

Abstract
PC12 cells exhibit precise temporal control of growth factor signaling in which stimulation with epidermal growth factor (EGF) leads to transient extracellular signal-regulated kinase (ERK) activity and cell proliferation, whereas nerve growth factor (NGF) stimulation leads to sustained ERK activity and differentiation. While cyclic AMP (cAMP)-mediated signaling has been shown to be important in conferring the sustained ERK activity achieved by NGF, little is known about the regulation of cAMP and cAMP-dependent protein kinase (PKA) in these cells. Using fluorescence resonance energy transfer (FRET)-based biosensors localized to discrete subcellular locations, we showed that both NGF and EGF potently activate PKA at the plasma membrane, although they generate temporally distinct activity patterns. We further show that both stimuli fail to induce cytosolic PKA activity and identify phosphodiesterase 3 (PDE3) as a critical regulator in maintaining this spatial compartmentalization. Importantly, inhibition of PDE3, and thus perturbation of the spatiotemporal regulation of PKA activity, dramatically increases the duration of EGF-stimulated nuclear ERK activity in a PKA-dependent manner. Together, these findings identify EGF and NGF as potent activators of PKA activity specifically at the plasma membrane and reveal a novel regulatory mechanism contributing to the growth factor signaling specificity achieved by NGF and EGF in PC12 cells.
INTRODUCTION
To ensure proper conversion of a specific environmental input into a distinct cellular output, cells exploit a number of molecular mechanisms to tightly regulate signal transduction in space and time. In PC12 cells, specific controls of the duration of the activity of extracellular signal-regulated kinase (ERK), a canonical mitogen-activated protein kinase (MAPK), are believed to help determine distinct cell fates (32). Specifically, activation of epidermal growth factor receptor (EGFR) by epidermal growth factor (EGF) leads to transient ERK activity and cell proliferation, whereas nerve growth factor (NGF) binding to and activating its receptor, TrkA, leads to sustained ERK activity and signals the cells to differentiate (23). An accepted model for the growth factor (GF) signaling specificity in these cells involves the activation of specific GTPases capable of activating the Raf family of kinases, which activate MEK, the upstream activator of ERK. In particular, while both EGF and NGF can transiently activate the GTPase Ras to recruit Raf to the plasma membrane, where it can be activated, only NGF activates Rap1, a cyclic AMP (cAMP)-regulated GTPase also capable of activating Raf (23). Since this NGF-induced Rap1 activation is sustained, it is suggested that the selective activation of Rap1 by NGF but not EGF leads to the sustained phase of ERK activity and the initiation of neurite outgrowth. Furthermore, EGF-stimulated ERK negatively regulates Raf activity, whereas NGF-stimulated ERK exerts positive feedback on Raf activity, further contributing to the transient and sustained duration of ERK activity as a result of the respective stimuli (11, 22).
As an added level of complexity in signal transduction regulation, many canonical signaling cascades are subject to cross talk, in which the molecular players of one pathway alter the state of another. For example, it is known that the ERK pathway and the cAMP-mediated signaling pathway are intricately connected (28). While the precise regulation that these pathways have on one another is complex and cell type specific (28), it is widely accepted that in PC12 cells, two cAMP effectors, namely, cAMP-dependent protein kinase (PKA) and exchange protein directly activated by cAMP (Epac), can indirectly activate the Raf/MEK/ERK cascade (4, 5, 28).
Intracellular cAMP is enzymatically produced from ATP by adenylyl cyclases, either transmembrane adenylyl cyclase (tmAC) or soluble adenylyl cyclase (sAC) (10), and degraded by phosphodiesterases (PDEs), of which there are 11 known isoforms (2). In PC12 cells, NGF binds to TrkA, which activates sAC to produce cAMP (26). Subsequently, activated PKA and Epac converge to activate Rap1 (26), the aforementioned mediator of sustained ERK activity (36, 37). In contrast, EGF was not known to increase cAMP or activate PKA in PC12 cells (14). Interestingly, a number of studies have shown that when EGF is used in conjunction with cAMP-elevating agents, neurite outgrowth can be induced in PC12 cells (9, 14, 15, 35). These studies suggest that cAMP-mediated signaling may play a role in GF signaling specificity in PC12 cells and point to a simple model showing that lack of cAMP-mediated cross-regulation specifies the transient ERK activity stimulated by EGF. However, GF-stimulated cAMP increases and PKA activities have not been characterized in this system, and this model remains to be tested.
Characterization of cAMP-mediated signaling in a number of cell systems has revealed complex spatiotemporal regulation that is often achieved by the formation of localized signaling complexes containing the key regulatory and effector molecules involved in cAMP signaling. One critical component of these complexes, which are frequently formed with the help of scaffolding proteins known as A-kinase anchoring proteins (AKAPs), are PDEs (1). Localization of PDEs to distinct intracellular locales results in the formation of discrete cAMP gradients which are able to control the signaling of PKA and Epac with high specificity (1). Proper compartmentalization of PKA- and Epac-mediated signaling is necessary for normal cellular function and regulates processes such as excitation and contraction of cardiomyocytes and insulin secretion in pancreatic β cells (13, 29). In hippocampal neurons, PDEs play an important role in establishing localized pools of cyclic nucleotides in undifferentiated neurites, which ultimately regulates the process of axon and dendrite formation (24). Although cAMP-mediated signaling contributes to specifying the cell fates of PC12 cells as discussed above, the spatiotemporal regulation of cAMP signaling and its functional roles in this cell system have not been fully characterized.
In this study we investigated the spatiotemporal regulation of GF-mediated PKA activity in PC12 cells by employing genetically encoded fluorescence resonance energy transfer (FRET)-based biosensors targeted to distinct subcellular locales. In doing so, we observed that both NGF and EGF stimulate PKA activity at the plasma membrane with discrete temporal activity patterns, but both stimuli fail to activate the cytosolic pool of PKA. Using pharmacological perturbations, we identify PDE3 to be a critical regulator of this GF-stimulated compartmentalized PKA activity. Moreover, PKA and PDE3 are shown to regulate both the onset and duration of GF-stimulated ERK activity. When PDE3 is inhibited and the spatiotemporal regulation of PKA is disrupted, PC12 cells treated with EGF display heightened durations of nuclear ERK activity. Together these findings reveal the distinct NGF- and EGF-induced spatiotemporal patterns of PKA activity as a novel level of regulation underlying the precise GF signaling specificity in PC12 cells.
MATERIALS AND METHODS
Cellular culture and transfection.
PC12 cells were grown in Dulbecco modified Eagle medium (DMEM) cell culture medium supplemented with 10% fetal bovine serum (FBS) and 5% donor horse serum (DHS) at 37°C with 5% CO2. Cells were plated onto 35-mm glass-bottom dishes for imaging or 15-cm culture dishes for fractionation studies. When necessary, cells were transfected at 60 to 70% confluence via Lipofectamine 2000 (Invitrogen). Prior to treatment, cells were grown in reduced serum medium (5% FBS, 1% DHS) for 24 h.
Cellular imaging and analysis.
For imaging, cells were washed once and then imaged in Hanks' balanced salt solution on a Zeiss Axiovert 200 M microscope with a cooled charge-coupled-device camera (MicroMAX BFT512; Roper Scientific, Trenton, NJ) controlled by METAFLUOR software (Molecular Devices, Sunnyvale, CA). Dual emission ratio imaging was performed using a 420DF20 excitation filter and a 450DRLP dichroic mirror and appropriate emission filters, 475DF40 for cyan fluorescent protein (CFP) and 535DF25 for yellow fluorescent protein (YFP). Cells were treated with drug as indicated, and images were analyzed using ImageJ software (NIH).
PC12 cell fractionation and Western blots.
For PC12 cell fractionation, cells were harvested in TNE buffer containing 2 mM each of phenylmethylsulfonyl fluoride (PMSF), NaF, and NaVO4, 10 nM calyculin A, and a protease inhibitor cocktail (Roche) and lysed in a Dounce homogenizer. Lysate was spun at 5,000 × g for 10 min, and the supernatant was then spun at 13,000 × g for 35 min. The supernatant (cytosolic fraction) was separated from the pellet (membrane fraction), and the pellet was solubilized in TNE containing 1% Triton X-100. Bicinchoninic acid (BCA) assay (Pierce) was used to detect the total protein concentration in each fraction. Each fraction contained 40 μg of total protein, was separated on 7.5% SDS-PAGE, and then was transferred to nitrocellulose membranes. For Western blots, 24 h prior to treatment, PC12 cells were switched into reduced serum medium and then treated as indicated. Cells were then harvested and lysed in radioimmunoprecipitation assay (RIPA) buffer containing 2 mM each of PMSF, NaF, and NaVO4, 10 nM calyculin A, and a protease inhibitor cocktail. Lysate was incubated on ice for 30 min and then spun at 13,000 × g at 4°C for 30 min. The total protein concentration was detected by BCA assay. Proteins were separated on 7.5% SDS-PAGE and then transferred to nitrocellulose membranes. In both cases, membranes were blocked in 5% BSA and incubated overnight with primary antibody. Membranes were washed and incubated with secondary antibody, and ECL Western blotting substrate (Pierce) was used for detection. Densitometric analysis was performed using ImageJ (NIH). For the fractionation studies, the total PDE3 in each fraction was quantified by summing the PDE3 band intensity from each fraction.
RESULTS
NGF and EGF induce distinct spatiotemporal patterns of PKA activity.
To begin our studies, we looked at the activation of PKA downstream of TrkA and EGFR at the plasma membranes of PC12 cells, where sAC is expressed at high levels (26). To do so, we used a variant of the genetically encoded FRET-based biosensor A-kinase activity reporter 4 (AKAR4) targeted to the plasma membrane with the farnesylation lipid modification sequence derived from K-Ras (AKAR4-Kras) (Fig. 1A) (3). This biosensor detects an increase in plasma membrane-localized PKA activity as an increase in the yellow/cyan emission ratio, and the maximal response of AKAR4-Kras, as determined by simultaneous addition of the tmAC activator forskolin (Fsk) and the general PDE inhibitor 3-isobutyl-1-methylxanthine (IBMX), is 14.5% ± 2.9% (n = 6) [mean ± standard error of the mean (SEM) (n = number of cells)] (Fig. 1B, open squares). Since sAC expression is high at the plasma membrane in PC12 cells (26), activation of TrkA with NGF and subsequent sAC activation were expected to induce robust PKA activity near the plasma membrane. Indeed, when PC12 cells expressing AKAR4-Kras were treated with NGF, there was rapid and robust PKA activation, as is indicated by an increase in the yellow/cyan emission ratio of 12.6% ± 1.4% (n = 11) (Fig. 1C, purple diamonds, and D, top). We next wanted to examine the effect of EGF on PKA activity in PC12 cells. Since EGF was shown not to increase cAMP or activate PKA in PC12 cells (14, 36), it was unexpected when EGF induced an AKAR4-Kras response of 9.1% ± 1.2% (n = 12) (Fig. 1E, purple diamonds, and F, top). However, in contrast to the sustained PKA activity observed with NGF stimulation, EGF-induced PKA activity at the plasma membrane was transient, reversing back to basal levels within 15 min following treatment (Fig. 1E, purple diamonds, F, and G). Together these observations identify NGF and EGF as potent activators of PKA at the plasma membrane, although they induce temporally distinct patterns of PKA activity.
Fig. 1.

GF-induced PKA activity is spatiotemporally regulated in PC12 cells. (A) Schematic representation of AKAR4-Kras and AKAR4-NES. (B) PC12 cells expressing AKAR4-NES (n = 3) or AKAR4-Kras (n = 6) were treated with Fsk (50 μM) plus IBMX (100 μM). Y/C, yellow/cyan emission ratio. (C) Time courses depicting the NGF (200 ng/ml)-induced responses of AKAR4-Kras (n = 5) and AKAR4-NES (n = 5). (D) YFP images and ratiometric images of AKAR4-Kras (top) and AKAR4-NES (bottom) before NGF addition (0 min) and at 5 min and 12.5 min after NGF addition. (E) Time courses depicting the EGF (100 ng/ml)-induced responses of AKAR4-Kras (n = 6) and AKAR4-NES (n = 3). (F) YFP images and ratiometric images of AKAR4-Kras (top) and AKAR4-NES (bottom) before EGF addition (0 min) and at 5 min and 12.5 min after EGF addition. (G) Bar graph depicting the AKAR4-Kras responses at 2.5, 5, 10, and 12.5 min following NGF (n = 11) or EGF (n = 12) treatment. n.s, no statistically significant difference between indicated treatments; **, P ≤ 0.01; ***, P ≤ 0.005. All data are shown as means ± SEMs.
We next wanted to examine the effect that both GFs had on cytosolic PKA activity by using an AKAR4 variant targeted to the cytosol with a nuclear export signal (NES) (AKAR4-NES), which displays a maximal yellow/cyan emission ratio increase of 51.3% ± 1.8% (n = 5) in response to Fsk and IBMX in PC12 cells (Fig. 1A and B, solid squares). Surprisingly, in cells expressing AKAR4-NES, NGF treatment elicited a negligible AKAR4-NES response (0.7% ± 0.2%; n = 18), suggesting that NGF is incapable of activating cytosolic PKA (Fig. 1C and D). Similarly, no cytosolic PKA activity was observed when cells expressing AKAR4-NES were treated with EGF (Fig. 1E and F), showing that EGF-induced PKA activity is also subject to precise spatial compartmentalization. Since this trend of GF-stimulated plasma membrane-compartmentalized PKA activity was observed with both NGF and EGF, these observations point to a molecular signaling barrier that is responsible for localizing GF-induced PKA activity to the plasma membrane while restricting the activation of cytosolic PKA.
PDE3 is a major isoform in PC12 cells.
Since PDEs are negative regulators of PKA signaling and are known to serve as a signaling barrier or sink to help establish cAMP gradients in cells (1), we investigated if PDEs contribute to the plasma membrane (PM)-compartmentalized PKA activity downstream of growth factor receptor activation. When cells expressing AKAR4-Kras or AKAR4-NES were treated with the general PDE inhibitor IBMX, an increase in the yellow/cyan emission ratio of 18.4% ± 2.4% (n = 13) and 27.3% ± 6.3% (n = 9), respectively, was observed (Fig. 2A to C, black), indicating that PDEs play a role in regulating basal cAMP levels and thus PKA activity in PC12 cells. Next, we used the PDE3 isoform-selective inhibitor milrinone and the PDE4-selective inhibitor rolipram to examine the roles of PDE3 and PDE4 on regulating PKA activity in these cells. First, at both the membrane and cytosol, PDE3 inhibition with milrinone activated PKA to levels comparable to those induced by IBMX, with responses of 13.1% ± 0.9% (n = 16) for AKAR4-Kras and 23.7% ± 4.2% (n = 7) for AKAR4-NES (Fig. 2A to C, gray). Conversely, cells that were treated with rolipram showed negligible changes in PKA activity at the PM (3.4% ± 0.7%; n = 9) and in the cytosol (1.9% ± 0.5%; n = 15) (Fig. 2A to C). Using a cytosolic biosensor for cAMP levels, Epac1-camps (16, 18), we observed effects of PDE inhibition on cAMP levels similar to those that were observed on PKA activity, specifically that IBMX and milrinone generate similar levels of cAMP accumulation while rolipram generates less cAMP (Fig. 2D). In agreement with previous studies (34), together these data suggest that PDE3, in contrast to PDE4, is a major isoform in PC12 cells and regulates basal PKA activity in these cells.
Fig. 2.
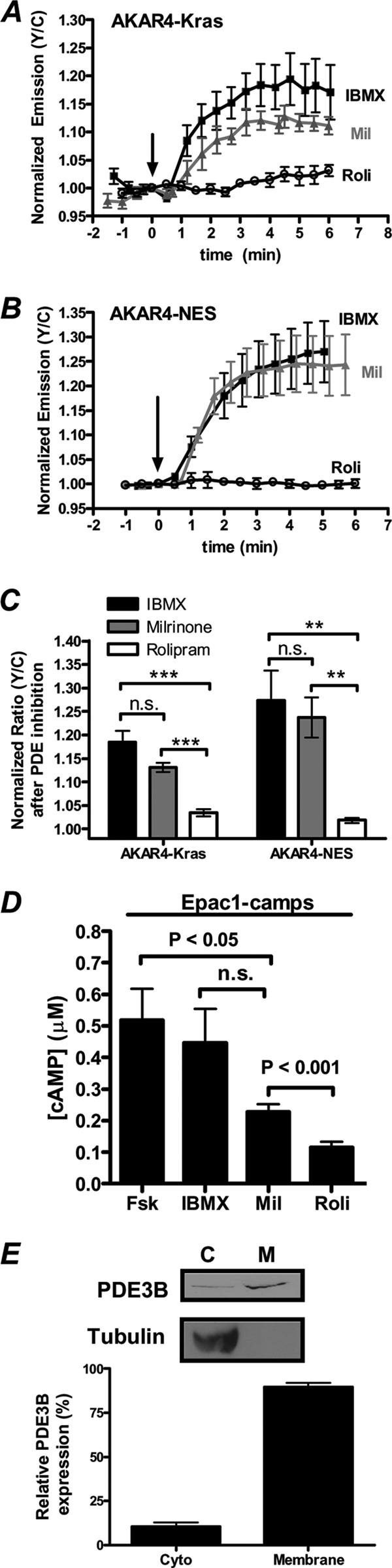
PDE3, but not PDE4, regulates both PM and cytosolic PKA activity in PC12 cells. (A) PC12 cells expressing AKAR4-Kras were treated with 100 μM IBMX (n = 4), 10 μM milrinone (Mil) (n = 5), or 1 μM rolipram (Roli) (open circles; n = 5), and PKA activity was monitored. (B) Cytosolic PKA activity was detected in cells expressing AKAR4-NES treated with 100 μM IBMX (n = 4), 10 μM Mil (n = 5), or 1 μM Roli (n = 6). (C) Responses of AKAR4-Kras and AKAR4-NES induced by IBMX (Kras [n = 13], NES [n = 9]), Mil (Kras [n = 16], NES [n = 7]), and Roli (Kras [n = 9], NES [n = 15]). (D) Bar graph depicting the cAMP concentration ([cAMP]) induced by various treatments, as determined by Epac1-camps [50 nM Fsk (n = 11), 100 μM IBMX (n = 8), 10 μM Mil (n = 15), 1 μM Roli (n = 11)]. (E) PC12 cells were separated into cytosolic (C) and membrane (M) fractions by centrifugation, and each fraction was separated by SDS-PAGE. Immunoblot analysis (top) is representative of one fractionation, and quantification of the relative expression of PDE3B in each fraction (bottom) is representative of four independent fractionation experiments and subsequent immunoblot analysis. Tubulin serves as a control for clean separation of the membrane and cytosolic fractions. All data are presented as means ± SEMs; “n.s.” indicates no statistically significant difference between indicated treatments. **, P < 0.01; ***, P < 1E−05.
Next, to confirm the expression of PDE3 in PC12 cells, we performed immunoblot analysis using a PDE3B antibody. Since PDE3B is known to be a membrane-associated protein (2), we sought to quantify the relative expression of PDE3B in the cytosolic and membrane compartments of PC12 cells. To do so, we separated the cytosolic and membrane fractions of PC12 cells via centrifugation and subsequently separated the proteins present in each fraction by SDS-PAGE. When both fractions were probed with an anti-tubulin antibody, the signal was observed predominantly in the cytosolic fraction, indicative of clean separation of the membrane from the cytosol. In contrast, when both fractions were probed for PDE3B, we observed 89.6% ± 2.6% of the total PDE3B in the membrane fraction (Fig. 2E). These data signify that PDE3B not only is present in PC12 cells but also is primarily membrane associated.
PDE3 is a critical regulator of the GF-stimulated spatiotemporal dynamics of PKA activity.
We next sought to investigate whether PDE3 contributes to the spatiotemporally regulated PKA activities downstream of TrkA and EGFR activation. To begin, we examined if PDE3 contributes to the spatial compartmentalization of GF-stimulated PKA to the plasma membrane. To do so, we concurrently treated cells expressing AKAR4-NES with GF and a low dose of milrinone (5 μM). Notably, while 1 μM milrinone generates a maximal response from AKAR4-Kras (data not shown), 5 μM milrinone induces a negligible response of 0.7% ± 0.6% from AKAR4-NES, indicating that this dose of milrinone does not activate cytosolic PKA (Fig. 3A, gray). Importantly, this observation is consistent with the notion that PDE3, while playing a role in regulating PKA activity at the plasma membrane in the basal state, is potent at restricting PKA activation in the cytosol. When NGF and a low dose of milrinone (5 μM) were simultaneously added to cells, an increase in PKA activity was observed (2.2% ± 0.3%; n = 12) (Fig. 3A and B). Similarly, an AKAR4-NES response of 2.6% ± 0.4% (n = 6) was detected when cells were concurrently treated with EGF and low-dose milrinone (Fig. 3A and C). Similar trends were observed using Epac1-camps; specifically, EGF and NGF did not produce detectable levels of cytosolic cAMP unless PDE3 was partially inhibited (Fig. 3D), and these trends were confirmed using cAMP enzyme immunoassays (EIA) (data not shown). Together these data suggest that cAMP produced by TrkA and EGFR activation is rapidly degraded by PDE3 and is only capable of diffusing into the cytosol when PDE3 is partially inhibited. In contrast to cells cotreated with GF and a low dose of milrinone, those cells that were simultaneously treated with GF and a submaximal dose of rolipram (0.5 μM) did not display detectable cytosolic PKA activity (Fig. 3E) or cAMP increases shown by cAMP EIA (data not shown). Taken with the observation that NGF and EGF are robust activators of PKA at the plasma membrane, the finding that partial pharmacological inhibition of PDE3 is necessary for both NGF and EGF to activate cytosolic PKA suggests that PDE3 functions to restrict the diffusion of plasma membrane-generated cAMP into the cytosol, where it could activate cytosolic PKA. As inhibition of PDE3 also alters the kinetics of EGF-induced PKA activity at the plasma membrane (Fig. 3F), these findings also show that pharmacological manipulation of PDE3 activity can be used as a tool to disrupt the spatiotemporal regulation of GF-mediated PKA signaling.
Fig. 3.

GFs activate cytosolic PKA when PDE3 is partially inhibited. (A) Bar graph depicting the negligible AKAR4-NES response induced by GF alone (white bars; NGF [n = 4] and EGF [n = 12]) and a subsaturating dose of milrinone (5 μM) (gray bars; n = 10). In contrast, when cells are simultaneously treated with GF and 5 μM milrinone (black bars; NGF [n = 12] and EGF [n = 6]), an increase in cytosolic PKA activity is observed. (B) Time course of PC12 cells expressing AKAR4-NES treated with NGF alone (n = 5) or NGF plus 5 μM milrinone (submil n = 3). (C) Time course depicting the AKAR4-NES response of cells treated with EGF alone (n = 3) or EGF plus 5 μM milrinone (n = 3). (D) Bar graph depicting the cAMP concentration induced by various treatments, as determined by the response of Epac1-camps (NGF [n = 8], EGF [n = 7], 5 μM Mil [n = 15], NGF plus 5 μM Mil [n = 14], and EGF plus 5 μM Mil [n = 10]). (E) A submaximal dose of rolipram (0.5 μM) does not activate cytosolic PKA in the absence (gray bars; n = 5) or presence (black bars; NGF [n = 11] and EGF [n = 9]) of GF. (F) The EGF-induced PKA activity at the plasma membrane (n = 12) is sustained in the presence of PDE3 inhibition with milrinone (10 μM) (n = 8) but not in the presence of PDE4 inhibition with rolipram (1 μM) (n = 6). All data are shown as means ± SEMs. *, P < 0.05.
cAMP-mediated signaling regulates GF-stimulated cytosolic ERK activity.
Given that cAMP and ERK signaling are intricately connected in PC12 cells (28), we next investigated how PDE3 and other components of cAMP signaling influence GF-induced ERK activity. To this end, we used the ERK activity reporter (EKAR), a FRET-based biosensor that specifically detects ERK activity as an increase in the yellow/cyan emission ratio (7), and first tested the effect of PKA-mediated signaling on the regulation of GF-stimulated ERK activity. In PC12 cells expressing the cytosolic EKAR (EKARcyto), NGF treatment results in a 23.6% ± 1.3% (n = 8) increase in the yellow/cyan emission ratio, with a time to reach half-maximal activation (t1/2) of 5.0 ± 0.1 min (mean ± SEM) (Fig. 4A and B). EGF generated a similar response, with a maximal emission ratio increase of 19.8% ± 0.6% (n = 23) and a t1/2 of 4.6 ± 0.2 min (Fig. 4A and C). In both cases, the GF-induced ERK activity was abolished when cells were pretreated with 20 μM MEK inhibitor U0126 (data not shown). To test the effect of PKA on regulating GF-induced ERK activity, cells expressing EKARcyto were pretreated with the relatively selective PKA inhibitor H89 for 10 min, followed by treatment with either NGF or EGF. In both cases, inhibition of PKA significantly slowed the onset of ERK activation induced by NGF and EGF, increasing the t1/2 values to 10.0 ± 1.2 min (n = 14) and 5.9 ± 0.4 min (n = 9), respectively (Fig. 4A to C). As similar increases in t1/2 were observed in cells expressing the PKA peptide inhibitor PKIα (Fig. 4A to C), these findings show that PKA plays a role in activating ERK following activation of both TrkA and EGFR, although the effect is more pronounced in the case of TrkA activation. Furthermore, disruption of PKA signaling by addition of St-Ht31, a peptide which disrupts PKA/AKAP interactions (12), decreases the magnitude of both NGF- and EGF-induced ERK activity and significantly delays the onset of NGF-induced ERK activity (Fig. 4D and E). These data not only further establish a role of PKA in regulating GF-induced ERK activity but also suggest that this regulation is achieved through a pool of PKA anchored by AKAP.
Fig. 4.

cAMP-mediated signaling regulates ERK activity in PC12 cells. (A) Bar graph depicting the time to reach half-maximal activation [(t(1/2)] of EKARcyto in response to various treatments (NGF alone [n = 12], NGF after H89 pretreatment [n = 14], NGF in the presence of PKIα expression [n = 5], EGF alone [n = 23], EGF after H89 pretreatment [n = 13], and EGF in the presence of PKIα expression [n = 5]). (B) Time courses of PC12 cells expressing EKARcyto treated with NGF (Cntl; n = 8), NGF after 10 μM H89 pretreatment (n = 7), or NGF in the presence of PKIα expression (n = 5). (C) Time courses depicting the EKARcyto response to EGF (cntl; n = 8), EGF after H89 pretreatment (n = 4), and EGF in the presence of PKIα expression (n = 3). (D and E) NGF (D)- and EGF (E)-induced time courses of ERK activity in cells in which PKA anchoring was disrupted via pretreatment with Ht31 (1 μM). (F) Bar graph depicting the time to reach half-maximal activation of EKARcyto in response to various treatments (NGF alone [n = 12], NGF after KH7 pretreatment [n = 14], EGF alone [n = 23], and EGF after KH7 pretreatment [n = 13]). (G) Time courses of PC12 cells expressing EKARcyto treated with NGF (Cntl; n = 8) or NGF after 30 μM KH7 pretreatment (n = 7). (H) Time courses depicting the EKARcyto response to EGF (Cntl; n = 8) and EGF after KH7 pretreatment (n = 7). (I) PC12 cells were pretreated with vehicle, 30 μM KH7 (K), or 10 μM H89 (H) for 20 min and subsequently treated with NGF (N) or EGF (E) for 5 min or 15 min. Control cells (Cntl) were treated with vehicle only. Immunoblots of p-ERK1/2 and tubulin (loading control) represent a single experiment. (J) Quantifications of p-ERK1/2 over tubulin for the NGF-treated samples are plotted and normalized to the vehicle-treated sample at each time point (n = 3 to 5 independent experiments). (K) Quantifications of p-ERK1/2 over tubulin for the EGF-treated samples are plotted and normalized to the vehicle-treated sample at each time point (n = 3 to 5 independent experiments). All imaging and immunoblot data are shown as means ± SEMs. *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.d., not detected.
Next, to examine the effect of sAC on GF-induced ERK activity, cells were pretreated with a sAC inhibitor, KH7, and subsequently treated with GF. The onset of ERK activity was delayed in the case of NGF-treated cells (t1/2 = 6.12 ± 0.4 min; n = 10), whereas sAC inhibition had no effect on EGF-stimulated ERK activity (t1/2 = 4.1 ± 0.2 min; n = 13) (Fig. 4F to H). Conversely, cells which were pretreated with 10 μM MDL-12330A, a tmAC inhibitor, showed a dramatic reduction in EGF-stimulated ERK activity compared to cells which were not pretreated (data not shown). Together these findings suggest that both NGF- and EGF-induced activation of ERK is dependent on cellular levels of cAMP, although the effects of the two GFs may be differentially regulated by sAC and tmAC.
Live-cell imaging with FRET-based kinase activity reporters is a sensitive method to monitor the spatiotemporal regulation of kinase activity in real time, but in some cases, these probes are so sensitive that they can be easily saturated when a kinase is robustly activated, making it difficult to discern discrepancies in the magnitude of kinase activity. Since we observed a slight reduction in the GF-induced EKAR response in the presence of PKA inhibition with H89 and PKIα (Fig. 4B and C), we sought to further investigate the effect of KH7 and H89 on GF-stimulated ERK activity with immunoblot analysis, a technique which is less prone to signal saturation. Therefore, to monitor the effect of sAC and PKA inhibition on the magnitude of GF-stimulated ERK activity, we monitored the levels of phosphorylated ERK1/2 (p-ERK) in a population of PC12 cells in response to various treatments. In doing so, we first showed that inhibition of sAC by pretreatment for 20 min with KH7 significantly reduces the levels of p-ERK induced by NGF 5 min after treatment but had little effect on p-ERK levels 15 min post-NGF treatment (Fig. 4I and J), confirming that NGF-stimulated ERK activity is dependent on sAC activity, especially at earlier time points. Similarly, we also showed that inhibition of PKA with H89 dramatically reduced the levels of p-ERK induced by NGF at 5 min following NGF treatment, but at 15 min post-NGF treatment, there was no detectable difference in p-ERK levels in the presence or absence of H89 treatment, confirming our findings with EKAR imaging (Fig. 4I and J).
In the case of EGF, we found that sAC inhibition with KH7 decreased the amplitude of EGF-stimulated p-ERK levels 5 min after EGF treatment (Fig. 4I and K). This finding is in contrast to what was observed in imaging experiments using EKAR, and this discrepancy is likely due to the difference in these two assays in which EKAR is saturated more easily, making it more difficult to detect changes in the magnitude of ERK signaling. However, when taken with the observation that the inhibition of tmAC can dramatically reduce the magnitude of EGF-induced ERK activity (data not shown), the observation that sAC inhibition affects the levels of EGF-induced p-ERK suggests that EGF-induced ERK activity is largely dependent on the levels of cAMP in the cell. Finally, we also showed that inhibition of PKA decreases the levels of p-ERK 5 min following EGF treatment (Fig. 4I and K). Regardless of the pretreatment, p-ERK levels were undetectable 15 min after EGF treatment. Together with our imaging data, the results of the immunoblot analysis confirm that cAMP/PKA signaling is a critical factor in the regulation of GF-induced ERK activity.
Next, to test the involvement of PDE3 in regulating GF-induced ERK activity, we pretreated cells with 10 μM PDE3 inhibitor milrinone for 10 min and then stimulated cells with either of the GFs. Under these conditions, there is a reduction in the onset of both NGF- and EGF-stimulated ERK activity, with t1/2s of 3.6 ± 0.3 min (n = 14) and 3.7 ± 0.3 min (n = 12), respectively, compared to those of cells that were not pretreated with milrinone (Fig. 5A to C), and the EKAR responses were abolished in the presence of MEK inhibition (data not shown). These findings indicate that PDE3, via regulation of GF-stimulated cAMP and PKA dynamics, also regulates GF-stimulated ERK activity. In agreement with this, in some cells, pharmacological inhibition of PDE3 alone was sufficient to activate ERK (data not shown), but these cells were not included in the analysis. Furthermore, in contrast to cells pretreated with milrinone, those that were pretreated with rolipram did not show a change in GF-stimulated ERK activity (Fig. 5A), consistent with the finding that PDE4 inhibition does not potently activate PKA in these cells.
Fig. 5.

PDE3, but not PDE4, regulates ERK activity in PC12 cells. (A) Bar graph depicting the t1/2s of various treatments (NGF alone [n = 12], NGF after milrinone [M] pretreatment [n = 14], NGF after pretreatment with 1 μM rolipram [R] [n = 7], EGF alone [n = 23], EGF after milrinone pretreatment [n = 12], and EGF after rolipram pretreatment [n = 16]). (B) Time courses depicting the EKARcyto response from cells treated with NGF (Cntl; n = 8) and NGF after pretreatment with 10 μM milrinone (Mil; n = 8). (C) Time courses depicting the EKARcyto response of cells treated with EGF (Cntl; n = 8) and EGF after pretreatment with milrinone (n = 6). (D) PC12 cells were pretreated with vehicle or 10 μM milrinone (M) for 20 min and subsequently treated with NGF or EGF for 2.5 min, 5 min, or 15 min. Control cells (Cntl) were treated with vehicle only. Immunoblots of p-ERK1/2 and tubulin represent a single experiment. (E) Quantifications of p-ERK1/2 over tubulin for the NGF-treated samples are plotted and normalized to the vehicle-treated sample at each time point (n = 3 independent experiments). (F) Quantifications of p-ERK1/2 over tubulin for the EGF-treated samples are plotted and normalized to the vehicle-treated sample at each time point (n = 3 independent experiments). All imaging and immunoblot data are shown as means ± SEMs. *, P < 0.05; **, P < 0.01; n.d., not detected.
Again, we sought to confirm our imaging data and investigate differences in the magnitude of ERK signaling using immunoblot analysis. In the case of NGF, we observed that inhibition of PDE3 resulted in significantly greater levels of p-ERK at 2.5 min following NGF treatment (Fig. 5D and E), similar to that observed with EKAR imaging. After NGF treatment for 5 or 15 min, however, the discrepancy in p-ERK levels in the presence or absence of milrinone was not as pronounced (Fig. 5D and E), again in agreement with our imaging data. We also confirmed that EGF-induced p-ERK levels are enhanced by inhibition of PDE3 at 2.5 min following EGF treatment, and PDE3 inhibition had less of an effect on p-ERK levels 5 min after EGF treatment (Fig. 5D and F). Together with the imaging data, the immunoblot data show that cAMP, along with PKA and PDE3, are critical regulators of ERK signaling in PC12 cells, such that subtle increases in intracellular cAMP can prime both the NGF- and EGF-mediated signaling to more rapidly activate ERK.
Disruption of the spatiotemporal regulation of PKA activity alters EGF-stimulated ERK activity in the nucleus.
We next wanted to investigate if PDE3 and spatiotemporally controlled PKA activity contribute to the specific growth factor signaling observed in PC12 cells by monitoring the effect of PDE3 inhibitor on the duration of nuclear ERK activity, where we have found it easiest to discern discrepancies between the duration of ERK activity induced by EGF and NGF (data not shown). To begin, we used a nucleus-localized EKAR (EKARnuclear) as a tool to monitor the differential growth factor signaling that results from NGF and EGF. As expected, in cells expressing EKARnuclear, we observed sustained and transient durations of nuclear ERK activity in response to NGF and EGF, respectively, with the corresponding time to reverse to half-maximal ERK activity [t1/2(Rev)] of 53.1 ± 4.5 min (n = 19) after NGF addition and 14.6 ± 0.9 min (n = 20) after EGF addition (Fig. 6A to C), and the kinetics of ERK activity observed with EKARnuclear correspond with translocation of ERK into and out of the nucleus (data not shown). The involvement of PKA in promoting the sustained phase of NGF-stimulated ERK activity was tested by simultaneously treating cells with NGF and the PKA inhibitor H89. Under these conditions, a more transient EKARnuclear response was observed, with a t1/2(Rev) value of 30.3 ± 3.8 min (n = 9) (Fig. 6D, orange). As a similar effect was observed in cells expressing PKIα (Fig. 6D, green), these observations confirm that PKA is at least partially responsible for sustaining ERK activity as a result of NGF treatment. Also in support of a role of cAMP/PKA signaling in promoting sustained ERK activity, compared to cells treated with EGF alone, cells treated with EGF and increasing doses of Fsk, which correlate with increased levels of cAMP produced (data not shown), show a dose-dependent increase in the duration of nuclear ERK activity (Fig. 6E and F). Specifically, EGF in conjunction with 0.05, 0.5, 5.0, or 50 μM Fsk show t1/2(Rev) values of 19.1 ± 1.2 min (n = 16), 23.8 ± 2.6 min (n = 17), 27.4 ± 1.1 min (n = 10), or 32.4 ± 1.9 min (n = 7), respectively (Fig. 6B, E, and F). While these data are in agreement with the early observation that cAMP in conjunction with EGF can cause sustained ERK activation on a global level (35), EKARnuclear allows for the specific detection of nuclear ERK activity, in which we have observed more dramatic temporal differences in NGF- versus EGF-stimulated ERK activity relative to the cytosol (data not shown). Since it is clear that a high concentration of cAMP, like that generated by Fsk (Fig. 2D), can sustain EGF-stimulated ERK activity in the nucleus, we wanted to test the effect of PDE3 inhibition alone, which induces less cAMP than Fsk at tested doses (Fig. 2D) yet perturbs the compartmentalization of PKA activity (Fig. 3). When cells expressing EKARnuclear were stimulated with EGF in conjunction with 10 μM milrinone, there was a significantly longer t1/2(Rev) (26.5 ± 3.0 min; n = 20) compared to that of cells treated only with EGF (Fig. 6A, B, and G), and a similar effect was observed with IBMX (Fig. 6B and H) but not rolipram (Fig. 6B and I), illustrating a role for PDE3 in the regulation of EGF-stimulated nuclear ERK activity. We did not, however, observe an extended duration of NGF-stimulated nuclear ERK activity in the presence of milrinone or 50 μM Fsk, suggesting that NGF alone can achieve the maximal duration of nuclear ERK activity (data not shown). We further showed that the extended duration of EGF-induced nuclear ERK activity that results from PDE3 inhibition is largely dependent on PKA activity since simultaneous treatment with EGF and milrinone in the presence of H89 did not result in sustained nuclear ERK activity with a t1/2(Rev) of 14.3 ± 1.0 min (n = 8) (Fig. 6A, B, and J, green), and a similar trend was observed in cells expressing PKIα (Fig. 6J, pink). Together with our observation that PDE3 regulates the plasma membrane compartmentalization of GF-stimulated PKA activity and the duration of EGF-stimulated PKA activity in that locale (Fig. 3), our studies with EKARnuclear show that PDE3 controls the spatiotemporal patterns of EGF-stimulated PKA activity and in turn acts as a critical regulator of nuclear ERK activity in PC12 cells.
Fig. 6.

PDE3 inhibition extends the duration of EGF-induced nuclear ERK in a PKA-dependent manner. All experiments are done in PC12 cells expressing EKARnuclear. (A) YFP direct (left) and ratiometric images depicting differential GF-induced nuclear ERK activities in the presence and absence of PDE3 inhibitor (Mil) and/or PKA inhibitor (H89). (B) Graphical representation of the time needed for nuclear ERK activity to reduce to 50% [t1/2(Rev)]. (C) NGF-induced nuclear ERK activity (n = 9) is more sustained than EGF-induced nuclear ERK activity (n = 10). (D) Time course of NGF-induced nuclear ERK activity in the presence of H89 (n = 8) and PKIα (n = 5) expression. (E) tmAC activation with increasing doses of Fsk extends the duration of EGF-stimulated ERK activity in the nucleus (0.05 μM Fsk, n = 8; 0.5 μM Fsk, n = 9; 5.0 μM Fsk, n = 10; 50 μM Fsk, n = 7. (F) Bar graph depicting the increase in reversal of nuclear ERK activity [t1/2(Rev)] following various treatments (EGF plus the following doses of Fsk: 0.05 μM, n = 16; 0.5 μM, n = 17; 5.0 μM, n = 10; 50 μM Fsk, n = 7). (G) PDE3 inhibition with 10 μM Mil (n = 7) extends the duration of EGF-stimulated ERK activity. (H and I) 100 μM IBMX increases the duration of EGF-stimulated nuclear ERK activity (n = 8) (H), while 1 μM Roli does not (n = 7) (I). (J) In the presence of milrinone and inhibition of PKA through H89 pretreatment (n = 8) or PKIα expression (n = 5), there is no increase in the duration of EGF-induced nuclear ERK activity. All data are shown as means ± SEMs. δδ, P < 0.01 compared to EGF. **, P ≤ 0.01 compared to NGF.
To test if the duration of nuclear ERK activity induced by various treatments correlates with neurite extension, we treated PC12 cells expressing a plasma membrane-targeted YFP for 3 days with various treatments and monitored their effects on neurite outgrowth. Membrane-targeted YFP is used to highlight the cell membrane and facilitate the quantification of neurite lengths. Consistent with the literature, we see that NGF induces significant neurite outgrowth (neurites were defined as processes with lengths greater than or equal to the length of the cell body), whereas EGF treatment does not. We also observe that EGF plus 5 μM Fsk induces neurite outgrowth (35), although the effect on neurite outgrowth is not as robust as that of NGF treatment, in terms of number of neurites or neurite length. Also consistent with the literature, we see that PKA inhibition with H89 does not reverse the effect of NGF on neurite outgrowth, whereas the effect of EGF plus Fsk is somewhat reversed upon addition of H89 (35). In the case of EGF plus milrinone, IBMX, H89, or milrinone and H89, we do not see significant effects on neurite outgrowth compared to cells treated with EGF alone (Fig. 7). Interestingly, our studies with EKARnuclear (Fig. 6) clearly show that the following treatments have indistinguishable effects on the duration of nuclear ERK activity: NGF plus H89, EGF plus milrinone, EGF plus IBMX, and EGF plus 5 μM Fsk. On the other hand, it is clear that these treatments show different effects on neurite outgrowth, specifically that NGF plus H89 and EGF plus Fsk induce neurite outgrowth, whereas EGF plus milrinone and EGF plus IBMX do not. These data demonstrate the complexity of the integrated controls by the cAMP and ERK signaling systems on cellular processes and suggest that additional signaling players are likely involved in regulating the differentiation process in PC12 cells. Future studies are needed to identify these signaling molecules and to characterize the role that the differential spatiotemporal activity patterns of GF-induced PKA activities have on these signaling molecules and their role in the differentiation of PC12 cells.
Fig. 7.

The duration of nuclear ERK activity does not precisely correlate with neurite outgrowth. PC12 cells expressing a plasma membrane-targeted YFP were treated for 3 days as indicated, and the number of cells expressing neurites (defined as a cellular extension longer than the length of the cell body) was quantified. n = 25 to 105 cells per treatment. Representative images for each treatment are shown.
DISCUSSION
In this study, the utilization of FRET-based biosensors, which have proven to be powerful tools for analyzing dynamic and spatially compartmentalized signaling activities (8, 38), has led to a few new observations. While it is known that NGF activates sAC to produce cAMP in PC12 cells (26) and that the activation of Rap1 and ERK via NGF treatment is PKA dependent (19), it has been difficult to directly detect the activation of PKA by NGF treatment (19, 36). One possible reason is that the averaged signals obtained when the NGF-induced PKA activity was monitored in whole cells result in low sensitivity. In contrast, localized FRET-based biosensors allow for the local detection of kinase activity and have led to the observation that NGF is indeed a robust activator of PKA activity at the plasma membrane but not in the cytosol. Moreover, using the targeted FRET-based biosensor approach, we were able to detect EGF-induced PKA activity in PC12 cells, uncovering a previously unknown mode of activation of PKA by EGF in this cell model. In the same vein, subcellularly targeted ERK biosensors revealed localized ERK activity patterns stimulated by different GFs. Specifically, we observed larger discrepancies in the transient versus sustained ERK activity induced by EGF and NGF in the nucleus than in the cytoplasm, suggesting that the temporally controlled ERK activities by different GFs can be further amplified by spatial constraints (21).
One critical finding from our study is the compartmentalization of GF-stimulated PKA activity to the plasma membrane. Previous studies have identified the plasma membrane to be a unique site of GF-regulated signaling. The signaling sensitivity appears to be high at this location, such that even low-level input signals can activate the Raf-MEK-ERK pathway (6, 17, 33). Since PKA is an upstream activator of Raf, our finding that GF-induced PKA activity is localized to the plasma membrane suggests a PKA-dependent mechanism for this observation and further supports the role of the plasma membrane as an important signaling platform in PC12 cells. While we have identified PDE3 to be critical in restricting NGF- and EGF-induced PKA activity to the plasma membrane, it is likely that there are other molecular players that help to confer this precise compartmentalization. For instance, our observation that disruption of PKA/AKAP interactions with Ht31 decreases the magnitude and onset of GF-stimulated ERK activity (Fig. 4D and E) suggests that specific AKAPs (25) could play a role in localizing the molecular machinery involved in PKA and ERK cross talk to the plasma membrane to maintain this compartmentalization.
Interestingly, although both EGF and NGF stimulate PKA activities that are restricted to the plasma membrane, the temporal patterns of the stimulated activities differ. One potential mechanism may be via differential regulation involving different sources of cAMP. Additional factors may also contribute. For instance, since EGFR is known to desensitize shortly after activation, it is possible that activation of tmACs via EGFR is transient due to the internalization of the receptor and desensitization of the signaling pathway (20, 31). It is also possible that activated EGFR and TrkA could differentially activate other signaling molecules, such as protein kinase C (PKC) (22), which could contribute to the transient and sustained EGF- and NGF-stimulated PKA activity at the plasma membrane. Future studies will focus on further elucidating the underlying molecular mechanisms which control the specific temporal patterns of EGF- and NGF-stimulated PKA activity at the plasma membrane.
While several signaling components are known to be involved in the temporal regulation of ERK, thereby influencing the cell fate specification achieved by EGF and NGF in PC12 cells (14, 15, 20, 22, 23, 30, 34), our study revealed the precise spatiotemporal regulation of GF-stimulated PKA activity, which is tightly controlled by PDE3, as a new level of regulation underlying the growth factor signaling specificity in these cells. Specifically, while NGF and EGF stimulate sustained and transient nuclear ERK responses, respectively, pharmacological inhibition of PDE3 resulted in a nearly a 2-fold increase in the duration of EGF-stimulated nuclear ERK activity. Since the amount of cAMP produced by PDE3 inhibition is small (Fig. 2D), this effect is likely to be mediated by the altered spatiotemporal pattern of PKA activity. As PKA is an established upstream activator of Rap1 and Raf, both previously identified to regulate the differential responses of EGF and NGF at the level of ERK activation (27, 23), it will be interesting to monitor the effect of altering PKA spatiotemporal dynamics has on these molecules. Importantly, while our findings cannot distinguish whether the nearly 2-fold increase in the duration of nuclear ERK activity in the presence of PDE3 inhibition is due to active cytosolic PKA or sustained PKA activity at the plasma membrane, they illustrate the importance of spatiotemporally regulated PKA activity in contributing to the control of specific signaling effects of EGF and NGF. However, at the functional level, we have shown that EGF in the presence of the tmAC activator and EGF in the presence of the PDE3 inhibitor, two conditions that cause similar effects on the duration of nuclear ERK activity, do not have the same effect on neurite outgrowth (Fig. 6 and 7). This finding that the duration of nuclear ERK activity do not precisely correlate with the induction of neurite outgrowth illustrates the complexity of integrated control by the cAMP and ERK signaling systems on cell differentiation. The distinct signaling activity patterns and gene expression profiles induced by NGF and EGF will be further analyzed in future studies.
Together, the findings in this study have led to an enhanced understanding of the molecular mechanisms underlying the GF signaling specificity in this classical cell model system. Furthermore, an illustrative example is presented in which the precise spatiotemporal control of one signaling pathway can strongly influence the spatiotemporal regulation of another signaling cascade. As regulation of any cellular processes is achieved by an integrated network of signaling molecules, this type of spatiotemporal cross-regulation is likely to be widespread.
ACKNOWLEDGMENTS
We thank Lonny R. Levin for the PC12 cell line and KH7 and Ronald Schnaar for suggestions and assistance. We also thank Viacheslav Nikolaev for providing Epac1-camps and Karal Svoboda for providing both EKAR variants.
This work was funded by NIH grant R01 DK073368 (to J.Z.).
Footnotes
Published ahead of print on 1 August 2011.
REFERENCES
- 1. Baillie G. S. 2009. Compartmentalized signalling: spatial regulation of cAMP by the action of compartmentalized phosphodiesterases. FEBS J. 276:1790–1799 doi:10.1111/j.1742-4658.2009.06926.x [DOI] [PubMed] [Google Scholar]
- 2. Bender A. T., Beavo J. A. 2006. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol. Rev. 58:488–520 doi:10.1124/pr.58.3.5 [DOI] [PubMed] [Google Scholar]
- 3. Depry C., Allen M. D., Zhang J. 2011. Visualization of PKA activity in plasma membrane microdomains. Mol. Biosyst. 7:52–58 doi:10.1039/c0mb00079e [DOI] [PubMed] [Google Scholar]
- 4. Dumaz N., Marais R. 2005. Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. Based on the anniversary prize of the Gesellschaft fur Biochemie und Molekularbiologie Lecture delivered on 5 July 2003 at the Special FEBS Meeting in Brussels. FEBS J. 272:3491–3504 doi:10.1111/j.1742-4658.2005.04763.x [DOI] [PubMed] [Google Scholar]
- 5. Grewal S. S., York R. D., Stork P. J. 1999. Extracellular-signal-regulated kinase signalling in neurons. Curr. Opin. Neurobiol. 9:544–553 doi:10.1016/S0959-4388(99)00010-0 [DOI] [PubMed] [Google Scholar]
- 6. Harding A., Tian T., Westbury E., Frische E., Hancock J. F. 2005. Subcellular localization determines MAP kinase signal output. Curr. Biol. 15:869–873 doi:10.1016/j.cub.2005.04.020 [DOI] [PubMed] [Google Scholar]
- 7. Harvey C. D., et al. 2008. A genetically encoded fluorescent sensor of ERK activity. Proc. Natl. Acad. Sci. U. S. A. 105:19264–19269 doi:10.1073/pnas.0804598105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Herbst K. J., Ni Q., Zhang J. 2009. Dynamic visualization of signal transduction in living cells: from second messengers to kinases. IUBMB Life 61:spcone. doi:10.1002/iub.259 [DOI] [PubMed] [Google Scholar]
- 9. Hilborn M. D., Rane S. G., Pollock J. D. 1997. EGF in combination with depolarization or cAMP produces morphological but not physiological differentiation in PC12 cells. J. Neurosci. Res. 47:16–26 [PubMed] [Google Scholar]
- 10. Kamenetsky M., et al. 2006. Molecular details of cAMP generation in mammalian cells: a tale of two systems. J. Mol. Biol. 362:623–639 doi:10.1016/j.jmb.2006.07.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kholodenko B. N. 2007. Untangling the signalling wires. Nat. Cell Biol. 9:247–249 doi:10.1038/ncb0307-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Klussmann E., Maric K., Wiesner B., Beyermann M., Rosenthal W. 1999. Protein kinase A anchoring proteins are required for vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J. Biol. Chem. 274:4934–4938 [DOI] [PubMed] [Google Scholar]
- 13. Lissandron V., Zaccolo M. 2006. Compartmentalized cAMP/PKA signalling regulates cardiac excitation-contraction coupling. J. Muscle Res. Cell Motil. 27:399–403 doi:10.1007/s10974-006-9077-2 [DOI] [PubMed] [Google Scholar]
- 14. Mark M. D., Liu Y., Wong S. T., Hinds T. R., Storm D. R. 1995. Stimulation of neurite outgrowth in PC12 cells by EGF and KCl depolarization: a Ca(2+)-independent phenomenon. J. Cell Biol. 130:701–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mark M. D., Storm D. R. 1997. Coupling of epidermal growth factor (EGF) with the antiproliferative activity of cAMP induces neuronal differentiation. J. Biol. Chem. 272:17238–17244 [DOI] [PubMed] [Google Scholar]
- 16. Mironov S. L., et al. 2009. Imaging cytoplasmic cAMP in mouse brainstem neurons. BMC Neurosci. 10:29 doi:10.1186/1471-2202-10-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mochizuki N., et al. 2001. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature 411:1065–1068 doi:10.1038/35082594 [DOI] [PubMed] [Google Scholar]
- 18. Nikolaev V. O., Bunemann M., Hein L., Hannawacker A., Lohse M. J. 2004. Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 279:37215–37218 doi:10.1074/jbc.C400302200 [DOI] [PubMed] [Google Scholar]
- 19. Obara Y., Labudda K., Dillon T. J., Stork P. J. 2004. PKA phosphorylation of Src mediates Rap1 activation in NGF and cAMP signaling in PC12 cells. J. Cell Sci. 117:6085–6094 doi:10.1242/jcs.01527 [DOI] [PubMed] [Google Scholar]
- 20. Orton R. J., Sturm O. E., Gormand A., Wolch W., Gilbert D. R. 2008. Computational modelling reveals feedback redundancy within the epidermal growth factor receptor/extracellular-signal regulated kinase signalling pathway. IET Syst. Biol. 2:173–183 doi:10.1049/iet-syb:20070066 [DOI] [PubMed] [Google Scholar]
- 21. Rubinfeld H., Seger R. 2005. The ERK cascade: a prototype of MAPK signaling. Mol. Biotechnol. 31:151–174 doi:10.1385/MB:31 [DOI] [PubMed] [Google Scholar]
- 22. Santos S. D., Verveer P. J., Bastiaens P. I. 2007. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat. Cell Biol. 9:324–330 doi:10.1038/ncb1543 [DOI] [PubMed] [Google Scholar]
- 23. Sasagawa S., Ozaki Y., Fujita K., Kuroda S. 2005. Prediction and validation of the distinct dynamics of transient and sustained ERK activation. Nat. Cell Biol. 7:365–373 doi:10.1038/ncb1233 [DOI] [PubMed] [Google Scholar]
- 24. Shelly M., et al. 2010. Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science 327:547–552 doi:10.1126/science.1179735 [DOI] [PubMed] [Google Scholar]
- 25. Smith F. D., et al. 2010. AKAP-Lbc enhances cyclic AMP control of the ERK1/2 cascade. Nat. Cell Biol. 12:1242–1249 doi:10.1038/ncb2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stessin A. M., et al. 2006. Soluble adenylyl cyclase mediates nerve growth factor-induced activation of Rap1. J. Biol. Chem. 281:17253–17258 doi:10.1074/jbc.M603500200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stork P. J. 2005. Directing NGF's actions: it's a Rap. Nat. Cell Biol. 7:338–339 doi:10.1038/ncb0405-338 [DOI] [PubMed] [Google Scholar]
- 28. Stork P. J., Schmitt J. M. 2002. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 12:258–266 [DOI] [PubMed] [Google Scholar]
- 29. Szaszak M., Christian F., Rosenthal W., Klussmann E. 2008. Compartmentalized cAMP signalling in regulated exocytic processes in non-neuronal cells. Cell. Signal. 20:590–601 doi:10.1016/j.cellsig.2007.10.020 [DOI] [PubMed] [Google Scholar]
- 30. Traverse S., Gomez N., Paterson H., Marshall C., Cohen P. 1992. Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem. J. 288 (Pt. 2):351–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Traverse S., et al. 1994. EGF triggers neuronal differentiation of PC12 cells that overexpress the EGF receptor. Curr. Biol. 4:694–701 [DOI] [PubMed] [Google Scholar]
- 32. Vaudry D., Stork P. J., Lazarovici P., Eiden L. E. 2002. Signaling pathways for PC12 cell differentiation: making the right connections. Science 296:1648–1649 doi:10.1126/science.1071552 [DOI] [PubMed] [Google Scholar]
- 33. Wang Z., et al. 2006. Rap1-mediated activation of extracellular signal-regulated kinases by cyclic AMP is dependent on the mode of Rap1 activation. Mol. Cell. Biol. 26:2130–2145 doi:10.1128/MCB.26.6.2130-2145.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu T. R., et al. 2010. Inferring signaling pathway topologies from multiple perturbation measurements of specific biochemical species. Sci. Signal. 3:ra20. [PubMed] [Google Scholar]
- 35. Yao H., et al. 1995. Cyclic AMP can convert epidermal growth factor into a differentiating factor in neuronal cells. J. Biol. Chem. 270:20748–20753 [DOI] [PubMed] [Google Scholar]
- 36. Yao H., York R. D., Misra-Press A., Carr D. W., Stork P. J. 1998. The cyclic AMP-dependent protein kinase (PKA) is required for the sustained activation of mitogen-activated kinases and gene expression by nerve growth factor. J. Biol. Chem. 273:8240–8247 [DOI] [PubMed] [Google Scholar]
- 37. York R. D., et al. 1998. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 392:622–626 doi:10.1038/33451 [DOI] [PubMed] [Google Scholar]
- 38. Zaccolo M., et al. 2005. Imaging the cAMP-dependent signal transduction pathway. Biochem. Soc. Trans. 33:1323–1326 doi:10.1042/BST20051323 [DOI] [PubMed] [Google Scholar]