

Abstract
The control of mRNA stability is an important component of regulation in bacteria. Processing and degradation of mRNAs are initiated by an endonucleolytic attack, and the cleavage products are processively degraded by exoribonucleases. In many bacteria, these RNases, as well as RNA helicases and other proteins, are organized in a protein complex called the RNA degradosome. In Escherichia coli, the RNA degradosome is assembled around the essential endoribonuclease E. In Bacillus subtilis, the recently discovered essential endoribonuclease RNase Y is involved in the initiation of RNA degradation. Moreover, RNase Y interacts with other RNases, the RNA helicase CshA, and the glycolytic enzymes enolase and phosphofructokinase in a degradosome-like complex. In this work, we have studied the domain organization of RNase Y and the contribution of the domains to protein-protein interactions. We provide evidence for the physical interaction between RNase Y and the degradosome partners in vivo. We present experimental and bioinformatic data which indicate that the RNase Y contains significant regions of intrinsic disorder and discuss the possible functional implications of this finding. The localization of RNase Y in the membrane is essential both for the viability of B. subtilis and for all interactions that involve RNase Y. The results presented in this study provide novel evidence for the idea that RNase Y is the functional equivalent of RNase E, even though the two enzymes do not share any sequence similarity.
INTRODUCTION
mRNAs transmit the genetic information from DNA to proteins. In contrast to the high stability of tRNA and rRNA, bacterial mRNAs are readily degraded. This crucial feature ensures the ability of bacteria to respond quickly to changing environmental conditions by adding an extra layer of control, in addition to the regulation of transcription and enzymatic activities of proteins (1). Moreover, mRNA processing allows the adjustment of different expression levels for genes encoded in one operon, as observed for the ilv operon and the glycolytic gapA operon of the Gram-positive soil bacterium Bacillus subtilis (24, 40, 45).
The enzymes that process and degrade RNAs are called RNases. In the two model organisms Escherichia coli and B. subtilis, a set of about 20 RNases has been described thus far, but only a small fraction of these are common to both bacteria (12). Some of these RNases (e.g., RNase M5 or RNase-Mini III in B. subtilis) are necessary for the maturation of one specific target, whereas others (e.g., RNase J1 or RNase Y in B. subtilis) are more promiscuous and have a more global impact on RNA metabolism (12, 43, 58). The individual RNases differ not only in their respective amino acid sequences and in their number of substrates but also in their enzymatic mode of action. Two different classes of RNases can be defined: exoribonucleases, which remove RNA nucleotides one at a time from either the 3′ or the 5′ end, and endoribonucleases, which cleave within the RNA molecule. For the degradation of mRNAs, the concerted action of both enzyme classes is necessary, as 5′ ends are protected by their native triphosphates, whereas decay from the 3′ end is often impeded by RNA secondary structural elements (e.g., transcriptional terminators) (9). For instance, in E. coli the endoribonuclease RNase E acts processively on the mRNA transcript, producing smaller fragments that are subsequently cleaved by exoribonucleases like polynucleotide phosphorylase (PNPase) or RNase II (33). The key step in this process is the initial endonucleolytic cleavage by RNase E, perhaps explaining the essential nature of this enzyme for E. coli.
RNase E is a large protein of 1,061 amino acids comprising two distinct functional domains. The N-terminal half (residues 1 to 530) comprises the catalytic core. This part of the protein is structured and well conserved among different prokaryotes. The structure of the catalytic domain is already known (6). In contrast, the C-terminal half of RNase E has little intrinsic order, rendering its structure solution impossible. The unstructured part of RNase E is very important for function, as it serves as an organizing center for the binding of the exoribonuclease PNPase, the DEAD-box RNA helicase RhlB, and the glycolytic enzyme enolase (7). Together, these four proteins form a protein complex called the RNA degradosome, a molecular machine devoted to RNA degradation. The importance of processing and degradation of RNA by protein complexes is underlined by the fact that degradosome-like complexes are found in bacteria and also in archaea and eukaryotes, where they are called exosomes (7, 15).
An RNA degradosome was found only recently in B. subtilis, and it consists of the novel essential endoribonuclease RNase Y, the essential endo/exoribonuclease RNase J1 and its nonessential paralog RNase J2, the polynucleotide phosphorylase PNPase, the DEAD-box RNA helicase CshA, and the two glycolytic enzymes enolase and phosphofructokinase (11, 35). The relevance of a degradosome in B. subtilis is also supported by the fact that RNase Y, RNase J1, and PNPase are all involved in the turnover of identical targets, such as the rpsO mRNA or the yitJ riboswitch (58, 65). The discovery of an RNA degradosome in B. subtilis was somewhat surprising, as the genomes of Gram-positive bacteria do not encode homologues of RNase E. However, the newly identified RNase Y in B. subtilis appears to be the functional counterpart of RNase E in E. coli, even though the two RNases do not share any sequence similarity. The functional similarity between the two enzymes is intriguing: both enzymes are essential endoribonucleases preferring 5′ monophosphorylated substrates (9, 58), and they are the only RNases shown, thus far, to have a major impact on bulk mRNA degradation in their host organisms (2, 58). Finally, both are central members of their respective RNA degradosomes, and both are integral membrane proteins (25, 31, 39).
While RNase E has been the subject of intensive investigation for several years (see reference 8 for a recent review), the analysis of RNase Y is still in the initial stages. RNase Y is required to control the degradation of bulk mRNA in B. subtilis, and the mRNAs of rpsO and the gapA operon as well as the yitJ riboswitch have been identified as targets (11, 58, 65). On the basis of the initial target analyses, it was suggested that RNase Y requires 5′ monophosphorylated RNA and prefers substrates with downstream secondary structures (58). From the amino acid sequence, RNase Y seems to comprise at least three domains. First, a short N-terminal transmembrane domain mediates the membrane localization of the protein (25). Second, a KH domain is required for RNA binding, and finally, an HD domain contains the catalytic apparatus (12, 25). The importance of the HD domain has already been demonstrated, as a strain carrying a single point mutation in the catalytic site is not viable (25).
In this work, we have studied the domain organization of RNase Y and its interactions with the other components of the RNA degradosome. Using biochemical techniques, we have shown that RNase Y is a natively disordered protein that forms dimers in solution. We have refined further the domain analysis in silico. On the basis of this new domain definition, we have analyzed the contribution of the single domains in the context of protein-protein interactions and cell viability. Our observation that a variant of RNase Y without the transmembrane domain (which is catalytically competent in vitro) could not complement in vivo revealed that membrane localization is essential for RNase Y activity in vivo. Using bacterial two-hybrid (B2H) analysis, we also demonstrated that the intact, full-length protein was required to interact with the other components of the RNA degradosome. Together, our new data provide further compelling evidence that RNase Y is the functional equivalent of RNase E.
MATERIALS AND METHODS
Bacterial strains, oligonucleotides, and growth conditions.
The B. subtilis strains were derived from the laboratory strain 168 (trpC2), and they are listed in Table 1. E. coli DH5α, BTH101, and BL21(DE3) (30, 56) were used for cloning experiments, B2H analyses, and protein overproduction, respectively. B. subtilis and E. coli were grown in LB medium. LB and sporulation medium (SP) plates were prepared by addition of 17 g Bacto agar/liter (Difco) to LB and SP, respectively (32, 56).
Table 1.
Bacillus subtilis strains used in this study
| Strain | Genotype | Source or referencea |
|---|---|---|
| 168 | trpC2 | Laboratory collection |
| GP193 | ΩymdA::pGP774 (PxylA-rny cat) | 11 |
| GP1012 | trpC2 rny-Strep ΔymdB::cat | See experimental procedures in text |
| GP1013 | trpC2 rny-Strep ΔymdB::cat amyE::pGP1354 (Ppgk::ymdB) | pGP1354 → GP1012 |
| GP1016 | trpC2 rny-Strep ΔymdB::cat amyE::pGP1354 (Ppgk::ymdB) pnpA-3×FLAG | pGP1376 → GP1013 |
| GP1022 | trpC2 rny-Strep ΔymdB::cat amyE::pGP1354 (Ppgk::ymdB) cshA-3×FLAG | pGP1333 → GP1013 |
| GP1023 | trpC2 rny-Strep ΔymdB::cat amyE::pGP1354 (Ppgk::ymdB) pfkA-3×FLAG | pGP1375 → GP1013 |
| GP1024 | trpC2 rny-Strep ΔymdB::cat amyE::pGP1354 (Ppgk::ymdB) rnjA-3×FLAG | pGP1376 → GP1013 |
| GP1025 | trpC2 rny-Strep ΔymdB::cat amyE::pGP1354 (Ppgk::ymdB) ytsJ-3×FLAG | pGP1758 → GP1013 |
| GP1033 | trpC2 rny-Strep spc | pGP1391 → 168 |
| GP1039 | trpC2 rny-Strep ΔymdB::cat amyE::pGP1354 (Ppgk::ymdB) rnjB-3×FLAG | pGP1851 → GP1013 |
| GP1040 | trpC2 rny-Strep ΔymdB::cat amyE::pGP1354 (Ppgk::ymdB) eno-3×FLAG | pGP1264 → GP1013 |
| GP1091 | trpC2 lacA::xylR aphA3 | pGP884 → 168 |
| GP1092 | trpC2 lacA::xylR aphA3 ΩymdA::pGP774 (PxylA-rny cat) | pGP774 → GP1091 |
Arrows indicate construction by transformation.
DNA manipulation.
Transformation of E. coli and plasmid DNA extraction were performed using standard procedures (56). All commercially available plasmids, restriction enzymes, T4 DNA ligase, and DNA polymerases were used as recommended by the manufacturers. DNA fragments were purified from agarose gels using a Nucleospin extraction kit (Macherey and Nagel, Germany). All primer sequences are provided in Table S1 in the supplemental material. DNA sequences were determined using the dideoxy chain termination method (56). Chromosomal DNA of B. subtilis was isolated as described previously (32).
Transformation and phenotypic analysis.
Standard procedures were used to transform E. coli (56), and transformants were selected on LB plates containing either ampicillin (100 μg/ml) or kanamycin (50 μg/ml). B. subtilis was transformed with plasmid DNA according to the two-step protocol (32). Transformants were selected on SP plates containing erythromycin (2 μg/ml) plus lincomycin (25 μg/ml), chloramphenicol (5 μg/ml), or kanamycin (20 μg/ml). For colony architecture analysis, bacteria were precultured in LB medium containing 1% xylose until an optical density at 600 nm of about 1.0 was reached. Ten microliters of this culture was then spotted onto LB-xylose plates, and the plates were incubated at 22°C for 6 days.
Plasmid constructions.
All plasmids used in this study are listed in Table S2 in the supplemental material. RNase Y variants were expressed in B. subtilis under the control of the constitutively active degQ(Hy) promoter using the expression vector pBQ200 (44). Briefly, the desired rny alleles were generated by PCR using the oligonucleotides listed in Table S1 in the supplemental material. The PCR products were digested with BamHI and SalI and cloned into pBQ200 linearized with the same restriction enzymes. The resulting plasmids are listed in Table S2 in the supplemental material. The chimeric ezrA-rny fusion gene was obtained by amplifying the regions encoding the transmembrane domain of EzrA and RNase Y without the membrane segment, using primer pairs ML32/ML33 and ML34/FR6, respectively (see Table S1 in the supplemental material). The primers ML33 and ML34 had an overlap allowing fusion of the products in another round of PCR using ML32 and FR6 as amplification primers. The PCR product was then cloned into pBQ200 as described above, and the resulting plasmid was named pGP1352.
To fuse different RNase Y variants to a 3×FLAG tag and allow overexpression in B. subtilis, we constructed plasmid pGP1370 that allows multicopy expression of FLAG-tagged proteins. This plasmid was obtained by amplification of the FLAG tag-encoding region from pGP1331 (35) using the primer pair ML5/ML6 and insertion of the PCR product into the expression vector pBQ200 (44) linearized with HindIII. Next, we amplified the coding sequence of full-length RNase Y (FR7/ML44), RNase Y without the transmembrane helix (FR4/ML44), and the chimeric RNase Y containing the N-terminal domain of EzrA (ML32/ML44). The resulting PCR products as well as plasmid pGP1370 were digested with BamHI and SalI to allow subsequent ligation. The resulting plasmids were pGP1363 (full-length RNase Y), pGP1364 (RNase Y without the transmembrane helix), and pGP1365 (RNase Y with the transmembrane segment of EzrA).
To obtain the plasmids for the B2H analyses, defined regions of rny were amplified by PCR using chromosomal DNA of B. subtilis 168. The gene-specific primers are listed in Table S1 in the supplemental material. The PCR products were digested with XbaI and KpnI, and the resulting fragments were cloned into each of the four plasmids p25-N, pKNT25, pUT18, and pUT18c (10, 30) digested with the same enzymes. The resulting plasmids used for the B2H analyses are listed in Table S2 in the supplemental material. All plasmid inserts were verified by DNA sequencing.
Construction of a strain that allows controlled depletion of RNase Y.
In our previous studies, we noticed that the xylose-controlled expression of RNase Y was not completely tight in the absence of xylose. To address this problem, we constructed strain GP1092, which encodes an extra copy of the xylose repressor XylR, as follows. First, the xylR gene was inserted at the ectopic lacA site of the B. subtilis chromosome by transforming B. subtilis 168 with plasmid pGP884 (20). The resulting strain, GP1091, was then transformed with plasmid pGP774, which places the rny gene under the control of the XylR-controlled promoter of the xylose operon (11).
Construction of strains expressing tagged degradosome proteins.
To facilitate the detection of the degradosome components by Western blot analysis, we fused RNase J1, RNase J2, PNPase, CshA, PfkA, Eno, and YtsJ to a C-terminal triple FLAG tag. For this purpose we used the plasmids pGP1375 (pfkA), pGP1376 (rnjA), pGP1377 (pnpA), and pGP1758 (ytsJ) (35, 46). Eno and RNase J2 were fused to the FLAG tag using plasmid pGP1331 (35). Briefly, the corresponding genes eno and rnjB were amplified with the primer pairs FR86/FR90 and ML173/ML174, respectively, digested with BamHI and SalI, and cloned into pGP1331 linearized with the same enzymes. The resulting plasmids were pGP1264 (eno) and pGP1851 (rnjB). The designations of the resulting strains are listed in Table 1. For primers, see Table S1 in the supplemental material.
To express RNase Y fused to a C-terminal Strep-tag from its native locus, we first constructed the cloning vector pGP1389, which allows easy integration of the constructs into the chromosome. For this purpose, the annealed oligonucleotides BD10/BD11 (23) were inserted into plasmid pUS19 linearized with BamHI and HindIII. In the next step, the rny gene was amplified using the primer pair ML97/ML44. The PCR product was cloned between the BamHI and SalI sites of pGP1389, giving pGP1391. This plasmid was used to construct B. subtilis GP1033 expressing the Strep-tag-carrying RNase Y. To avoid interference between the two copies of integrated plasmids that are associated with the Strep-tag-carrying and the FLAG-tagged proteins, we next constructed strain GP1012, which carries the rny-Strep fusion construct linked to a chloramphenicol resistance gene, by transformation with PCR products constructed using oligonucleotides (see Table S1 in the supplemental material) to amplify DNA fragments flanking each target gene and an intervening antibiotic resistance cassette as described previously (63).
In vivo detection of protein-protein interactions.
The isolation of protein complexes from B. subtilis cells was performed by the SPINE technology (23). Briefly, growing cultures of B. subtilis were treated with formaldehyde (0.6% [wt/vol], 20 min) to facilitate cross-linking of interacting proteins (23). The Strep-tag-carrying proteins and their potential interaction partners were then purified from crude extracts using a Streptactin column (IBA, Göttingen, Germany) and desthiobiotin as the eluent. Interacting proteins were identified by Western blot analysis.
Bacterial two-hybrid assay.
Primary protein-protein interactions were identified by B2H analysis (30). The B2H system is based on the interaction-mediated reconstruction of Bordetella pertussis adenylate cyclase (CyaA) activity in E. coli. Functional complementation between two fragments (T18 and T25) of CyaA as a consequence of the interaction between bait and prey molecules results in the synthesis of cyclic AMP (cAMP), which is monitored by measuring the β-galactosidase activity of the cAMP-catabolite gene activator protein-dependent promoter of the E. coli lac operon. Plasmids pUT18 and p25N allow the expression of proteins fused to the N terminus of the T18 and T25 fragments of CyaA, respectively, whereas pUT18C and pKT25 allow the expression of proteins fused to the C terminus of the T18 and T25 fragments of CyaA, respectively (10, 30). The plasmids constructed for the B2H assay (see Table S2 in the supplemental material) were used for cotransformation of E. coli BTH101, and the protein-protein interactions were then analyzed by plating the cells on LB plates containing 100 μg/ml ampicillin, 50 μg/ml kanamycin, 40 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal), and 0.5 mM (isopropyl-β-d-thiogalactopyranoside (IPTG), respectively. The plates were incubated for a maximum of 36 h at 30°C.
Recombinant protein production.
To generate RNase Y constructs for recombinant protein production, coding sequences were amplified by PCR with Phusion High Fidelity DNA polymerase (New England BioLabs) and the primers listed in Table S1 in the supplemental material using B. subtilis 168 genomic DNA as the template. The PCR product encoding residues 25 to 520 of the RNase Y open reading frame, designed so as to remove the predicted N-terminal transmembrane helix, was cloned into pET24a by restriction with NdeI and NotI for the overexpression of an in-frame C-terminal hexahistidine-tagged protein. The RNase Y constructs were transformed into E. coli BL21(DE3) or Rosetta(DE3) for protein overexpression. Cultures were grown in liquid LB medium supplemented with kanamycin at 37°C until the optical density at 600 nm reached 0.6 to 0.8, at which point overexpression was induced with the addition of IPTG to a final concentration of 0.1 mM and the culture was left overnight at 25°C before harvesting by centrifugation.
RNase Y was purified by immobilized metal affinity chromatography using a His-Trap column (GE Healthcare), equilibrated in 50 mM Tris, pH 8.0, 20 mM imidazole, and eluted with a linear gradient of 20 to 250 mM imidazole. Fractions containing RNase Y were pooled, concentrated, and loaded onto a Superdex 200 gel filtration column that had been preequilibrated in 50 mM Na/HEPES, pH 7.0, 500 mM NaCl. To avoid the precipitation of RNase Y in low-ionic-strength solutions at high protein concentrations, 500 mM NaCl was included in all buffers to stabilize the protein. The final yield of RNase Y was 0.3 mg protein per gram cell paste. The final protein purity was estimated to be greater than 90% by SDS-PAGE, and its concentration was determined by measuring the absorbance at 280 nm.
AUC.
Sedimentation velocity experiments were carried out in a Beckman Coulter XL-I analytical ultracentrifuge (Palo Alto, CA) using an 8-hole AnTi50 rotor and using both absorbance and interference optics. Analytical ultracentrifugation (AUC) runs were carried out at a rotation speed of 50,000 rpm using a 400-μl sample volume, with protein concentrations ranging from 0.1 to 2 mg/ml. The density and viscosity of the buffers were calculated from their composition using the program SEDENTERP (34). The velocity data were analyzed with the program SEDFIT using a diffusion deconvoluted continuous sedimentation coefficient distribution c(s) model (57).
Western blot analysis.
For Western blot analysis, B. subtilis cell extracts were separated on 12.5% SDS-polyacrylamide gels. After electrophoresis, the proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad) by electroblotting. RNase Y and RNase Y-FLAG were detected using antibodies raised against RNase Y (66) and the FLAG tag (Sigma), respectively. The primary antibodies were visualized by using anti-rabbit IgG-alkaline phosphatase secondary antibodies (Promega) and a CDP* detection system (Roche Diagnostics).
RESULTS
In silico analysis of RNase Y.
An in silico analysis of the domain structure of RNase Y detected an N-terminal transmembrane domain (residues 5 to 24) and two central KH and HD domains (residues 210 to 280 and 330 to 430, respectively), in agreement with previous analyses (11, 58). The relevance of the transmembrane domain of RNase Y is supported by the membrane localization of the protein (25, 66). Similarly, the importance of the catalytic residues in the HD domain has been demonstrated both in vivo and in vitro (25, 58). Our analysis additionally revealed the presence of a coiled-coil domain with the potential to form a leucine zipper, spanning residues 30 to 150 (Fig. 1), and a C-terminal domain (residues 430 to 520) that has not been described previously but that is conserved in all RNases Y.
Fig. 1.

Domain arrangement of RNase Y. The N-terminal 30 amino acids of RNase Y form a transmembrane-spanning α helix. Using the program COILS, a coiled-coil region is predicted between amino acids 30 and 150. The KH domain (RNA binding) and HD domain (catalytic activity) span from amino acids 210 to 280 and 330 to 430, respectively. No functional domain is predicted for the C terminus of the protein (amino acids 430 to 520); however, this sequence is well conserved in RNase Y proteins from different bacteria.
To extend the domain analysis of RNase Y, we have submitted the amino acid sequence to the web servers PONDR-FIT (64) and metaPrDOS (26), which predict specifically the presence of naturally disordered regions of proteins, an important and still emerging facet of protein function (14). PONDR-FIT and metaPrDOS are both meta predictors which incorporate the results of several individual bioinformatic tools to output a consensus score with greater accuracy than the best-performing individual tools (64). As can be seen from Fig. 2A, both tools predict a single, large contiguous stretch of disorder near the N terminus of RNase Y with high disorder propensity. The exact boundaries of this region vary depending on both the type of predictor and the strictness of criteria used, with estimates of length ranging from 158 residues (amino acids 34 to 192) to just 76 residues (amino acids 70 to 146), with the general consensus being in the range of 100 amino acids. A sharp decrease in the disorder propensity can be observed at about residue 190, which we believe to be a good estimate of the upper limit of the disordered region, with the lower limit being less well defined. A number of the individual bioinformatic tools predicted additional shorter disordered regions which, importantly, lie in between the ordered domains identified in silico. Thus, it is possible that additional regions of RNase Y are disordered, but the shorter length and inconsistent predictions mean that little confidence can be assigned to these predictions. Charge-hydropathy analysis (62) of the amino acid sequence using the domain boundaries previously identified shows that the HD, KH, and C-terminal domains clearly fall in the region of the plot occupied by folded proteins, which is in contrast to the N-terminal disordered region, which clearly belongs to the distribution of disordered proteins (Fig. 2B).
Fig. 2.

In silico and in vitro analysis of RNase Y. (A) Disorder propensity analysis of RNase Y calculated using the program PONDR and the VL-XT neural network predictor and plotted as a function of the RNase Y amino acid sequence. Continuous regions of disorder >30 amino acids long are considered significant. (B) Hydropathy-charge analysis of RNase Y domains. The mean net charge is plotted as a function of the mean hydrophobicity for a set of 105 ordered and 54 disordered proteins. Disordered proteins are expected to possess a higher proportion of charged residues and a lower content of hydrophobic amino acids, causing them to cluster toward the upper left-hand side of this plot. The individual domains of RNase Y are plotted on the graph as indicated in the key. The solid line represents a boundary dividing ordered and disordered regions. (C) Output from the COILS coiled-coil prediction server for RNase Y, plotted as a function of residue number.
The N-terminal disordered region overlaps significantly with the predicted coiled-coil domain identified using the COILS program (41), which predicts a strong tendency to form coiled coils (>0.92 confidence score throughout) for residues 31 to 153 (Fig. 2C). The high confidence levels assigned to this region by both programs and the accuracy for disorder prediction algorithms for the prediction of long disordered regions (a <0.4% error rate for 40 consecutive residues [13]) lead us to suggest that this region may form a flexible coiled-coil-like structure that is also significantly disordered and is able to occupy many different conformational states. Indeed, purified RNase Y is highly sensitive to proteolysis and is rapidly and completely digested over the time course of a limited trypsinolysis experiment (data not shown).
RNase Y is disordered in solution and forms dimers.
Previous interaction studies with RNase Y using the bacterial two-hybrid system had suggested that the protein is capable of self-interaction (11). However, the nature of these oligomers, whether they were dimers, trimers, or higher-level oligomers, was unknown. To address this question, we have analyzed purified protein that lacks the N-terminal transmembrane domain by size exclusion chromatography. A significant proportion of the protein seemed to be in an aggregated form, with the molecular mass of the nonaggregated form estimated to be 280 kDa (Fig. 3A). To characterize RNase Y in solution further, we analyzed the hydrodynamic properties of the enzyme by sedimentation velocity AUC (Fig. 3B). Analysis of the velocity data using a continuous c(s) distribution model revealed that a mixture of two species is present in solution, with a major peak at an S value of 4.5 and a minor fraction at an S value of 7.6, corresponding to molecular masses of 100 and 200 kDa, which most likely represent dimers and tetramers, respectively (Fig. 3C). The relative abundance of these two species does not appear to change with protein concentration under the conditions tested, between 0.37 and 1.5 mg/ml. The value of the frictional ratio, f/f0shape, was 1.41, which is significantly greater than the value (1.0) expected for a perfectly spherical particle. These data indicate that RNase Y adopts an extended, nonglobular conformation in solution, a property common to intrinsically disordered proteins and to proteins with coiled-coil domains. The apparent discrepancy between the major 100-kDa dimeric species identified by AUC and the apparent mass of RNase Y of 280 kDa observed by size exclusion chromatography can also be explained by the fact that RNase Y adopts an extended nonglobular structure in solution with a larger Stokes radius than a globular protein of equal mass. The elongated nature of RNase Y would cause it to elute earlier in size exclusion, with a consequent overestimation of its apparent molecular mass. It is also not clear at present whether this single peak corresponds to a very elongated dimer or a slightly elongated tetramer. The sedimentation data are also consistent with the in silico predictions that RNase Y contains extensive regions of intrinsic disorder, which, presumably and classically, will become ordered upon the interaction with other proteins.
Fig. 3.

Hydrodynamic analysis of RNase Y in solution. (A) Gel filtration chromatography of purified RNase Y. The nonaggregated form was found to elute at ∼59 ml, which corresponds to an expected molecular mass of 280 kDa for a globular protein (calculated from calibration curve shown in the inset) Ve, elution volume; Vo, column void volume. (B) Sedimentation velocity AUC of RNase Y. (Top) The raw absorbance data overlaid with the fit using a continuous distribution for sedimentation coefficient model c(s); (bottom) the residuals. (C) Normalized sedimentation coefficient values calculated from the data, with different dilutions represented as colored lines. RNase Y appears to exist as two species in solution, a major smaller species (s20, w = 4.5, corresponding to an approximate molecular mass of 100 kDa) and a minor larger species (s20,w = 7.6, corresponding to an approximate molecular mass of 200 kDa); these two species would correspond to dimers and tetramers of RNase Y, respectively.
Complementation analysis of the domains of RNase Y.
To assess the functional properties of RNase Y in vivo, we performed a complementation assay using truncated fragments of the RNase. The rny gene encoding RNase Y is essential for the growth of B. subtilis (25). We have constructed a B. subtilis strain, GP1092, in which rny expression is controlled by the xylose repressor, and, therefore, this strain is unable to grow in the absence of xylose. Strain GP1092 was transformed with a series of plasmids that encode the full-length or truncated variants of the rny gene, and growth of the bacteria was recorded. As shown in Fig. 4, transformation of GP1092 with the empty vector pBQ200 allowed growth of the bacteria only in the presence of xylose, confirming the essentiality of RNase Y for B. subtilis in this genetic background. Transformation with plasmid pGP1201, which allows expression of the full-length rny gene, resulted in xylose-independent growth, suggesting that the plasmid-borne RNase Y effectively complements the chromosomal copy that is repressed in the absence of xylose. In contrast, all truncated variants of the rny gene were unable to complement RNase Y depletion (Fig. 4). The lack of growth might be due to poor expression or instability of the truncated proteins or to their functional inactivity. To distinguish between these possibilities, we studied the accumulation of the different RNase Y variants by Western blot analysis. As shown in Fig. 4C, all truncated variants were expressed, as detected with antibodies raised against RNase Y. However, using the anti-Rny antibody, it was difficult to distinguish chromosomally expressed RNase Y from plasmid-encoded variants for those proteins that were similar in size to the wild-type protein. Therefore, we constructed additional plasmids that expressed FLAG-tagged variants of those proteins and detected their expression using anti-FLAG antibodies (Fig. 4D). Again, these proteins were expressed in B. subtilis. Taken together, the data demonstrate that both the transmembrane and the C-terminal domains of RNase Y are essential for the function of the protein in the organism. As a consequence, we then assessed the roles of the various domains of RNase Y in membrane association, for self-assembly into dimers, and for the interaction with other proteins in the degradosome.
Fig. 4.

Functional complementation by truncated RNase Y proteins in vivo. RNase Y was truncated N and C terminally with respect to the protein domains annotated in Fig. 1. The truncated proteins were expressed from an overexpression vector in strain GP1016. In this strain, the chromosomal copy of rny is under the control of a xylose-inducible promoter. Fresh cells (optical density, 1.0) were spotted in 10-fold dilution on LB agar plates with (A) and without xylose (B) to facilitate or impede chromosomal rny expression. To verify the expression of the RNase Y variants, Western blot analyses were performed with anti-RNase Y (C) and anti-FLAG (D) antibodies. A band that results from the nonspecific interaction of one protein with the antibodies raised against RNase Y is marked by an asterisk (C). Abbreviations: TM, transmembrane domain; CC, coiled-coil/disordered domain; KH, KH domain; HD, HD domain; CT, C-terminal domain.
The transmembrane domain of RNase Y can be replaced by a heterologous domain.
The transmembrane domain of RNase Y comprises residues 5 to 24 (Fig. 1) and is predicted to form a single transmembrane α helix, but its amino acid sequence does not conform to the signal peptide paradigm (60, 61) (Fig. 5A), consistent with RNase Y being a membrane rather than a secreted protein. Furthermore, our complementation data indicated that the transmembrane domain is crucial for the function of RNase Y in B. subtilis (Fig. 4). To probe the membrane association of this domain of RNase Y, we replaced the transmembrane region with the analogous domain of an unrelated protein, the divisome protein EzrA (37). First, we tested the ability of the chimeric protein to replace RNase Y in the rny depletion strain, GP1092. As shown in Fig. 4, transformants of GP1092 with a plasmid expressing the EzrA-RNase Y chimera grew well in both the presence and the absence of xylose. In contrast, the RNase Y lacking the N-terminal transmembrane domain was unable to grow when the wild-type rny gene was depleted by xylose limitation. The strain that expressed the EzrA-RNase Y chimera grew at higher dilution rates than the strain that expressed the wild-type RNase Y (Fig. 4), suggesting that the chimeric EzrA-RNase Y might be even more active and/or more stable than wild-type RNase Y. Indeed, Western blot analyses on the accumulation of the RNase Y variants demonstrated that the EzrA-RNase Y protein was present at higher levels than the full-length, wild-type protein (Fig. 4D).
Fig. 5.

Influence of a heterologous N-terminal domain in RNase Y on colony morphology. (A) Alignment of the first amino acids of RNase Y and EzrA. The first 26 amino acids of RNase Y (comprising the transmembrane segment of the enzyme) were replaced by the N terminus of the unrelated EzrA protein. The arrows at amino acid 26 of RNase Y mark the endpoint of the domain exchange. (B) Phenotypic characterization of the strain GP193 (Pxyl-rny) harboring the empty vector pBQ200, an rny overexpression plasmid, and a variant of rny with a heterologous transmembrane segment. This chimeric protein is more abundant in the cell (see the Western blot in Fig. 4D), leading to the formation of a more pronounced biofilm. All strains were spotted on LB agar plates supplemented with xylose and inoculated at 22°C for 6 days.
Recently, we have also shown that RNase Y is required for the full expression of the epsA-epsO and tapA-sipW-tasA operons that encode the proteins required for extracellular polysaccharide synthesis and the matrix protein TasA, i.e., the major contributors to biofilm formation (36). Moreover, overexpression of RNase Y was sufficient to induce biofilm formation even in the laboratory strain B. subtilis 168 (36). We therefore examined the morphology of B. subtilis GP1092, a derivative of B. subtilis 168, in the presence of the plasmid pGP1352, which expresses the EzrA-RNase Y chimera. The strain carrying the empty vector, pBQ200, did not form complex colonies (Fig. 5B). In good agreement with a previous report (36), overexpression of wild-type RNase Y resulted in the formation of complex colonies (Fig. 5B). The complexity of the colonies was even more pronounced in the presence of the EzrA-RNase Y chimera (Fig. 5B). In conclusion, the transmembrane domain of the divisome protein EzrA can replace the cognate transmembrane domain of RNase Y, and the functional, chimeric protein seems to accumulate to higher levels than the wild-type protein.
Contributions of the domains to dimerization of RNase Y.
The ability of RNase Y to form both dimers and tetramers led us to investigate the contributions of the individual domains to oligomerization. For this purpose, the N- and C-terminal domains of Bordetella pertussis adenylate cyclase were fused to the domains of RNase Y, and the restoration of adenylate cyclase activity upon interaction of the fusion partners was investigated. As shown in Fig. 6, and consistent with the sedimentation velocity data (Fig. 3), full-length RNase Y showed a strong self-interaction. Moreover, all individual domains, with the exception of the KH domain, were capable of interacting with the full-length protein, suggesting that all the domains contribute to dimerization (Fig. 6). The analysis of the interaction of the isolated domains with each other revealed a different picture: only the transmembrane and the coiled-coil domains showed self-interactions, suggesting that the KH, the HD, and the C-terminal domains make only minor contributions to the oligomerization (Fig. 6). The observation that the coiled-coil domains showed the strongest self-association is in good agreement with the well-established role of these domains for the self-interaction of many proteins (4).
Fig. 6.
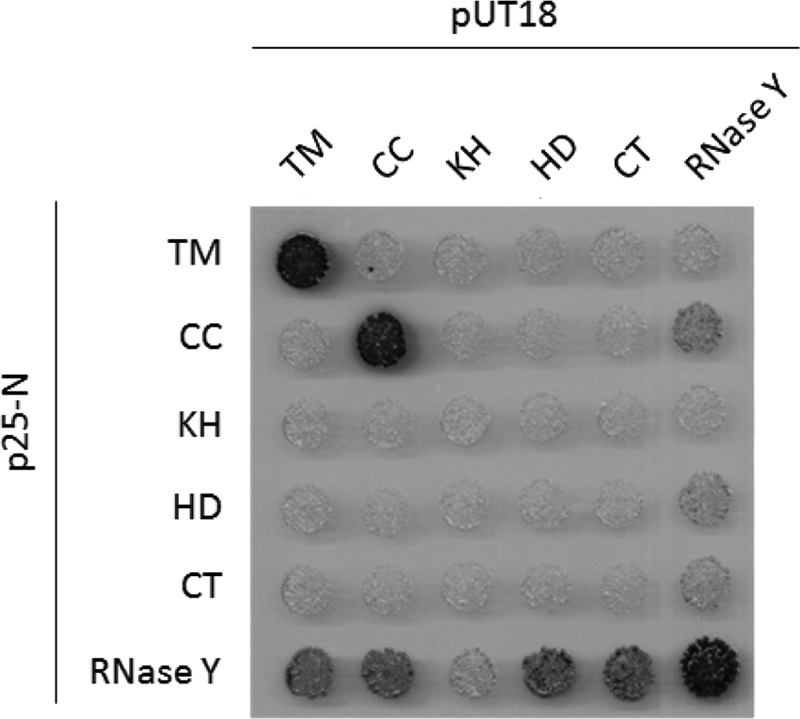
Bacterial two-hybrid analysis to study the interactions among the single domains of RNase Y and with full-length protein. All genes were cloned in the plasmids pUT18, pUT18C, p25-N, and pKT25. Plasmids pUT18 and pUT18C allow the expression of the selected truncations fused to either the N or C terminus of the T18 domain of the B. pertussis adenylate cyclase, respectively. Plasmids p25-N and pKT25 allow the expression of the selected enzymes fused to either the N or C terminus of the T25 domain of the adenylate cyclase, respectively. The E. coli transformants were incubated for 48 h at 30°C. The degradation of X-Gal (blue) indicates the presence of a functional adenylate cyclase owing to the interaction of the two proteins of interest. Abbreviations: TM, transmembrane domain; CC, coiled-coil/disordered domain; KH, KH domain; HD, HD domain; CT, C-terminal domain. For further details, see the legend to Fig. 1.
In vivo evidence for the interaction of RNase Y with the other components of the degradosome.
Previously, we have observed interactions between several RNases, the RNA helicase CshA, and the glycolytic enzymes enolase and phosphofructokinase in a bacterial two-hybrid assay and in affinity copurification experiments with glycolytic enzymes and CshA (11, 35). The binary interactions in the degradosome have also been the focus of recent in vitro biochemical and biophysical studies (J. Newman et al., unpublished results). The observation of multiple protein-protein complexes has led us to propose that these proteins form a complex that is the B. subtilis equivalent of the RNA degradosome, with the novel RNase Y being the central component. To provide evidence for the in vivo interaction of RNase Y with other members of the degradosome, we attempted to purify RNase Y together with its interaction partners. To facilitate detection of components of the degradosome, strains expressing RNase Y with a C-terminal Strep-tag under the control of its native promoter and the potential degradosome components carrying a C-terminal triple FLAG tag were constructed. A strain expressing the malic enzyme, YtsJ, fused to a FLAG tag was used as a control. As shown in Fig. 7, all potential degradosome proteins as well as YtsJ were expressed under the conditions employed to isolate the protein complexes. With Strep-tag-carrying RNase Y as the bait, the polynucleotide phosphorylase PNPase, the RNases J1 and J2, the DEAD-box RNA helicase CshA, and the glycolytic enzymes Eno and PfkA were copurified after cross-linking. In contrast, a very faint signal was observed for YtsJ, suggesting that the degradosome proteins do indeed form a complex with RNase Y, whereas YtsJ does not (Fig. 7). These findings are in excellent agreement with previous results of bacterial two-hybrid assays that suggested binary protein-protein interactions of RNase Y with all components of the degradosome (11, 35), with the common association of the degradosome proteins to the cytoplasmic membrane (21, 35), and with a recent study on an RNase Y homologue from Streptococcus pyogenes, which was demonstrated to interact with enolase (29).
Fig. 7.

Confirmation of the in vivo interaction of RNase Y with the degradosome components. RNase Y fused to a C-terminal Strep-tag and expressed at its native locus was purified with its interaction partners from various B. subtilis strains (indicated in the first column) carrying triple FLAG tags attached to the putative interaction partners. All strains were grown in LB medium. Twenty-five microliters of the first fraction eluting from each purification was loaded onto a 12.5% SDS-polyacrylamide gel. After electrophoresis and blotting onto a polyvinylidene difluoride membrane, interaction partners were detected by FLAG tag polyclonal antibodies. YtsJ was used as a negative control.
Contributions of RNase Y domains to interactions with partners in the degradosome.
Our data are consistent with RNase Y forming a mixture of mostly dimers, but also some tetramers (Fig. 3), and suggest that this self-association is driven primarily by the transmembrane and coiled-coil domains (Fig. 6). We have extended the domain analysis of RNase Y to address the contribution of these domains for the interaction with several other proteins that are found in a protein complex called the RNA degradosome (11, 35). The truncated variants of RNase Y described above were used in an in vivo complementation experiment. In good agreement with previous reports (35), full-length RNase Y interacted with all the different members of the degradosome (Fig. 8). In contrast, truncations of RNase Y at the N or C terminus resulted in a loss of interaction with the other degradosome proteins. To exclude the possibility that a lack of interaction reflected poor expression or stability of the truncated variants, we tested the interaction of the truncated versions with full-length RNase Y. All truncated variants showed a strong interaction with RNase Y, suggesting that the truncated proteins retain the ability to interact (Fig. 8). Thus, the loss of interaction seen upon deletion of domains from either the N or the C terminus of RNase Y suggests that these domains are essential for the interactions with other degradosome partners but not for dimerization of RNase Y. In contrast, and quite unexpectedly, the N-terminal transmembrane region is required in vivo for all the properties of RNase Y tested, for cell viability, and for the interaction with the other components in the degradosome.
Fig. 8.
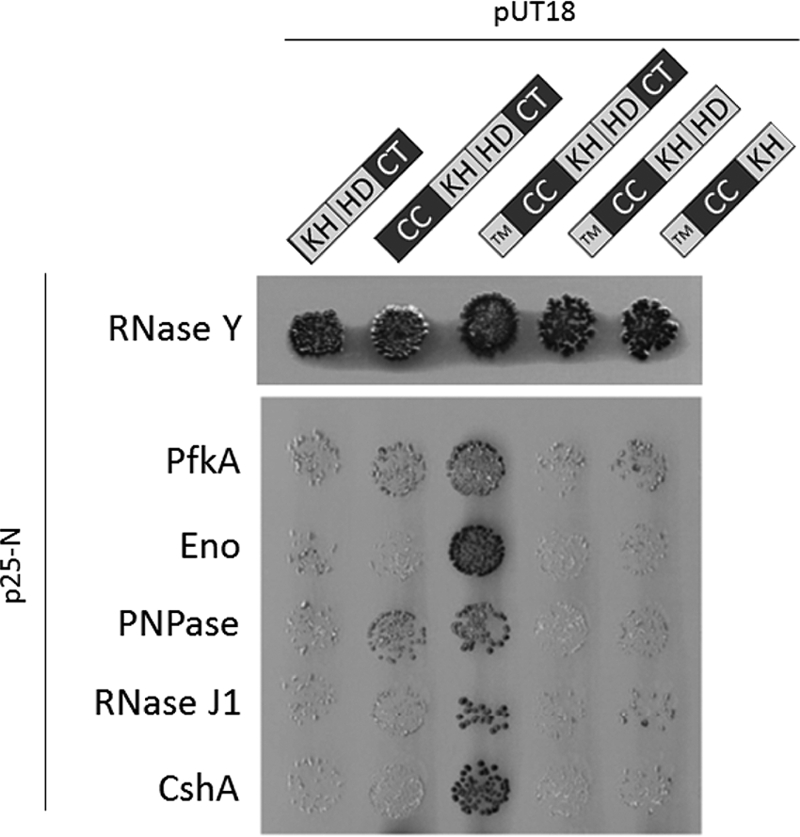
Bacterial two-hybrid analysis to study the interactions among full-length RNase Y and its fragments with the other components of the degradosome. The gene encoding RNase Y and its fragments were cloned into p25N, whereas the coding sequences of the other degradosome components, PfkA (phosphofructokinase), Eno (enolase), PNPase (polynucleotide phosphorylase), RNase J1, and CshA, were cloned into pUT18. For further details of the B2H analysis, see the legend to Fig. 6.
DISCUSSION
Although it was discovered only 2 years ago (11, 58), the accumulating evidence suggests that RNase Y is a key player in RNA processing and degradation in B. subtilis and most likely also in other Gram-positive bacteria. This is supported by the notion that RNase Y shares a growing list of similarities with the RNase E from E. coli and other Gram-negative bacteria, even though the two proteins do not share any sequence similarity. Moreover, RNase Y is found to occur in more bacterial phyla that lack RNase E than any other endoribonuclease (28). The list of similarities between RNase Y and RNase E is extended by the results reported in this study. Both proteins are endoribonucleases with a preference for 5′ monophosphorylated mRNA ends (42, 58), both have a major impact on the global RNA stability (2, 58), both are attached to the membrane (25, 31, 35, 66), and both interact with other proteins involved in RNA processing in a protein complex called the RNA degradosome (7, 11, 55). This work provides evidence for the importance of the membrane localization of RNase Y and for the presence of disordered regions of RNase Y. Both of these features are shared with RNase E, although both the way in which membrane localization is achieved and the distribution of ordered and disordered domains are distinct between the two enzymes.
The prediction by bioinformatics and the observation by direct experimentation that RNase Y contains disordered regions will fundamentally alter our understanding of this important enzyme. Disordered regions within proteins have been found to play key roles in interaction networks, as they allow rapid evolution of protein recognition microdomains without the structural and thermodynamic constraints of protein folding and stability. They also allow the evolution of high-specificity, low-affinity interactions where the free energy of the interaction is used for protein folding (14); it is thus likely that RNase Y adopts an ordered structure upon binding to its interaction partners. This disorder-to-order transition is not uncommon in proteins (17). For instance, a C-terminal region of the HPr kinase/phosphorylase adopts an ordered structure only upon binding its target protein, phosphorylated HPr (16). The discovery of unstructured regions of RNase Y in the B. subtilis degradosome extends the already extensive functional similarities with RNase E in the E. coli degradosome. In RNase E, the unstructured C terminus is used as a scaffold to facilitate and to coordinate the organization and activities of the enzymes in the E. coli degradosome (27); we suggest an equivalent role for the unstructured regions of RNase Y.
The results of our domain analysis suggest that the coiled-coil domain may be important for oligomerization. The involvement of coiled-coil domains in protein self-interaction is very common (19, 52), and oligomerization of RNases is widespread (5, 59). In the case of RNase Y, self-association may be of special importance for the assembly of the RNA degradosome and for contacting other degradosome components. The formation of higher oligomeric structures extends significantly the potential interaction interface for binding other proteins.
Previous reports of the interactions of RNase Y with the members of the RNA degradosome of B. subtilis were mainly based on two-hybrid studies in a heterologous host (11, 35). To verify that RNase Y physically interacts with these proteins inside the cell, we froze the in vivo physical interactions by chemical cross-linking. This analysis confirmed that RNase Y does indeed interact with RNase J1, with polynucleotide phosphorylase, with the DEAD-box RNA helicase CshA, and with the two glycolytic enzymes enolase and phosphofructokinase (Fig. 7). While the formation of a complex of different endo- and exoribonucleases with an RNA helicase might be intuitive, it is less so for the involvement of the glycolytic enzymes. However, metabolic enzymes are commonly found in RNA-degrading complexes, as enolase is also a component of the E. coli RNA degradosome (48) and the degradosome of Caulobacter crescentus contains aconitase, an RNA-binding enzyme of the tricarboxylic acid cycle (22). These seemingly unusual connections imply that RNA degradation might be controlled by fluctuations in metabolite concentrations. This hypothesis is supported by the recent finding that the activity of E. coli polynucleotide phosphorylase, a component of the RNA degradosome, is controlled by citrate, the first intermediate of the tricarboxylic acid cycle (51). Recently, association of RNase Y with enolase was also reported for Streptococcus pyogenes and Staphylococcus aureus (29, 55).
The mandatory requirement for RNase Y to have a functional transmembrane domain and therefore be membrane localized is intriguing and may reflect that RNA turnover is somehow compartmentalized in the cell. Even though RNase Y is catalytically active without its membrane segment in vitro, proper cellular localization is essential in vivo. That membrane localization of RNases is physiologically important is an emerging area in RNA biology (see reference 15 for a review). Early studies using membrane fractionation experiments in E. coli suggested that RNase E, RNase III, and RNase P were associated with the membrane (47). Membrane localization of RNA-degrading protein complexes was also recently observed in other bacteria and even in archaea (54). Even though RNase E lacks an obvious transmembrane domain like that found in RNase Y, it has been demonstrated that RNase E contains an amphipathic α helix which binds to the phospholipid bilayer (31). The importance of the membrane localization of RNase E was demonstrated by the deletion of the membrane binding segment, which severely compromises growth. This phenotype is most likely not due to impaired catalytic activity but is due to impaired cellular localization. Similarly, RNase Y still exhibits catalytic activity when the transmembrane domain is deleted (58), but the consequences in vivo are very drastic, as a strain depending only on a copy of RNase Y without the transmembrane segment is not viable. Again, the mandatory need for RNase Y to be membrane located in Bacillus subtilis provides additional evidence for the importance of protein compartmentalization in bacteria. The use of green fluorescent protein (GFP) technology in B. subtilis has revealed that the RNA polymerase is primarily associated with the nucleoid, whereas ribosomes occupy the cytoplasmic space outside the nucleoid (38). Therefore, bacteria may exhibit a certain level of spatial organization to separate transcription, translation, and perhaps also RNA degradation. Consistent with this hypothesis, the GFP tagging of mRNAs in E. coli and C. crescentus showed that even mRNAs are not distributed equally throughout the cell (49, 50). Therefore, despite lacking organelle-like structures, bacteria may organize spatially the synthesis, translation, and degradation of mRNAs.
The work presented here indicates that RNase Y is the central component of the B. subtilis RNA degradosome. The hypothesis that disorder-order transitions may play a role in its interactions with its partner proteins paves the way for the structural analysis of this key component of the RNA-degrading machinery in Gram-positive bacteria.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jan-Maarten van Dijl for raising antibodies against RNase Y.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB860) to J.S. and from the United Kingdom BBSRC to R.J.L.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
Published ahead of print on 29 July 2011.
REFERENCES
- 1. Arraiano C. M., et al. 2010. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol. Rev. 34:883–923 [DOI] [PubMed] [Google Scholar]
- 2. Bernstein J. A., Lin P. H., Cohen S. N., Lin-Chao S. 2004. Global analysis of Escherichia coli RNA degradosome function using DNA microarrays. Proc. Natl. Acad. Sci. U. S. A. 101:2758–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reference deleted.
- 4. Burkhard P., Stetefeld J., Strelkov S. V. 2001. Coiled coils: a highly versatile protein folding motif. Trends Cell Biol. 11:82–88 [DOI] [PubMed] [Google Scholar]
- 5. Callaghan A. J., et al. 2003. Quaternary structure and catalytic activity of the Escherichia coli RNase E amino-terminal catalytic domain. Biochemistry 42:13848–13855 [DOI] [PubMed] [Google Scholar]
- 6. Callaghan A. J., et al. 2005. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature 437:1187–1191 [DOI] [PubMed] [Google Scholar]
- 7. Carpousis A. J. 2007. The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu. Rev. Microbiol. 61:71–87 [DOI] [PubMed] [Google Scholar]
- 8. Carpousis A. J., Luisi B. F., McDowall K. J. 2009. Endonucleolytic initiation of mRNA decay in Escherichia coli. Prog. Mol. Biol. Transl. Sci. 85:91–135 [DOI] [PubMed] [Google Scholar]
- 9. Celesnik H., Deana A., Belasco J. G. 2007. Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol. Cell 27:79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Claessen D., et al. 2008. Control of the cell elongation-division cycle by shuttling of PBP1 protein in Bacillus subtilis. Mol. Microbiol. 68:1029–1046 [DOI] [PubMed] [Google Scholar]
- 11. Commichau F. M., et al. 2009. Novel activities of glycolytic enzymes in Bacillus subtilis: interactions with essential proteins involved in mRNA processing. Mol. Cell. Proteomics 8:1350–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Condon C. 2003. RNA processing and degradation in Bacillus subtilis. Microbiol. Mol. Biol. Rev. 67:157–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dunker A. K., et al. 2001. Intrinsically disordered protein. J. Mol. Graph. Model. 19:26–59 [DOI] [PubMed] [Google Scholar]
- 14. Dyson H. J., Wright P. E. 2005. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6:197–208 [DOI] [PubMed] [Google Scholar]
- 15. Evguenieva-Hackenberg E., Roppelt V., Lassek C., Klug G. 2011. Subcellular localization of RNA degrading proteins and protein complexes in prokaryotes. RNA Biol. 8:49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fieulaine S., et al. 2002. X-ray structure of a bifunctional protein kinase in complex with its protein substrate HPr. Proc. Natl. Acad. Sci. U. S. A. 99:13437–13441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fong J. H., Shoemaker B. A., Garbuzynskiy S. O., Lobanov M. Y., Pancheko A. R. 2009. Intrinsic disorder in protein interactions: insights from a comprehensive structural analysis. PLoS Comput. Biol. 5:e1000316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reference deleted.
- 19. Grigoryan G., Keating A. E. 2008. Structural specificity in coiled-coil interactions. Curr. Opin. Struct. Biol. 18:477–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gunka K. 2011. Der Einfluss der Glutamatdehydrogenasen auf die Verknüpfung des Kohlenstoff- und Stickstoffstoffwechsels in Bacillus subtilis. Ph.D. thesis University of Göttingen, Göttingen, Germany [Google Scholar]
- 21. Hahne H., Wolff S., Hecker M., Becher D. 2008. From complementarity to comprehensiveness—targeting the membrane proteome of growing Bacillus subtilis by divergent approaches. Proteomics 8:4123–4136 [DOI] [PubMed] [Google Scholar]
- 22. Hardwick S. W., Chan V. S. Y., Broadhurst R. W., Luisi B. 2010. An RNA degradosome assembly in Caulobacter crescentus. Nucleic Acids Res. 39:1449–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Herzberg C., et al. 2007. SPINE: a method for the rapid detection and analysis of protein-protein interactions in vivo. Proteomics 7:4032–4035 [DOI] [PubMed] [Google Scholar]
- 24. Homuth G., Mogk A., Schumann W. 1999. Post-transcriptional regulation of the Bacillus subtilis dnaK operon. Mol. Microbiol. 32:1183–1197 [DOI] [PubMed] [Google Scholar]
- 25. Hunt A., Rawlins J. P., Thomaides H. B., Errington J. 2006. Functional analysis of 11 putative essential genes in Bacillus subtilis. Microbiology 152:2895–2907 [DOI] [PubMed] [Google Scholar]
- 26. Ishida T., Kinoshita K. 2008. Prediction of disordered regions in proteins based on the meta approach. Bioinformatics 24:1344–1348 [DOI] [PubMed] [Google Scholar]
- 27. Kaberdin V. R., et al. 1998. The endonucleolytic N-terminal half of Escherichia coli RNase E is evolutionarily conserved in Synechocystis sp. and other bacteria but not the C-terminal half, which is sufficient for degradosome assembly. Proc. Natl. Acad. Sci. U. S. A. 95:11637–11642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaberdin V. R., Singh D., Lin-Chao S. 2011. Composition and conservation of the mRNA-degrading machinery in bacteria. J. Biomed. Sci. 18:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kang S. O., Caparon M. G., Cho K. H. 2010. Virulence gene regulation by CvfA, a putative RNase: the CvfA-enolase complex in Streptococcus pyogenes links nutritional stress, growth-phase control, and virulence gene expression. Infect. Immun. 78:2754–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karimova G., Pidoux J., Ullmann A., Ladant D. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. U. S. A. 95:5752–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khemici V., Poljak L., Luisi B. F., Carpousis A. J. 2008. The RNase E of Escherichia coli is a membrane binding protein. Mol. Microbiol. 70:799–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kunst F., Rapoport G. 1995. Salt stress is an environmental signal affecting degradative enzyme synthesis in Bacillus subtilis. J. Bacteriol. 177:2403–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kushner S. R. 2002. mRNA decay in Escherichia coli comes of age. J. Bacteriol. 184:4658–4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Laue T., Shah B., Ridgeway T., Pelletier S. 1992. Computer-aided interpretation of analytical sedimentation data for proteins. In Harding S., Rowe A., Horton J. (ed.). Royal Society of Chemistry, Cambridge, United Kingdom [Google Scholar]
- 35. Lehnik-Habrink M., et al. 2010. The RNA degradosome in Bacillus subtilis: identification of CshA as the major RNA helicase in the multi-protein complex. Mol. Microbiol. 77:958–971 [DOI] [PubMed] [Google Scholar]
- 36. Lehnik-Habrink M., et al. RNA processing in Bacillus subtilis: identification of targets of the essential RNase Y. Mol. Microbiol., in press doi:10.1111/j.1365-2011.07777.x [DOI] [PubMed] [Google Scholar]
- 37. Levin P. A., Kurtser I. G., Grossman A. D. 1999. Identification and characterization of a negative regulator of FtsZ ring formation in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 96:9642–9647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lewis P. J., Thaker S. D., Errington J. 2000. Compartmentalization of transcription and translation in Bacillus subtilis. EMBO J. 19:710–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liou G. G., Jane W. N., Cohen S. N., Lin N. S., Lin-Chao S. 2001. RNA degradosomes exist in vivo in Escherichia coli as multicomponent complexes associated with the cytoplasmic membrane via the N-terminal region of RNase E. Proc. Natl. Acad. Sci. U. S. A. 98:63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ludwig H., et al. 2001. Transcription of glycolytic genes and operons in Bacillus subtilis: evidence for the presence of multiple levels of control of the gapA operon. Mol. Microbiol. 41:409–422 [DOI] [PubMed] [Google Scholar]
- 41. Lupas A., van Dyke M., Stock J. 1991. Predicting coiled coils from protein sequences. Science 252:1162–1164 [DOI] [PubMed] [Google Scholar]
- 42. Mackie G. A. 1998. RNase E is a 5′-end-dependent endonuclease. Nature 395:720–723 [DOI] [PubMed] [Google Scholar]
- 43. Mäder U., Zig L., Kretschmer J., Homuth G., Putzer H. 2008. mRNA processing by RNases J1 and J2 affects Bacillus subtilis gene expression on a global scale. Mol. Microbiol. 70:183–196 [DOI] [PubMed] [Google Scholar]
- 44. Martin-Verstraete I., Débarbouillé M., Klier A., Rapoport G. 1994. Interaction of wild-type truncated LevR of Bacillus subtilis with the upstream activating sequence of the levanase operon. J. Mol. Biol. 241:178–192 [DOI] [PubMed] [Google Scholar]
- 45. Meinken C., Blencke H. M., Ludwig H., Stülke J. 2003. Expression of the glycolytic gapA operon in Bacillus subtilis: differential syntheses of proteins encoded by the operon. Microbiology 149:751–761 [DOI] [PubMed] [Google Scholar]
- 46. Meyer F. M., et al. 2011. Physical interactions between tricarboxylic acid cycle enzymes in Bacillus subtilis: evidence for a metabolon. Metab. Eng. 13:18–27 [DOI] [PubMed] [Google Scholar]
- 47. Miczak A., Srivastava R. A., Apirion D. 1991. Location of the RNA-processing enzymes RNase III, RNase E and RNase P in the Escherichia coli cell. Mol. Microbiol. 5:1801–1810 [DOI] [PubMed] [Google Scholar]
- 48. Miczak A., Kaberdin V. R., Wei C. L., Lin-Chao S. 1996. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc. Natl. Acad. Sci. U. S. A. 93:3865–3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Montero Llopis P., et al. 2010. Spatial organization of the flow of genetic information in bacteria. Nature 466:77–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nevo-Dinur K., Nussbaum-Schochat A., Ben-Yehuda S., Amster-Choder O. 2011. Translation-independent localization of mRNA in E. coli. Science 331:1081–1084 [DOI] [PubMed] [Google Scholar]
- 51. Nurmohamed S., et al. 2011. Polynucleotide phosphorylase activity may be modulated by metabolites in Escherichia coli. J. Biol. Chem. 286:14315–14323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rigden M. D., et al. 2008. Identification of the coiled-coil domains of Enterococcus faecalis DivIVA that mediate oligomerization and their importance for biological function. J. Biochem. 144:63–76 [DOI] [PubMed] [Google Scholar]
- 53. Reference deleted.
- 54. Roppelt V., et al. 2010. The archaeal exosome localizes to the membrane. FEBS Lett. 584:2791–2795 [DOI] [PubMed] [Google Scholar]
- 55. Roux C. M., DeMuth J. P., Dunman P. M. 2011. Characterization of components of the Staphylococcus aureus mRNA degradosome holoenzyme-like complex. J. Bacteriol. 193:5520–5526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sambrook J., Fritsch E. F., Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 57. Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78:1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shahbabian K., Jamalli A., Zig L., Putzer H. 2009. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 28:3523–3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shi Z., Yang W. Z., Lin-Chao S., Chak K. F., Yuan H. S. 2008. Crystal structure of Escherichia coli PNPase: central channel residues are involved in processive RNA degradation. RNA 14:2361–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tjalsma H., et al. 2004. Proteomics of protein secretion by Bacillus subtilis: separating the “secrets” of the secretome. Microbiol. Mol. Biol. Rev. 68:207–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tjalsma H., van Dijl J. M. 2005. Proteomics-based consensus prediction of protein retention in a bacterial membrane. Proteomics 5:4472–4482 [DOI] [PubMed] [Google Scholar]
- 62. Uversky V. N., Gillespie J. R., Fink A. L. 2000. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41:415–427 [DOI] [PubMed] [Google Scholar]
- 63. Wach A. 1996. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12:259–265 [DOI] [PubMed] [Google Scholar]
- 64. Xue B., DunBrack R. L., Williams R. W., Dunker A. K., Uversky V. N. 2010. PONDR-Fit: a meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 1804:996–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yao S., Bechhofer D. H. 2010. Initiation of decay of Bacillus subtilis rpsO mRNA by endoribonuclease RNase Y. J. Bacteriol. 192:3279–3286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zweers J. C., Wiegert T., van Dijl J. M. 2009. Stress-responsive systems set specific limits to the overproduction of membrane proteins in Bacillus subtilis. Appl. Environ. Microbiol. 75:7356–7364 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.