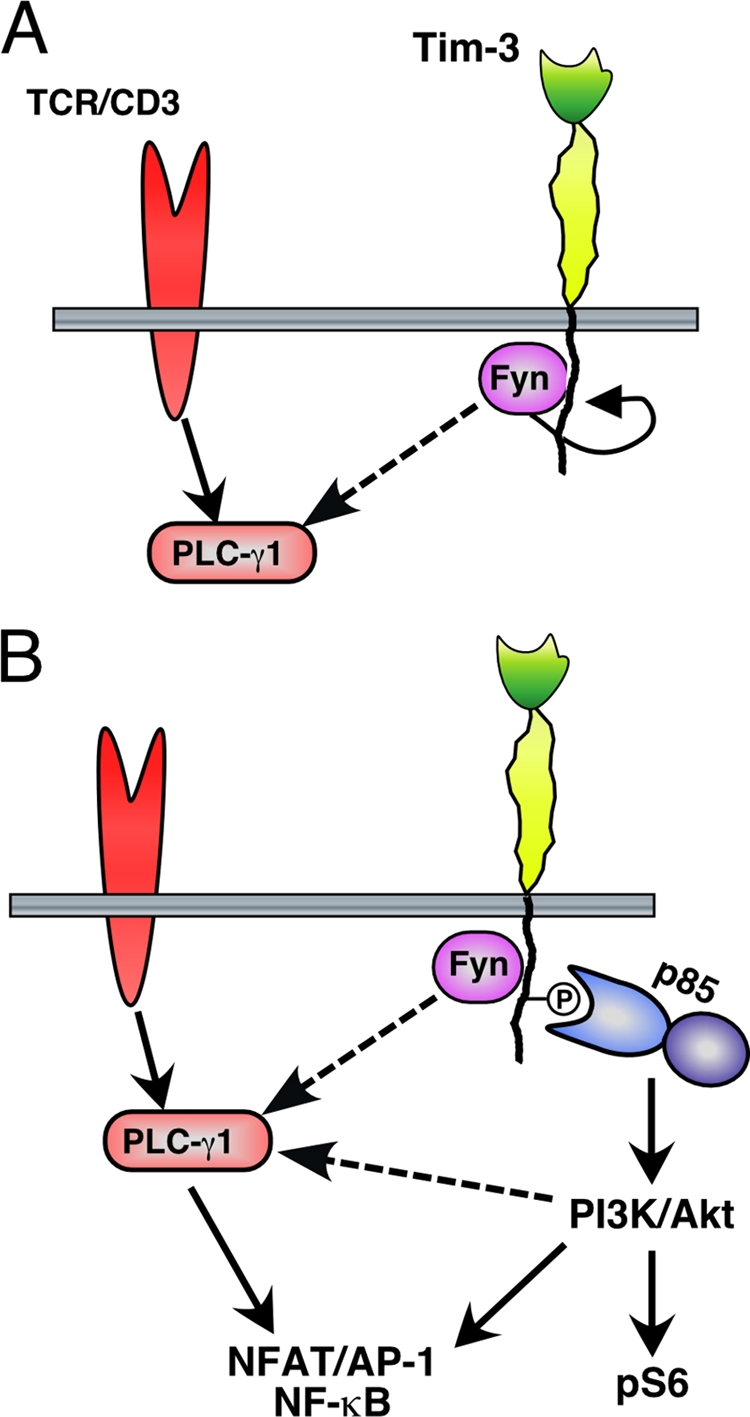
Model for acute enhancement of TCR signaling and T-cell activation by Tim-3.
The requirements for Src kinases and for ZAP-70 and SLP-76 in Tim3-mediated activation suggest that Tim-3 intersects closely with TCR signaling pathways. This finding is reminiscent of previous work on another Tim family member, Tim-1. For example, we showed that Tim-1 also requires proximal T-cell receptor signaling machinery in order to activate NFAT/AP-1 (13). In addition, it has been reported that in human T cells, Tim-1 can be cocapped with the TCR/CD3 complex (5). There may therefore be some similarities in the signaling pathways employed by Tim-1 and Tim-3. Nonetheless, while Tim-1 augments activation of NFAT/AP-1 but not NF-κB (14), we show here that Tim-3 can activate all these pathways. We do not believe that our results can be attributed merely to overexpression in a particular cell line. First, our previous studies of the Tim family proteins Tim-1 and Tim-2 with this system have yielded results largely consistent with those obtained with other approaches (12, 14, 19, 30). Also, although many of our reporter activation studies were initially carried out with the Jurkat human leukemic line, we have validated our findings with a nontransformed murine T-cell clone (D10) that, like Jurkat, does not express endogenous Tim-3. Finally, we have performed retroviral infection of primary murine T cells, which yielded findings very much in line with those obtained using the cell lines.
How can our results be reconciled with reported findings that Tim-3 contributes to T-cell exhaustion? One possibility is that early during the induction of T-cell exhaustion, Tim-3 enhances T-cell activation, which may augment or accelerate the acquisition of an exhausted phenotype. Gene expression array studies show that of the genes differentially upregulated in exhausted CD8+ T cells, a greater number of these genes overlap with effector T cells than with memory T cells (34). This suggests that exhausted T cells are effector T cells that have not completely differentiated into memory cells. Thus, the accentuated TCR/CD28-based signaling that occurs in the presence of Tim-3 may lead to exhaustion through the extension of an effector type phenotype and the corresponding frustration of memory development. From our studies here, such signaling appears to be mediated at least in part by tyrosine kinase recruitment and activation, although we cannot rule out other mechanisms at this point.
It is also possible that Tim-3 might have both inhibitory and stimulatory activities, as proposed for the related protein Tim-1. In the case of Tim-1, these opposing effects can be produced with two antibodies that both target Tim-1's IgV domain but with varying avidities. The “stimulatory” antibody 3B3 binds Tim-1 with an avidity 17 times higher than that of the “inhibitory” antibody RMT1-10 (36). It is postulated that 3B3 costimulates T-cell activation by bringing Tim-1 closer to the TCR/CD3 complex and allowing the formation of large macromolecular signaling clusters. RMT1-10, however, does not engage Tim-1 efficiently enough to have such stabilizing effects and may instead mediate abortive T-cell activation, akin to partial or antagonistic TCR ligands. Tim-3 has been shown to bind to both galectin-9 and at least one other ligand that remains to be identified (8). Thus, it is possible that depending on the ligand it associates with, Tim-3 might have either an inhibitory or stimulatory effect. This hypothesis is also consistent with the observation that although Tim-3 is upregulated on viral antigen-specific CD8+ T cells in both acutely and chronically infected mice, only viral antigen-specific Tim-3+ CD8+ T cells in chronically infected mice become exhausted (16). As discussed above, exhausted T cells appear to derive from effector T cells that are unable to completely differentiate into memory T cells. Receptors and transcription factors associated with T-cell exhaustion (e.g., PD-1 and Blimp-1) are also upregulated during acute infection (28) but do not seem to drive exhaustion unless there is persistent antigen stimulation. This suggests that during chronic inflammation, global changes occur to induce and maintain exhaustion in antigen-specific T cells. Therefore, it is distinctly possible that the ligands available to Tim-3 on the surface of T cells can vary between the different stages of activation leading up to exhaustion.
In the maintenance/late phase of T-cell exhaustion, exhausted T cells exhibit high basal levels of phosphorylated Erk1/2, p38, and STAT-5, which likely reflects continuous engagement of the TCR and CD28. However, these cells are also unable to respond to secondary stimulation, suggesting the active delivery of an inhibitory signal. Coblockade of PD-1 and Tim-3 has a greater restorative effect on exhausted T cells than blocking each receptor alone. This suggests that PD-1 and Tim-3 are both involved in maintaining exhaustion in T cells. PD-1 ligation inhibits T-cell effector function by inducing the recruitment of the phosphatase SHP-2, which dephosphorylates signaling mediators proximal to the TCR and CD28. How the putative “blocking” antibodies to Tim-3 work to restore T-cell function is not yet clear, but our studies strongly suggest that it is through a mechanism quite distinct from the blocking of PD-1.
ACKNOWLEDGMENTS
This work was supported by grants from the NIH to L.P.K. (AI067544 and AI073748), V.K.K. (AI073748), and T.E.S. (GM077629). This project was also supported by the Ragon Institute of MGH, MIT, and Harvard (to V.K.K.).
Footnotes
Published ahead of print on 1 August 2011.
REFERENCES
- 1. Anderson A. C., et al. 2007. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 318:1141–1143 [DOI] [PubMed] [Google Scholar]
- 2. Angelosanto J. M., Wherry E. J. 2010. Transcription factor regulation of CD8+ T-cell memory and exhaustion. Immunol. Rev. 236:167–175 [DOI] [PubMed] [Google Scholar]
- 3. Apetoh L., et al. 2010. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 11:854–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barber D. L., et al. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–687 [DOI] [PubMed] [Google Scholar]
- 5. Binne L. L., Scott M. L., Rennert P. D. 2007. Human TIM-1 associates with the TCR complex and up-regulates T cell activation signals. J. Immunol. 178:4342–4350 [DOI] [PubMed] [Google Scholar]
- 6. Blackburn S. D., Shin H., Freeman G. J., Wherry E. J. 2008. Selective expansion of a subset of exhausted CD8 T cells by anti-PD-L1 blockade. Proc. Natl. Acad. Sci. U. S. A. 105:15016–15021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blackburn S. D., et al. 2009. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 10:29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cao E., et al. 2007. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity 26:311–321 [DOI] [PubMed] [Google Scholar]
- 9. Dardalhon V., et al. 2010. Tim-3/galectin-9 pathway: regulation of Th1 immunity through promotion of CD11b+Ly-6G+ myeloid cells. J. Immunol. 185:1383–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Day C. L., et al. 2006. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–354 [DOI] [PubMed] [Google Scholar]
- 11. DeKruyff R. H., et al. 2010. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J. Immunol. 184:1918–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Souza A. J., Kane L. P. 2006. Immune regulation by the TIM gene family. Immunol. Res. 36:147–155 [DOI] [PubMed] [Google Scholar]
- 13. de Souza A. J., et al. 2008. T cell Ig and mucin domain-1-mediated T cell activation requires recruitment and activation of phosphoinositide 3-kinase. J. Immunol. 180:6518–6526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Souza A. J., Oriss T. B., O'Malley K., Ray A., Kane L. P. 2005. TIM-1 is expressed on in vivo-activated T cells and provides a co-stimulatory signal for T cell activation. Proc. Natl. Acad. Sci. U. S. A. 102:17113–17118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fourcade J., et al. 2010. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 207:2175–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jin H. T., et al. 2010. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. U. S. A. 107:14733–14738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jones R. B., et al. 2008. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 205:2763–2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kane L. P., Shapiro V. S. S., Stokoe D., Weiss A. 1999. Induction of NF-κB by the Akt/PKB kinase. Curr. Biol. 9:601–604 [DOI] [PubMed] [Google Scholar]
- 19. Knickelbein J. E., de Souza A. J., Tosti R., Narayan P., Kane L. P. 2006. Cutting edge: inhibition of T cell activation by TIM-2. J. Immunol. 177:4966–4970 [DOI] [PubMed] [Google Scholar]
- 20. Kuchroo V. K., Meyers J. H., Umetsu D. T., DeKruyff R. H. 2006. TIM family of genes in immunity and tolerance. Adv. Immunol. 91:227–249 [DOI] [PubMed] [Google Scholar]
- 21. Nakayama M., et al. 2009. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 113:3821–3830 [DOI] [PubMed] [Google Scholar]
- 22. Narayan P., Holt B., Tosti R., Kane L. P. 2006. CARMA1 is required for Akt-mediated NF-kappaB activation in T cells. Mol. Cell. Biol. 26:2327–2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Obenauer J. C., Cantley L. C., Yaffe M. B. 2003. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31:3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Obenauer J. C., Yaffe M. B. 2004. Computational prediction of protein-protein interactions. Methods Mol. Biol. 261:445–468 [DOI] [PubMed] [Google Scholar]
- 25. Okazaki T., Honjo T. 2007. PD-1 and PD-1 ligands: from discovery to clinical application. Int. Immunol. 19:813–824 [DOI] [PubMed] [Google Scholar]
- 26. Riley J. L. 2009. PD-1 signaling in primary T cells. Immunol. Rev. 229:114–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sakuishi K., et al. 2010. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 207:2187–2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shin H., et al. 2009. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 31:309–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Straus D. B., Weiss A. 1992. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell 70:585–593 [DOI] [PubMed] [Google Scholar]
- 30. Su E. W., Lin J. Y., Kane L. P. 2008. TIM-1 and TIM-3 proteins in immune regulation. Cytokine 44:9–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trible R. P., Emert-Sedlak L., Smithgall T. E. 2006. HIV-1 Nef selectively activates Src family kinases Hck, Lyn, and c-Src through direct SH3 domain interaction. J. Biol. Chem. 281:27029–27038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van de Weyer P. S., et al. 2006. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem. Biophys. Res. Commun. 351:571–576 [DOI] [PubMed] [Google Scholar]
- 33. Virgin H. W., Wherry E. J., Ahmed R. 2009. Redefining chronic viral infection. Cell 138:30–50 [DOI] [PubMed] [Google Scholar]
- 34. Wherry E. J., et al. 2007. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27:670–684 [DOI] [PubMed] [Google Scholar]
- 35. Williams B. L., et al. 1998. Genetic evidence for differential coupling of Syk family kinases to the T-cell receptor: reconstitution studies in a ZAP-70-deficient Jurkat T-cell line. Mol. Cell. Biol. 18:1388–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiao S., et al. 2007. Differential engagement of Tim-1 during activation can positively or negatively costimulate T cell expansion and effector function. J. Exp. Med. 204:1691–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yablonski D., Kuhne M. R., Kadlecek T., Weiss A. 1998. Uncoupling of nonreceptor tyrosine kinases from PLC-gamma1 in an SLP-76-deficient T cell. Science 281:413–416 [DOI] [PubMed] [Google Scholar]
- 38. Zhu C., et al. 2005. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 6:1245–1252 [DOI] [PubMed] [Google Scholar]