

Abstract
Histone deacetylase inhibitors (HDACi) are promising antitumor drugs acting through reactivation of silenced tumor suppressor genes. Several HDACi are currently in clinical trials both for hematological and solid tissue malignancies. Cooperative action of HDACi and DNA methylation inhibitors (DNMTi) has been reported, making combined treatment an attractive choice for cancer therapy. There is some evidence that synergistic effects of HDACi and DNMTi are achieved by their action on common targets, including DNA methyltransferase 1 (DNMT1). To further analyze this interaction, we investigated the effect of the HDACi trichostatin A on global and gene-specific DNA methylation and applied methods with single molecule sensitivity, confocal laser scanning microscopy with avalanche photodiode detectors (APD imaging) and fluorescence correlation spectroscopy (FCS), to study its effect on the nuclear dynamics of DNMT1 in live cells. Our data show that trichostatin A treatment reduces global DNA methylation and the DNMT1 protein level and alters DNMT1 nuclear dynamics and interactions with chromatin. The mechanisms underlying these effects are apparently distinct from the mechanisms of action of the DNMT inhibitor 5-azacytidine. Our study sheds light on the molecular mechanisms underlying the synergistic action of HDACi and DNMTi and may also help to define improved policies for cancer treatment.
INTRODUCTION
Epigenetic modifications of chromatin play an important role in differentiation, aging, and tumorigenesis (41). Major epigenetic mechanisms include DNA methylation and covalent posttranslational modification of histone tails. These events switch chromatin between relatively “open” (expressed) and “closed” (suppressed) structures. Although there are several different histone modifications, the opposing activities of histone acetyl transferases (HATs) and histone deacetylases (HDACs) are considered important regulators of chromatin structure (28). DNA methylation is an important epigenetic modification in the vertebrate genome which occurs mainly on cytosines located in CpG dinucleotide sequences (2). DNA methylation patterns are established by DNA methyltransferase 3a (DNMT3a) and DNMT3b, which are de novo DNA methyltransferases (DNMTs), and are mainly propagated by DNMT1, a maintenance methyltransferase (39). DNA methyltransferases are involved in the aberrant DNA methylation patterns frequently found in cancer cells (21). In addition, there is growing evidence that inappropriate gene expression by epigenetic events is crucial for the initiation and progression of cancer. These epigenetic abnormalities, including epigenetic silencing of tumor suppressor genes by abnormal promoter methylation, are likely to play an important role in tumor growth and metastasis when a substantial number of genes can be inactivated by DNA methylation in a tumor (20). In addition to DNA methylation, genome-wide changes in histone modifications are also found in cancer cells. Loss of acetylation at lysine 16 and trimethylation at lysine 20 of histone H4 have been reported as common hallmarks of human cancer (11).
New insights into the high prevalence of epigenetic changes in cancer have led to novel therapeutic approaches in oncology that rely on the dynamic nature of epigenetic factors. The main idea behind these approaches is that abnormal epigenetic marks leading to gene silencing may be reversed. Two categories of drugs, affecting histone acetylation and DNA methylation, are currently considered in epigenetic therapy of cancer. In myelodysplastic syndrome (MDS) and cutaneous T-cell lymphoma, DNA methyltransferase inhibitors (DNMTi) and histone deacetylase inhibitors (HDACi) are already in clinical use (15, 23). HDACi can open up chromatin by causing hyperacetylation of histones H3 and H4 (49). DNMTi, such as 5-aza-deoxycytidine, can induce genomic DNA hypomethylation (35). However, some gene promoters are not readily activated with either of the two inhibitors alone, but there are reports showing that HDACi and DNMTi can act synergistically to reactivate silenced genes (12, 47). Therefore, the functional interaction between HDACs and DNMTs has been a key issue in epigenetic research. Trichostatin A (TSA), a general inhibitor for class I and II HDACs, has also been shown to reactivate methylation-silenced genes even in the absence of DNMT inhibitors (25). In addition, it has been reported that TSA decreases DNMT3b mRNA levels in endometrial cell lines (46) and DNMT1 levels in Jurkat T cells (19).
Ou et al. reported that TSA treatment reduced the genomic DNA methylation level in T24 cells (bladder carcinoma) and MDA-MB-231 cells (breast carcinoma) through an active demethylation process (29). The change in DNA methylation was demonstrated at the gene-specific and global levels. DNMT1 was reported to be decreased by TSA treatment both at the mRNA and protein levels (29). It has also been suggested that HDAC inhibition promotes DNMT1 degradation through the ubiquitin-dependent proteosomal pathway (50).
We set out to investigate TSA-induced changes in DNA methylation at gene-specific and global levels in the hepatoma cell line Hep3B and used methods with single molecule sensitivity, i.e., confocal laser scanning microscopy (CLSM) imaging with avalanche photodiode detectors (APD imaging) (43) and fluorescence correlation spectroscopy (FCS) (1, 8–10, 26, 45), to analyze the effects on DNMT1 interaction with nuclear components. This cell line was chosen since it has previously been used in this laboratory in studies of TSA effects on p21Waf1-driven apoptosis (40), in relation to cell density (17), and on changes in DNA methylation. The great complexity of the problem—a low number of DNMT1 molecules in comparison to the surrounding molecules (concentrations of the order 10 to 500 nM), fast molecular interactions (typically characterized by on-off rates of the order 105 to 108 M−1 s−1), and rapid molecular movement (diffusion coefficients of the order 10−12 to 10−10 m2 s−1)—requires ultrasensitive methods with high spatial (submicrometer) and temporal (submillisecond resolution and single molecule sensitivity). Two glioblastoma cell lines, U373MG and U343MG, were used for comparison.
MATERIALS AND METHODS
Antibodies.
Anti-p21waf1 (ab7960), anti-human HDAC1 (ab7028), anti-human HDAC2 (ab7029), and anti-human HDAC3 (ab7030) were all purchased from Abcam (Cambridge, United Kingdom). Anti-human DNMT-1 (M0231S) was obtained from New England Biolabs (MA). Goat anti-rabbit (Bio-Rad) and goat anti-mouse (Bio-Rad) horseradish peroxidase (HRP)-conjugated antibodies were used as secondary antibodies.
Cell culture and treatments.
The human hepatocellular carcinoma Hep3B cell line and two glioblastoma cell lines, U343MG and U373MG, were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% fetal calf serum (Gibco) and penicillin-streptomycin (Gibco). Hep3B cells were treated with 0 to 800 nM TSA (Sigma-Aldrich, Sweden) or 5 μM 5-azacytidine (Sigma-Aldrich, Sweden). U343MG and U373MG cells were treated with 500 nM TSA or 5 μM 5-azacytidine for 24 h before analysis.
Protein extraction and immunoblotting.
Cells were harvested by scraping in ice-cold phosphate-buffered saline (PBS) and lysed in lysis buffer containing 2% SDS, 10% glycerol, 250 mM Tris-HCl (pH 8.5), 25 mM phenylmethylsulfonyl fluoride (PMSF; Sigma, St. Louis, MO), and fresh protease inhibitor cocktail (Roche, Mannheim, Germany).
Samples were boiled for 3 min and then sonicated three times for 1 min, with 1-min intervals on ice. Samples were stored at −80°C prior to electrophoresis.
For cellular and nuclear fractionation, an NE-PER protein extraction kit (Pierce Biotechnology) was used according to the provided protocol. Protein concentrations were determined using a Micro BSA Protein Assay (Pierce Biotechnology), and proteins were separated on SDS-PAGE gels, transferred to nitrocellulose membrane (Schleicher and Schuell), and detected by immunoblotting according to standard protocols using an enhanced chemiluminescence detection system (Amersham-Bioscience, Sweden). The densities of protein bands on Western blots were determined using Bio-Rad Quantity One software on scanned films. Quantification of protein bands was done by first normalizing to β-actin, followed by calculation of relative band intensity against untreated control cells.
LUMA.
Genomic DNA was extracted using a GenElute mammalian genomic DNA miniprep kit (Sigma, St. Louis, MO). A luminometric methylation assay (LUMA) was performed as described earlier in detail by our group (22). Briefly, 200 to 500 ng of genomic DNA was digested by HpaII and EcoRI restriction endonucleases for 4 h at 37°C. The extent of DNA digestion was determined by a polymerase extension assay using a PSQ 96MA pyrosequencing platform (Biotage, Sweden). The DNA methylation level was defined as the HpaII/EcoRI ratio, where EcoRI served as an internal control for relative HpaII digestion (22).
5-Methylcytosine quantification by NNA.
CpG methylation levels were quantified by nearest-neighbor analysis (NNA) as described previously by Ramsahoye (32), using [α-32P]dGTP (Amersham Biosciences, Uppsala, Sweden) for labeling. The intensities of 5-methylcytosine and cytosine mononucleotide spots were measured using a phosphorimager (Fujifilm, Japan) and Image Gauge analysis software (Fujifilm, Japan). DNA methylation levels were defined as follows: [methylcytosine]/[methylcytosine plus cytosine] × 100.
Methylation of p21waf1 promoter by bisulfite sequencing.
Bisulfite modification of genomic DNA was performed as described previously (6). A 312-bp PCR product covering an upstream CpG island in the p21waf1 promoter was amplified through a seminested PCR. In the first reaction, bisulfite-treated DNA was amplified by primers p21UP-F1 (5′-GTG GTT TAT TTY GTG GGG AAA TGT-3′) and p21UP-R1 (5′-TAT CTA CCR CCR CTC TCT CAC CTC CTC-3′). In the second reaction the p21UP-F1 primer was replaced by p21UP-F2 (5′-GAG GAG GGA AGT GTT TTT TTG TAG-3′). The PCR product was purified using a QiaexII agarose gel extraction kit (Qiagen, Sweden) and cloned into the pCR2.1 TA cloning vector (Invitrogen, Carlsbad, CA), and 10 clones from each treatment were sequenced using M13-R primer and standard Sanger sequencing (Applied BioSystems).
Effect of TSA treatment on gene-specific promoter methylation.
Methylation-specific PCR (MS-PCR) was applied to analyze the methylation status of seven genes. Briefly, bisulfite-treated genomic DNA was amplified by methylation-specific primers for CHFR (checkpoint with forkhead and ring finger domains), E-cadherin, CDH13, death-associated protein kinase (DAPK), anaphase-promoting complex (APC), the glutathione S-transferase gene GSTP1, and RASSF1 promoters (Table 1). In vitro methylated human genomic DNA was used as a positive control in all reactions. Bisulfite treatment and genomic DNA input were normalized through amplification of a fragment of the β-actin coding gene as a control.
Table 1.
MS-PCR primers used to investigate promoter methylation of seven genes
| Gene | Primer name | Primer sequence | Annealing temp (°C) | Product size (bp) | Reference or source |
|---|---|---|---|---|---|
| CDH13 | Forward | 5′-TCGCGGGGTTCGTTTTTCGC-3′ | 60 | 243 | 38 |
| Reverse | 5′-GACGTTTTCATTCATACACGCG-3′ | ||||
| CHFR | Forward | 5′-GTCGGGTCGGGGTTC-3′ | 60 | 155 | 7 |
| Reverse | 5′-CCCAAAACTACGACGACG-3′ | ||||
| DAPK | Forward | 5′-GGATAGTCGGATGAGTTAACGTC-3′ | 64 | 98 | 14 |
| Reverse | 5′-CCCTCCCAAACGCCGA-3′ | ||||
| E-cadherin | Forward | 5′-GGTGAATTTTTAGTTAATTAGCGGTAC-3′ | 57 | 204 | 16 |
| Reverse | 5′-CATAACTAACCGAAAACGCCG-3′ | ||||
| RASSF1A | Forward | 5′-GTGTTAACGCGTTGCGTATC-3′ | 60 | 93 | 48 |
| Reverse | 5′-AACCCCGCGAACTAAAAACGA-3′ | ||||
| APC | Forward | 5′-TATTGCGGAGTGCGGGTC-3′ | 56 | 98 | This paper |
| Reverse | 5′-CCACATATCGATCACGTACG-3′ | ||||
| GSTP1 | Forward | 5′-GTCGTGATTTAGTATTGGGGC-3′ | 56 | 129 | This paper |
| Reverse | 5′-CTAATAACGAAAACTACGACGACG-3′ | ||||
| ß-Actin | Forward | 5′-AAGTTAAGTTTTGTTTTTATTTTT-3′ | 56 | 184 | This paper |
| Reverse | 5′-CAATAATCTCCTTCTACATCCTATC-3′ |
Cell preparations for measurements of DNMT1 mobility by fluorescent correlation spectroscopy.
The pDNMT1-RFP expression plasmid for DNMT1 tagged with the red fluorescent protein (RFP) was kindly provided by H. Leonhardt (Ludwig Maximilians University, Munich, Germany). The cells were cultured in chambered coverslips with eight wells (LabTek, Nunc) in phenol red-free DMEM (Gibco) supplemented with 10% fetal calf serum. The cells were transfected at 50% confluence, using Lipofectamine Plus reagents according to the manufacturer's protocol (Invitrogen). After 24 h, the cells were subjected to overnight treatment with 500 nM TSA or 5 μM 5-azacytidine and analyzed by APD imaging and FCS.
APD imaging and FCS.
APD imaging enables us to visualize DNMT1-RFP molecules in live cells expressing low, physiologically relevant levels of this construct. FCS enables us to measure the concentration of DNMT1-RFP and characterize its mobility and interactions in the cell nucleus. A theoretical background on FCS is provided as supplemental information SI 1 posted at http://www.cmm.ki.se/en/Research/Neurogenetics-and-Psychiatric-Diseases/Medical-Epigenetics/Medical-Epigenetics/Supplementary-Materials.
APD imaging and FCS measurements were performed on a uniquely modified ConfoCor3 instrument (Carl Zeiss, Jena, Germany) consisting of an inverted microscope for transmitted light and epifluorescence (Axiovert 200 M); a visible light (VIS)-laser module comprising the Ar/ArKr (458, 477, 488, and 514 nm), HeNe 543-nm, and HeNe 633-nm lasers; and the scanning module LSM 510 META. The instrument was modified to enable detection using silicon avalanche photodiodes (SPCM-AQR-1X; PerkinElmer) for APD imaging and FCS. Images were recorded at a 512- by 512-pixel resolution. A C-Apochromat 40× (numerical aperture, 1.2) water immersion UV-VIS-infrared (IR) objective was used throughout the study.
RFP fluorescence was excited using the HeNe 543-nm laser. Emitted fluorescence was collected using long-pass filter LP 560. Fluorescence intensity fluctuations were recorded in arrays of 10 consecutive measurements, with each measurement lasting 10 s. Averaged curves were analyzed using the FCS/CLSM running software for online data analysis or exported and fitted offline using IGOR Pro, version 5, data analysis software (WaveMetrics, Inc., Portland, OR). In either case, the nonlinear least-square fitting of the autocorrelation curve was performed using the Levenberg-Marquardt algorithm. The fitting of the FCS data was evaluated by residual analysis as described before (1, 45). Experiments were performed in triplicates with newly transfected and treated cells. At least 5 cells were analyzed in each run.
RESULTS
TSA influences the fate of the HDAC1 and HDAC2 proteins but not HDAC3.
The effect of TSA on HDACs in Hep3B cells was investigated by Western blotting. Although the overall amount of HDAC1 did not change significantly after TSA treatment, the amount of protein located in the cytoplasm apparently increased in a dose-dependent manner compared to the amount in the nucleus. Thus, in untreated cells, HDAC1 was hardly detected in the cytoplasm, but after TSA treatment the nuclear HDAC1 decreased significantly in a dose-dependent manner. The cytoplasmic fraction displayed an apparent parallel increase, but this did not reach statistical significance (Fig. 1).
Fig. 1.

Effect of TSA treatment on expression and subcellular localization of different HDACs. Hep3B cells were treated with different concentrations of TSA for 24 h. Cells were harvested, cytoplasmic and nuclear proteins were separated on SDS-PAGE gels, and HDACs were detected by Western blotting. The graphs show the average of triplicate experiments, with error bars denoting standard errors of the means. Significance was calculated with a Student's t test. ***, P < 0.001.
HDAC2 was present in both the nuclear and cytoplasmic fractions in untreated Hep3B cells. The expression level of the HDAC2 protein did not change significantly after TSA treatment. However, after treatment, a lower band appeared by Western blotting in both fractions, indicating a degradation of HDAC2 caused by TSA treatment (Fig. 1).
The HDAC3 protein pattern showed no significant changes after TSA treatment, neither by degradation nor in terms of cellular relocalization (Fig. 1). These data indicate that HDAC3 expression is not affected by TSA in Hep3B cells.
TSA treatment reduces nuclear DNMT1 in Hep3B cells.
Several investigators have reported synergistic effects of DNMTi and HDACi, and it has been suggested that TSA may affect the DNA methylation machinery (31, 37). Therefore, we examined the expression levels of DNMT1 in Hep3B cells by Western blotting after TSA treatment. Our analyses revealed a dose-dependent and significant decrease in nuclear DNMT1 expression levels after 24 h of TSA treatment (Fig. 2). These data are supported by several findings from different groups who have reported decreased DNMT expression, at both the protein and mRNA levels, after HDACi treatment (see Discussion).
Fig. 2.

Decreased expression of DNMT1 by TSA treatment. Hep3B cells were treated with TSA for 24 h. Cytoplasmic and nuclear proteins were resolved by SDS-PAGE, and DNMT1 was detected by Western blotting. Nuclear DNMT1 protein levels show a dose-dependent decrease. The graph shows the average of triplicate experiments, with error bars denoting standard errors of the means. Significance was calculated with a Student's t test. **, P < 0.01; ***, P < 0.001.
Global DNA methylation is reduced after TSA treatment.
Next, we sought to determine whether the reduced nuclear DNMT1 levels correlated with an effect on genomic DNA methylation. We assessed global genomic DNA methylation by two different methods, luminometric methylation assay (LUMA) (22) and nearest-neighbor analysis (NNA) (32). As shown in Fig. 3, a general decrease in genomic DNA methylation was evident by both methods. LUMA revealed a TSA-dependent decrease in genomic DNA methylation after TSA treatment. The average HpaII/EcoRI ratio increased significantly from 0.88 in untreated cells to 1.14 (P = 0.004) after treatment with 800 nM TSA, indicating substantial loss of methylation at CCGG sequences. NNA also revealed a 15% decrease in genomic methylated cytosine after TSA treatment. Numerical data from these experiments are shown in Table S1 posted at http://www.cmm.ki.se/en/Research/Neurogenetics-and-Psychiatric-Diseases/Medical-Epigenetics/Medical-Epigenetics/Supplementary-Materials. For clarification of the principle, a pyrogram from a LUMA is shown in Fig. S2 posted at the above URL.
Fig. 3.
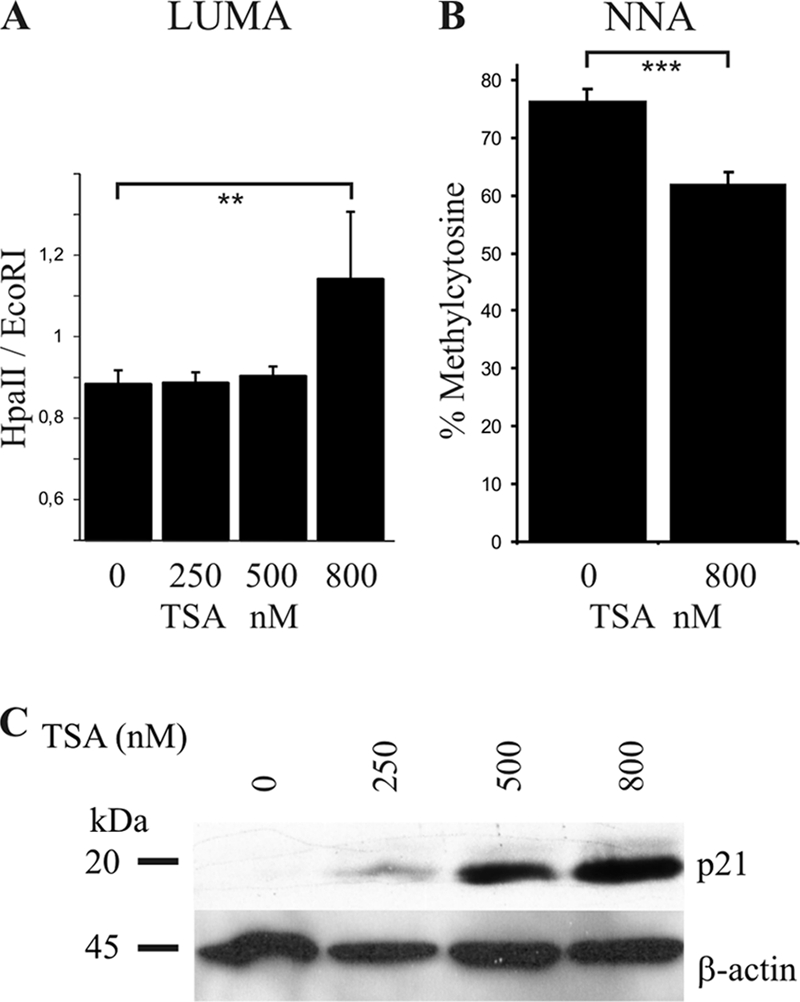
Decreased global DNA methylation by TSA treatment. Hep3B cells were treated by TSA for 24 h, and genomic DNA was extracted and analyzed for DNA methylation by LUMA (A) and NNA (B). Bars represent mean values ± standard deviations (triplicate experiments). Both assays revealed significant hypomethylation following TSA treatment. (C) Induction of p21waf1 by TSA treatment. Hep3B cells were treated by TSA for 24 h and then harvested and analyzed by Western blotting. P values were calculated using a Student's t test. **, P = 0.004; ***, P = 0.001.
p21waf1 induction by TSA does not involve demethylation.
Among HDAC inhibitor-responsive genes, p21waf1 is a common target which is induced quickly after HDACi treatment (34). To verify that our experimental conditions reproduced earlier studies, Western blotting of p21waf1 was performed after Hep3B cell incubation with TSA at various concentrations. As shown in Fig. 3C, p21waf1 expression was undetectable in untreated Hep3B cells but increased substantially after TSA treatment in a dose-dependent manner after 24 h, as shown previously (40). Furthermore, since TSA treatment results in global hypomethylation, we elucidated the role of DNA methylation for p21waf1 expression. The methylation pattern in the CpG island in the p21waf1 promoter before and after TSA treatment was investigated using bisulfite genomic sequencing, as described in Materials and Methods. This revealed no DNA methylation in the p21waf1 promoter in Hep3B cells in either untreated or treated cells. A typical sequencing profile of untreated and TSA-treated cells (one clone each) is shown in Fig. S3 posted at http://www.cmm.ki.se/en/Research/Neurogenetics-and-Psychiatric-Diseases/Medical-Epigenetics/Medical-Epigenetics/Supplementary-Materials. Therefore, changes in DNA methylation do not appear to be involved in the induction of p21waf1 expression following TSA treatment in these cells. Our results are in agreement with those of Chiba et al., who reported that p21waf1 is not methylated in six different hepatoma cell lines (3).
Gene-specific demethylation by TSA treatment.
To investigate genomic hypomethylation at a gene-specific level, seven arbitrary gene promoters were analyzed by MS-PCR. Four of these (E-cadherin, CDH13, DAPK, and RASSF1 promoters) were significantly demethylated after TSA treatment for 24 h, while there was no change in the promoters of the remaining three genes (Fig. 4). Demethylation of gene promoters by TSA has been reported in several studies (25), but there are also some contradictory reports showing no changes in promoter methylation (18). Our data indicate that genomic hypomethylation by TSA treatment is, indeed, a selective phenomenon which occurs at specific genomic loci.
Fig. 4.
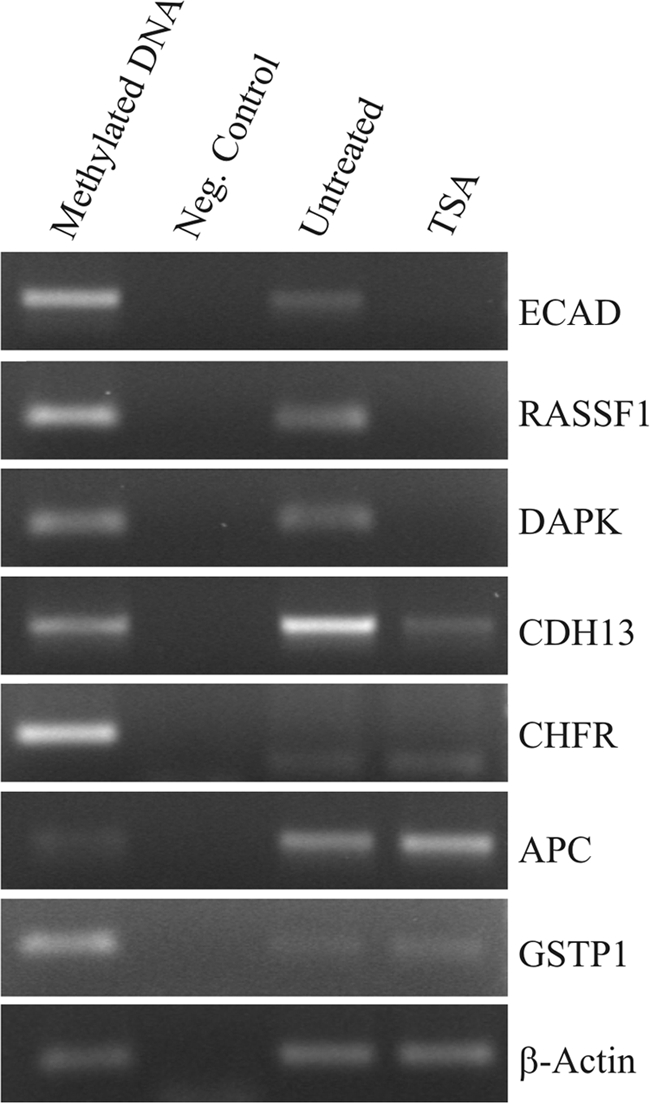
Gene-specific hypomethylation in Hep3B cells by TSA treatment. Hep3B cells were treated with TSA (800 nM) for 24 h. Genomic DNA was extracted and converted with bisulfite treatment. Methylation-specific PCR was applied to investigate promoter methylation in seven different genes. Four promoters showed decreased methylation after treatment.
Live-cell studies by APD imaging and FCS.
To investigate the effects of TSA on nuclear distribution and the dynamics of DNMT1 in living cells, genetically modified cells transiently expressing the fluorescent construct DNMT1-RFP were subjected to APD imaging and FCS analysis (Fig. 5, 6, 7, and 8).
Fig. 5.

(A) Autocorrelation curves recorded in the nucleus of U343MG cells using detection volume elements of different sizes. The amplitude of the autocorrelation curve decreases, indicating that the number of observed DNMT1 molecules increases as the detection volume element is enlarged. (B) Normalized autocorrelation curves [Gn(τ)] showing that both characteristic times, τ1 and τ2, increase when the detection volume element is enlarged. For clarity reasons, the fitted curves are shown in the inset. (C) The number of DNMT1 molecules in the detection volume element increases linearly with the median surface area of the detection volume element. (D) The characteristic times increase linearly with the square of the pinhole size, indicating that fluorescence intensity fluctuations are generated by molecular movement.
Fig. 6.

(A) CLSM images showing nuclear localization of DNMT1-RFP in human hepatoma Hep3B cells not treated (control) and treated with 5 μM 5-azacytidine or 500 nM TSA. Scale bar, 15 μm. (B) Autocorrelation curves recorded in the nucleus of untreated cells (control) and in the nucleoplasm (dark spots) in cells treated with 5-azacytidine and TSA. The autocorrelation curves are complex, with two characteristic times (τ1 and τ2). The actual values are given in Table 2. (C) Autocorrelation curves shown in panel B normalized to the same amplitude.
Fig. 7.

(A) CLSM images showing nuclear localization of DNMT1-RFP in the human glioblastoma U373MG cells not treated (control) and treated with 5 μM 5-azacytidine or 500 nM TSA. In cells treated with 5-azacytidine, inhomogeneous distribution of DNMT1 was observed in the nucleus, reflected by interspersed regions of high- and low-fluorescence intensity. Scale bar, 15 μm. (B) Autocorrelation curves recorded in the nucleus of nontreated cells (control) and in the nucleoplasm (dark spots) in cells treated with 5-azacytidine and TSA. (C) FCS measurements at the intensely fluorescent loci in 5-azacytidine-treated cells are compromised by severe photobleaching, indicating that DNMT1 mobility in these regions is low. (D) Autocorrelation curves shown in panel B normalized to the same amplitude.
Fig. 8.

(A) CLSM images showing nuclear localization of DNMT1-RFP in human glioblastoma U343MG cells not treated (control) and treated with 5 μM 5-azacytidine or 500 nM TSA. Scale bar, 15 μm. (B) Autocorrelation curves recorded in the nucleus of untreated cells (control) and in the nucleoplasm (dark spots) in cells treated with 5-azacytidine and TSA. (C) Autocorrelation curves shown in panel B normalized to the same amplitude.
(i) DNMT1 partitioning in the nucleus.
In FCS, fluctuations in fluorescence intensities are observed and analyzed in order to derive quantitative information about the investigated process (see supplemental information SI 1 posted at http://www.cmm.ki.se/en/Research/Neurogenetics-and-Psychiatric-Diseases/Medical-Epigenetics/Medical-Epigenetics/Supplementary-Materials for details). Temporal autocorrelation analysis of the recorded fluorescence intensity fluctuations yielded biphasic autocorrelation curves with two characteristic times, τ1 and τ2 (Fig. 5 to 8).
In the investigated system, two distinct processes may generate fluorescence intensity fluctuations: time-dependent changes in DNMT1-RFP localization and chemical interactions in the nucleus. The presence of chemical interactions between DNMT1-RFP and nuclear elements may contribute to fluorescence intensity fluctuations in several ways: chemical interactions may influence the apparent diffusion behavior of DNMT1-RFP, changes in RFP brightness may occur due to DNMT1-RFP binding, and bound DNMT1-RFP may reflect dynamic changes in chromatin structure. Therefore, we first needed to identify the process that leads to fluorescence intensity fluctuations in the investigated system. To this end, we performed a series of FCS measurements varying the size of the observation volume element by changing the pinhole size in front of the detector (30, 42, 44). This analysis (Fig. 5) revealed that both characteristic times, τ1 and τ2, increase linearly with the pinhole diameter squared, i.e., τ = f (ph2), which is proportional to the surface area of the waist of the observation volume element (Fig. 5D). This suggests that the observed fluorescence intensity fluctuations are generated by DNMT1-RFP movement in the nucleus such that the larger the observation volume element, the longer it takes for DNMT1-RFP molecules to traverse it. The biphasic shape of the autocorrelation curve indicates that two fractions of DNMT1-RFP with different mobilities are observed.
(ii) DNMT1-RFP mobility is affected by TSA treatment.
FCS analysis revealed that DNMT1-RFP mobility in live-cell nuclei is affected by TSA treatment. The DNA methylation inhibitor 5-azacytidine was used as a validation control for these experiments because of its direct action on DNA methylation. 5-Azacytidine induces marked redistribution of DNMT1-RFP in Hep3B (Fig. 6) and U373MG (Fig. 7) cells, but the concentration tested in this study did not affect its dynamics in the U343MG cells (Fig. 8). In the affected cells (Fig. 6 and 7), patchy, bright regions interspersed with dark, low-fluorescence intensity areas were observed. DNMT1-RFP dynamics in the bright and murky regions are different. FCS data recorded in the murky regions yielded biphasic autocorrelation curves with two characteristic times (Fig. 6 and 7, red curves). FCS measurements performed at the bright spots were severely compromised by photobleaching (Fig. 7C). Previous studies have shown that 5-azacytidine readily incorporates into the DNA, forming stable complexes with DNMTs (4). Excessive photobleaching (Fig. 7C) may indicate that DNMT1 mobility in the bright regions is low because of its binding to 5-azacytidine incorporated in chromatin-dense spots. For this reason, DNMT1 dynamics was investigated in the murky regions, presumably representing the nucleoplasm.
DNMT1 distribution in the nucleus of TSA-treated cells resembled its distribution in control cells (Fig. 6 to 8). FCS analysis yielded biphasic curves (Fig. 6 to 8, green curves) similar in shape to the ones observed in control, nontreated cells (Fig. 6 to 8, blue curves) but with shorter characteristic times, as evident from the shifting of the autocorrelation curve to the left, toward shorter times (Fig. 6).
DISCUSSION
In this study the effect of TSA treatment on DNA methylation was studied. First, we investigated effects by TSA on HDAC1, HDAC2, and HDAC3. The results suggest that following TSA treatment of Hep3B cells, HDAC1 is translocated from the nucleus to the cytoplasm, HDAC2 is degraded, and there is no change in HDAC3 localization or expression (Fig. 1). Subsequently, we analyzed the effect of TSA on the maintenance DNA methyltransferase, DNMT1, and we observed that nuclear DNMT1 protein is decreased at the protein level upon TSA treatment (Fig. 2). Results from several studies support our findings. Januchowski et al. reported that TSA treatment downregulates DNMT1 expression in Jurkat T cells (19), Xiong et al. reported DNMT1 to be downregulated by HDAC inhibitors in endometrial cells (46), and Ou et al. also reported changes in global DNA methylation following TSA treatment in T24 and MDA-MB-231 cells (bladder and breast cancer cells, respectively) (29). Zhou et al. also reported that DNMT1 degradation after HDACi treatment is mediated by ubiquitination (50).
Next, we analyzed the effect of TSA treatment on DNA methylation. Our analysis by both LUMA and NNA revealed significant global hypomethylation after TSA treatment. In addition, we investigated TSA effects on promoter-specific methylation in a subset of genes and found that the promoters in only four out of the seven studied genes became hypomethylated upon TSA treatment. This selective promoter hypomethylation can explain the inconsistent results by previous studies where both hypomethylation and no changes in methylation were reported following TSA treatment (18, 25). In an interesting paper, Gius et al. reported that the expression patterns in the HCT-116 cell line treated by 5-azacytidine or TSA are quite similar for both drugs, however, suggesting that they are targeting the same or converging cellular processes (13).
In addition to alterations in expression levels, we investigated the effect of TSA treatment on DNMT1 distribution and interactions with the genomic DNA. Confocal microscopy has already been reported to be a powerful method to study DNA methyltransferase activity in live cells following 5-azacytidine treatment (36). We applied APD imaging to visualize low levels of DNMT1 (300 nM to 2 μM) and investigated the interactions between DNMT1 and nuclear components in real time by FCS (Fig. 6 to 8). We identified two modes of DNMT1 movement with distinct characteristic times that are of the order 250 ± 150 μs for τ1 and 10 ± 5 ms for τ2. Both characteristic times, τ1 and τ2, depended on the size of the observation volume element (Fig. 5), suggesting that the fluorescence intensity fluctuations are generated by molecular movement. FCS analysis indicated that DNMT1 molecules are not undergoing free three-dimensional (3D) diffusion, as evident from the positive intercept in Fig. 5D (30, 42), and their movement is different in different domains in the nucleus. In the context of our studies, this may mean that DNMT1 partitions between the nucleoplasm and the chromatin-dense regions. DNMT1 mobility is faster in the nucleoplasm (characterized by the short average residence time τ1) and slows down in the chromatin-dense regions because of extensive interactions with the significantly larger biological targets, such as the genomic DNA (characterized by the long average residence time τ2). If the kinetics of DNMT1 interactions with the genomic DNA is sufficiently slow, the autocorrelation curves [G(τ)] can be modeled as two diffusing components (Fig. 5A and B) (9):
In this equation, N is the average number of molecules in the observation volume element, τ1 and τ2 are the apparent diffusion times or the apparent average residence times in the nucleoplasm and the chromatin-dense domains, respectively, y is the fraction of the bound DNMT1 wxy and wz are the e−2 beam radial and axial radii, respectively, of the detection volume approximated by a prolate ellipsoid, T is the average equilibrium fraction of RFP molecules in triplet state, and τT is the triplet correlation time, related to rate constants for intersystem crossing and the triplet decay of RFP. Results obtained by analyzing the FCS data using this model are summarized in Table 2. The results presented in Table 2 indicate that TSA affects DNMT1 mobility, reflecting changes in its interaction with the genomic DNA. However, we need to underline that autocorrelation analysis, when applied to systems with more than one component, has a practical detection limit that depends critically on the quantum yield, concentration, and differences in mobilities of the investigated molecules (27). Differences observed in this study are close to the detection limit of the methodology, necessitating measurements at even lower concentrations (DNMT1 concentration of <300 nM). At low concentrations the contribution of nonspecific interactions between DNMT1 and the nuclear components would be significantly reduced, enabling us to study the specific interactions of DNMT1 with its biological targets (44). DNMT1 mobility may also vary due to local differences in chromatin density, protein crowding, and pH, further complicating quantitative analysis. In spite of these difficulties, some tentative conclusions may be drawn from the presented FCS studies.
Table 2.
Changes in DNMT1 mobility under treatment with 5-azacytidine and TSA
| Treatment | Parametera | Value for the indicated cell line |
||
|---|---|---|---|---|
| Hep3B | U373MG | U343MG | ||
| Control | 1 − y | 0.4 ± 0.2 | 0.5 ± 0.2 | 0.5 ± 0.2 |
| τ1 (μs) | 250 ± 150 | 250 ± 150 | 250 ± 150 | |
| y | 0.6 ± 0.2 | 0.5 ± 0.2 | 0.5 ± 0.2 | |
| τ2 (ms) | 10 ± 5 | 20 ± 15 | 10 ± 5 | |
| 5-Azacytidine | 1 − y | 0.4 ± 0.2 | 0.7 ± 0.2 | 0.6 ± 0.2 |
| τ1 (μs) | 250 ± 150 | 250 ± 150 | 250 ± 150 | |
| y | 0.6 ± 0.2 | 0.3 ± 0.2 | 0.4 ± 0.2 | |
| τ2 (ms) | 10 ± 5 | 8 ± 5 | 8 ± 5 | |
| TSA | 1 − y | 0.7 ± 0.2 | 0.7 ± 0.2 | 0.6 ± 0.2 |
| τ1 (μs) | 250 ± 150 | 250 ± 150 | 150 ± 100 | |
| y | 0.3 ± 0.2 | 0.3 ± 0.2 | 0.4 ± 0.2 | |
| τ2 (ms) | 5 ± 3 | 8 ± 5 | 15 ± 10 | |
τ1 and τ2 are the apparent diffusion times or the apparent average residence times in the nucleoplasm and the chromatin-dense domains, respectively; y is the fraction of the bound DNMT1.
Our data show that DNMT1 mobility increases upon TSA treatment (Table 2). This observation may be interpreted in several ways, with one possible explanation being by HSP90 degradation, as suggested by Zhou et al. They reported that HSP90 bound to DNMT1 in untreated cells is degraded after TSA treatment, resulting in DNMT1 release from the complex (50). Another possibility is that mobility increases because of DNMT1 acetylation per se. In a proteomic study, Kim et al. reported that DNMT1 is acetylated on three lysines on a GK repeat around amino acid position 1010 (24). They suggested that this is an important linker region between the methyltransferase activity domain and the N-terminal regulatory domain of DNMT1. Therefore, HDACi treatment may induce hyperacetylation of DNMT1, resulting in a disturbance of DNMT1 binding to other nuclear components. It is also possible that the DNMT1 release is mediated by p21waf1 since p21waf1 is induced by HDACi. We have already reported that HDACi-induced apoptosis is partly mediated by p21waf1 (40). Also, it has been reported that p21waf1 and DNMT1 share the same binding site on proliferating cell nuclear antigen PCNA (5). It is thus possible that TSA-induced p21waf1 binds to PCNA and that DNMT1 is competed out and released from the complex. In addition, TSA-induced reduction in chromatin compaction (33) may also increase local DNMT1 mobility.
In conclusion, we have verified that TSA causes genomic hypomethylation and that this involves decreased nuclear DNMT1 levels. By using FCS, we have also shown that both the DNMT inhibitor 5-azacytidine and the HDAC inhibitor TSA change the nuclear kinetics of DNMT1. These data are important since they show that the effects of HDACi are not limited to direct hyperacetylation of histones but may indirectly affect other epigenetic factors, such as DNMT1 activity, through converging pathways. An increased knowledge about the effect of epigenetic modifiers on nuclear kinetics will be important for therapy using epigenetic drugs.
ACKNOWLEDGMENTS
This work was supported by the Swedish Cancer Society, the Knut and Alice Wallenberg Foundation, the Swedish Research Council, the Swedish Brain Foundation, and the Ministry of Sciences and Technological Development of Serbia (grant numbers 142025 and 142019).
The pDNMT1-RFP plasmid was kindly provided by Heinrich Leonhardt, Munich, Germany.
Footnotes
Published ahead of print on 25 July 2011.
REFERENCES
- 1. Bacia K., Schwille P. 2007. Fluorescence correlation spectroscopy. Methods Mol. Biol. 398:73–84 [DOI] [PubMed] [Google Scholar]
- 2. Bird A. 2002. DNA methylation patterns and epigenetic memory. Genes Dev. 16:6–21 [DOI] [PubMed] [Google Scholar]
- 3. Chiba T., et al. 2004. Identification of genes up-regulated by histone deacetylase inhibition with cDNA microarray and exploration of epigenetic alterations on hepatoma cells. J. Hepatol. 41:436–445 [DOI] [PubMed] [Google Scholar]
- 4. Christman J. K. 2002. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21:5483–5495 [DOI] [PubMed] [Google Scholar]
- 5. Chuang L. S., et al. 1997. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 277:1996–2000 [DOI] [PubMed] [Google Scholar]
- 6. Clark S. J., Harrison J., Paul C. L., Frommer M. 1994. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 22:2990–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Corn P. G., et al. 2003. Frequent hypermethylation of the 5′ CpG island of the mitotic stress checkpoint gene Chfr in colorectal and non-small cell lung cancer. Carcinogenesis 24:47–51 [DOI] [PubMed] [Google Scholar]
- 8. Ehrenberg M., Rigler R. 1974. Rotational Brownian-motion and fluorescence intensity fluctuations. Chem. Phys. 4:390–401 [Google Scholar]
- 9. Elson E. L. 2001. Fluorescence correlation spectroscopy measures molecular transport in cells. Traffic 2:789–796 [DOI] [PubMed] [Google Scholar]
- 10. Elson E. L., Magde D. 1974. Fluorescence correlation spectroscopy. 1. Conceptual basis and theory. Biopolymers 13:1–27 [DOI] [PubMed] [Google Scholar]
- 11. Fraga M. F., et al. 2005. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37:391–400 [DOI] [PubMed] [Google Scholar]
- 12. Ghoshal K., Majumder S., Jacob S. T. 2002. Analysis of promoter methylation and its role in silencing metallothionein I gene expression in tumor cells. Methods Enzymol. 353:476–486 [DOI] [PubMed] [Google Scholar]
- 13. Gius D., et al. 2004. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell 6:361–371 [DOI] [PubMed] [Google Scholar]
- 14. Gonzalez-Gomez P., et al. 2003. Aberrant methylation of multiple genes in neuroblastic tumours. Relationship with MYCN amplification and allelic status at 1p. Eur. J. Cancer 39:1478–1485 [DOI] [PubMed] [Google Scholar]
- 15. Gore S. D., Hermes-DeSantis E. R. 2009. Enhancing survival outcomes in the management of patients with higher-risk myelodysplastic syndromes. Cancer Control 16(Suppl.):2–10 [DOI] [PubMed] [Google Scholar]
- 16. Graff J. R., Herman J. G., Myohanen S., Baylin S. B., Vertino P. M. 1997. Mapping patterns of CpG island methylation in normal and neoplastic cells implicates both upstream and downstream regions in de novo methylation. J. Biol. Chem. 272:22322–22329 [DOI] [PubMed] [Google Scholar]
- 17. Gray S. G., Ekström T. J. 1998. Effects of cell density and trichostatin A on the expression of HDAC1 and p57Kip2 in Hep 3B cells. Biochem. Biophys. Res. Commun. 245:423–427 [DOI] [PubMed] [Google Scholar]
- 18. Horswill M. A., Narayan M., Warejcka D. J., Cirillo L. A., Twining S. S. 2008. Epigenetic silencing of maspin expression occurs early in the conversion of keratocytes to fibroblasts. Exp. Eye Res. 86:586–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Januchowski R., Dabrowski M., Ofori H., Jagodzinski P. P. 2007. Trichostatin A down-regulate DNA methyltransferase 1 in Jurkat T cells. Cancer Lett. 246:313–317 [DOI] [PubMed] [Google Scholar]
- 20. Jones P. A., Baylin S. B. 2007. The epigenomics of cancer. Cell 128:683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jones P. A., Baylin S. B. 2002. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 3:415–428 [DOI] [PubMed] [Google Scholar]
- 22. Karimi M., et al. 2006. LUMA (luminometric methylation assay)—a high throughput method to the analysis of genomic DNA methylation. Exp. Cell Res. 312:1989–1995 [DOI] [PubMed] [Google Scholar]
- 23. Kavanaugh S. M., White L. A., Kolesar J. M. 2010. Vorinostat: a novel therapy for the treatment of cutaneous T-cell lymphoma. Am. J. Health Syst. Pharm. 67:793–797 [DOI] [PubMed] [Google Scholar]
- 24. Kim S. C., et al. 2006. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23:607–618 [DOI] [PubMed] [Google Scholar]
- 25. Maass N., et al. 2002. Hypermethylation and histone deacetylation lead to silencing of the maspin gene in human breast cancer. Biochem. Biophys. Res. Commun. 297:125–128 [DOI] [PubMed] [Google Scholar]
- 26. Magde D., Elson E. L., Webb W. W. 1974. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers 13:29–61 [DOI] [PubMed] [Google Scholar]
- 27. Meseth U., Wohland T., Rigler R., Vogel H. 1999. Resolution of fluorescence correlation measurements. Biophys. J. 76:1619–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nightingale K. P., O'Neill L. P., Turner B. M. 2006. Histone modifications: signalling receptors and potential elements of a heritable epigenetic code. Curr. Opin. Genet. Dev. 16:125–136 [DOI] [PubMed] [Google Scholar]
- 29. Ou J. N., et al. 2007. Histone deacetylase inhibitor trichostatin A induces global and gene-specific DNA demethylation in human cancer cell lines. Biochem. Pharmacol. 73:1297–1307 [DOI] [PubMed] [Google Scholar]
- 30. Owen D. M., Williamson D., Rentero C., Gaus K. 2009. Quantitative microscopy: protein dynamics and membrane organisation. Traffic 10:962–971 [DOI] [PubMed] [Google Scholar]
- 31. Primeau M., Gagnon J., Momparler R. L. 2003. Synergistic antineoplastic action of DNA methylation inhibitor 5-AZA-2′-deoxycytidine and histone deacetylase inhibitor depsipeptide on human breast carcinoma cells. Int. J. Cancer 103:177–184 [DOI] [PubMed] [Google Scholar]
- 32. Ramsahoye B. H. 2002. Nearest-neighbor analysis. Methods Mol. Biol. 200:9–15 [DOI] [PubMed] [Google Scholar]
- 33. Rao J., Bhattacharya D., Banerjee B., Sarin A., Shivashankar G. V. 2007. Trichostatin-A induces differential changes in histone protein dynamics and expression in HeLa cells. Biochem. Biophys. Res. Commun. 363:263–268 [DOI] [PubMed] [Google Scholar]
- 34. Rocchi P., et al. 2005. p21Waf1/Cip1 is a common target induced by short-chain fatty acid HDAC inhibitors (valproic acid, tributyrin and sodium butyrate) in neuroblastoma cells. Oncol. Rep. 13:1139–1144 [PubMed] [Google Scholar]
- 35. Santi D. V., Norment A., Garrett C. E. 1984. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc. Natl. Acad. Sci. U. S. A. 81:6993–6997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schermelleh L., et al. 2005. Trapped in action: direct visualization of DNA methyltransferase activity in living cells. Nat. Methods 2:751–756 [DOI] [PubMed] [Google Scholar]
- 37. Shaker S., Bernstein M., Momparler R. L. 2004. Antineoplastic action of 5-aza-2′-deoxycytidine (Dacogen) and depsipeptide on Raji lymphoma cells. Oncol. Rep. 11:1253–1256 [PubMed] [Google Scholar]
- 38. Sun D., et al. 2007. Aberrant methylation of CDH13 gene in nasopharyngeal carcinoma could serve as a potential diagnostic biomarker. Oral Oncol. 43:82–87 [DOI] [PubMed] [Google Scholar]
- 39. Suzuki M. M., Bird A. 2008. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9:465–476 [DOI] [PubMed] [Google Scholar]
- 40. Svechnikova I., Ammerpohl O., Ekström T. J. 2007. p21waf1/Cip1 partially mediates apoptosis in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 354:466–471 [DOI] [PubMed] [Google Scholar]
- 41. Szyf M. 2009. Epigenetics, DNA methylation, and chromatin modifying drugs. Annu. Rev. Pharmacol. Toxicol. 49:243–263 [DOI] [PubMed] [Google Scholar]
- 42. Wawrezinieck L., Rigneault H., Marguet D., Lenne P. F. 2005. Fluorescence correlation spectroscopy diffusion laws to probe the submicron cell membrane organization. Biophys. J. 89:4029–4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vukojevic V., et al. 2008. Quantitative single-molecule imaging by confocal laser scanning microscopy. Proc. Natl. Acad. Sci. U. S. A. 105:18176–18181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vukojevic V., Papadopoulos D. K., Terenius L., Gehring W. J., Rigler R. 2010. Quantitative study of synthetic Hox transcription factor-DNA interactions in live cells. Proc. Natl. Acad. Sci. U. S. A. 107:4093–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vukojevic V., et al. 2005. Study of molecular events in cells by fluorescence correlation spectroscopy. Cell. Mol. Life Sci. 62:535–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xiong Y., et al. 2005. Histone deacetylase inhibitors decrease DNA methyltransferase-3B mRNA stability and down-regulate de novo DNA methyltransferase activity in human endometrial cells. Cancer Res. 65:2684–2689 [DOI] [PubMed] [Google Scholar]
- 47. Yang X., et al. 2001. Synergistic activation of functional estrogen receptor (ER)-α by DNA methyltransferase and histone deacetylase inhibition in human ER-α-negative breast cancer cells. Cancer Res. 61:7025–7029 [PubMed] [Google Scholar]
- 48. Yeo W., et al. 2005. High frequency of promoter hypermethylation of RASSF1A in tumor and plasma of patients with hepatocellular carcinoma. Liver Int. 25:266–272 [DOI] [PubMed] [Google Scholar]
- 49. Yoshida M., Kijima M., Akita M., Beppu T. 1990. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 265:17174–17179 [PubMed] [Google Scholar]
- 50. Zhou Q., Agoston A. T., Atadja P., Nelson W. G., Davidson N. E. 2008. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol. Cancer Res. 6:873–883 [DOI] [PMC free article] [PubMed] [Google Scholar]