

Abstract
Fimbrial ushers are the largest β-barrel outer membrane proteins (OMPs) known to date, which function in the polymerization of fimbriae and their translocation to the bacterial surface. Folding and assembly of these complex OMPs are not characterized. Here, we investigate the role of periplasmic chaperones (SurA, Skp, DegP, and FkpA) and individual components of the β-barrel assembly machinery (BAM) complex (BamA, BamB, BamC, and BamE) in the folding of the Escherichia coli FimD usher. The FimD level is dramatically reduced (∼30-fold) in a surA null mutant, but a strong cell envelope stress is constitutively activated with upregulation of DegP (∼10-fold). To demonstrate a direct role of SurA, FimD folding was analyzed in a conditional surA mutant in which SurA expression was controlled. In this strain, FimD is depleted from bacteria in parallel to SurA without significant upregulation of DegP. Interestingly, the dependency on SurA is higher for FimD than for other OMPs. We also demonstrate that a functional BAM complex is needed for folding of FimD. In addition, FimD levels were strongly reduced (∼5-fold) in a mutant lacking the accessory lipoprotein BamB. The critical role of BamB for FimD folding was confirmed by complementation and BamB depletion experiments. Similar to SurA dependency, FimD showed a stronger dependency on BamB than OMPs. On the other hand, folding of FimD was only marginally affected in bamC and bamE mutants. Collectively, our results indicate that FimD usher follows the SurA-BamB pathway for its assembly. The preferential use of this pathway for the folding of OMPs with large β-barrels is discussed.
INTRODUCTION
Gram-negative bacteria display on their surfaces adhesive multimeric protein fibers known as pili, fimbriae, or fibrillae that mediate attachment to host cells and tissues for colonization and infection (12, 24). The assembly of a large family of these appendages depends on the conserved chaperone-usher (CU) pathway (36, 49). The pilin subunits that form these fibers initially cross the cytoplasmic membrane through the Sec translocon to reach the periplasmic space, where they bind to their cognate chaperones, forming binary complexes. These complexes are specifically targeted to the usher, a ∼90-kDa integral outer membrane protein (OMP), that catalyzes the polymerization of pilin subunits in an ordered manner and secretes the resulting protein fiber to the bacterial surface (32, 34, 37). Type 1 (fim) and P (pap) fimbriae from uropathogenic Escherichia coli (UPEC) strains have been established as paradigms of the CU pathway, and their respective chaperones and ushers are studied as models of these types of proteins. Biochemical and structural studies of the usher proteins of type 1 and P fimbriae (named FimD and PapC, respectively) indicate that these proteins may form dimers in the OM and contain the largest β-barrel known to date in OMPs, with 24 antiparallel β-strands (18, 34, 37). In addition to the large β-barrel domain, FimD and PapC ushers contain several globular domains, including periplasmic N- and C-terminal domains that interact with the chaperone subunit complexes and participate in the catalysis of fiber formation and a central globular domain, also known as “the plug,” which is localized inside the large β-barrel and is displaced during secretion of the polymerized fimbrial subunits (11, 16, 31, 34).
Despite the large quantity of information regarding the complex structure of fimbrial ushers, little is known about the steps of folding and OM insertion and the specific periplasmic chaperones and OM assembly factors involved in these processes. Studies with other integral β-barrel OMPs from E. coli (e.g., OmpA, LamB, OmpF, and OmpC) indicate that upon their translocation through the inner membrane (IM), the nascent unfolded OMPs interact with periplasmic chaperones (e.g., SurA, DegP, and Skp) that assist their folding and/or prevent their aggregation and deliver them to a large protein complex in the OM, the BAM (from β-barrel assembly machinery) complex, which is the assembly site for OM insertion and folding of β-barrel OMPs (4, 15, 25).
The periplasmic chaperones SurA, DegP, and Skp form two distinct routes to transport nascent OMPs to the OM (i.e., the SurA pathway and DegP/Skp pathway), as supported by the synthetic lethality of E. coli mutants inactivating components of both pathways (surA degP or surA skp) (38). E. coli single null mutants in surA, degP, or skp are viable but show some OM permeability defects (e.g., higher sensitivity to detergents and/or antibiotics) and decreased amounts of integral β-barrel OMPs in the OM (6, 27, 39, 45). It has been proposed that SurA may be responsible for the assembly of most β-barrel OMPs in E. coli, whereas the DegP/Skp pathway would be a “rescue pathway” for OMPs that fall off the SurA pathway under stress conditions (44). In addition, SurA has been demonstrated to directly interact with the central component of the BAM complex (BamA) (2, 44), but, to our knowledge, there is no direct evidence that DegP or Skp interacts with the BAM complex. However, a global proteomic analysis of an E. coli surA mutant strain found that only a subset of OMPs were affected by the lack of SurA (47), suggesting that the DegP/Skp pathway also participates in the steady-state assembly of OMPs in E. coli (25).
The BAM complex from E. coli is constituted by a conserved β-barrel OMP, BamA (previously YaeT/Omp85), and four interacting lipoproteins anchored to the periplasmic side of the OM, named BamB (YfgL), BamC (NlpB), BamD (YfiO), and BamE (SmpA) (15, 25). BamA and BamD are essential proteins required for the function of the BAM complex, and loss of any of them completely blocks OM insertion of β-barrel OMPs, leading to E. coli cell death (10, 52, 53). Single null mutants in bamB, bamC, or bamE are viable but exhibit OM assembly defects (5, 33, 43). An altered pattern of OMPs is observed in mutants lacking BamB, with an ∼1.4- to 3-fold reduction of integral OMPs, like OmpA, OmpF, and LamB, but not in others, like OmpC (5). Mutants lacking only BamE showed some folding defects in certain OMPs (e.g., LamB) (43), and no significant alterations in the pattern of OMPs were observed in bamC mutants (33). Nonetheless, the importance of these accessory lipoproteins for OM biogenesis in E. coli is also manifested by the synthetic lethality of conditional bamB bamE double mutants and the severe phenotypes observed in bamB bamC and bamC bamE double mutants (5, 33). According to their nonessential function, these lipoproteins are considered accessory components that enhance the efficiency of the BAM complex. Although their actual role in the assembly of OMPs is unclear, it has been hypothesized that they may participate at different stages of the membrane insertion process as chaperones and/or docking sites for specific periplasmic chaperone-OMP complexes (25). In this sense, certain similarities between the phenotypes of surA and bamB mutants and the synthetic lethality observed in the conditional bamB degP double mutant, similar to the synthetic lethality of the surA degP double mutant, have suggested that SurA and BamB may constitute a distinct pathway for delivery of OMPs to the BAM complex (2, 5, 33).
Previous work has shown that E. coli K-12 and UPEC strains lacking the chaperone and peptidyl prolyl isomerase (PPIase) SurA, but not other periplasmic PPIases such as FkpA, produce low levels of type 1 and P fimbriae and have low levels of FimD and PapC in the OM, suggesting that the chaperone activity of SurA is involved in the biogenesis of fimbrial ushers (21, 51). However, the constitutive absence of SurA in E. coli strongly affects the overall OM composition, reducing the levels of major OMPs (e.g., OmpA, OmpC/OmpF, and LamB). This reduction is due, at least in part, to the activation of envelope stress responses (e.g., σE and Cpx signaling) in the surA null mutant, which upregulate the levels of proteases (e.g., the protease activity of DegP) and small RNAs that inhibit OMPs biosynthesis (33, 43, 47).
In this work we investigate the role of the individual components of the BAM complex and of the SurA and DegP/Skp pathways for the folding and insertion of the fimbrial usher FimD in E. coli. In contrast to previous studies, folding of FimD is evaluated without the induction of envelope stress responses by employing native expression levels from the single-copy chromosomal fimD gene and conditional mutant strains in surA. Our results demonstrate that the FimD usher, the largest β-barrel described so far, follows the SurA-BamB pathway for its insertion in the OM and that the participation of at least one of the other accessory proteins of the BAM complex (i.e., BamC or BamE) is required for efficient OM insertion of FimD.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
E. coli strains used for FimD analysis are listed in Table 1. The E. coli DH10B-T1R strain [F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara-leu)7697 galU galK λ− rpsL nupG tonA] (Invitrogen) was used for cloning and plasmid preparation. The E. coli BL21(DE3) strain omp8 [F− ompT hsdSB(rB− mB−) gal dcm (DE3) ΔlamB ompF::Tn5 ΔompA ΔompC] (35) was used for FimD purification. For expression of endogenous FimD in E. coli strains, cultures were grown under reported conditions for optimal production of type 1 fimbriae (29) in liquid brain heart infusion (BHI) medium using static growth at 37°C. Preinoculum cultures were grown for 16 h, diluted in fresh medium to a final optical density at 600 nm (OD600) of 0.05, and grown under identical conditions until mid-exponential growth phase for harvesting bacteria. When mentioned, growth medium was supplemented with d-glucose (0.4%, wt/vol), l-arabinose (0.4%, wt/vol), and appropriate antibiotics. Antibiotics were used at the following concentrations: ampicillin (Ap), 100 μg/ml; chloramphenicol (Cm), 25 μg/ml; kanamycin (Km), 50 μg/ml; zeocin (Zeo), 40 μg/ml. Zeo-resistant bacteria were selected in LB-agar medium with a low salt concentration (5 g/liter of NaCl). For the BamB complementation assay, bacteria carrying plasmid pBamB or pBAD30 were grown overnight at 37°C statically in liquid BHI medium with Ap and l-arabinose (0.4%, wt/vol). These cultures were diluted in fresh medium to a final OD600 of 0.05 in the presence of l-arabinose (0.4%, wt/vol) or d-glucose (0.4%, wt/vol) and grown until bacteria were harvested at the times indicated in the figures.
Table 1.
E. coli strains
| Strain | Description | Reference or source |
|---|---|---|
| UT5600 | K-12 F− λ− Δ(ompT-fepC)266 | 13 |
| UT5601 | UT5600 ΔaraC | 3 |
| UTfimD | UT5600 ΔfimD | This study |
| UTfkpA | UT5600 ΔfkpA::kan | 46 |
| UTskp | UT5600 Δskp::kan | 3 |
| UTsurA | UT5600 ΔsurA::kan | 3 |
| UTdegP | UT5600 ΔaraC ΔdegP::kan | 3 |
| UTPBAD::surA | UT5600 ΔaraC::kan ZeoraraC PBAD::surA | This study |
| UTdegP-PBAD::surA | UT5600 ΔaraC ΔdegP::kan ZeoraraC PBAD::surA | 3 |
| UTPBAD::bamA | UT5600 ΔaraC::kan ZeoraraC PBAD::bamA | 3 |
| UTbamB | UT5600 ΔaraC ΔbamB::kan | This study |
| UTbamC | UT5600 ΔaraC ΔbamC::kan | This study |
| UTbamE | UT5600 ΔaraC ΔbamE::kan | This study |
| UTbamC bamB | UT5600 ΔaraC ΔbamC ΔbamB::kan | This study |
| UTbamC bamE | UT5600 ΔaraC ΔbamC ΔbamE::kan | This study |
Bacterial strain constructions.
The mutant E. coli strains described in this work (Table 1) were obtained by one-step inactivation of chromosomal genes with PCR products obtained after amplification of the kanamycin (Kmr) resistance gene cassette from plasmid pKD4 (9) using specific oligonucleotides flanking the 5′ and 3′ ends of the coding sequence of each targeted gene: for the bamB null mutant, BamB_KO.FR (5′-tga aaa tta ata att tgt cca tct gag agg gac ccg ATG GTG TAG GCT GGA GCT GCT TC-3′) and BamB_KO.RE (5′-gtc cag gag ccg ttt tca aag tga acg aca gag acg aTT ACA TAT GAA TAT CCT CCT TAG T-3′); for the bamC null mutant, BamC_KO.FR (5′-agc atg ccg gtt tgc tgt aaa gtt tag gga gat ttg ATG GTG TAG GCT GGA GCT GCT TC-3′) and BamC_KO.RE (5′-tca gaa aaa agg gcc gga tga ttc cag ccc tgt att tTT ACA TAT GAA TAT CCT CCT TAG T-3′); for the bamE null mutant, BamE_KO.FR (5′-cac gta ctg ctc ggg ccc gaa aag gaa tca aat cac tAT GGT GTA GGC TGG AGC TGC TTC-3′) and BamE_KO.RE (5′-agc acc ttt ttt aac gtc ttt gag agc aac ttt att aTT ACA TAT GAA TAT CCT CCT TAG T-3′). The lowercase letters correspond to the DNA sequence hybridizing to upstream and downstream coding sequences of the bam genes. The uppercase letters correspond to the sequence hybridizing to the Kmr resistance gene cassette. Colonies resistant to Km were selected, and the insertion of the Kmr gene into bamB, bamC, and bamE genes was confirmed by PCR using the oligonucleotides BamB_before (5′-ATAAGCAAGGTGCGCGTAGT-3′) and BamB_after (5′-GGGTGCGAGTTAGACGGTTA-3′) for ΔbamB, BamC_before (5′-AGACCGGAAAGCAAAAGGTT-3′) and BamC_after (5′-CAGGCAACGAGCAGAAAAAT-3′) for ΔbamC, and BamE_before (5′-TGACACCAATCACCGACAGT-3′) and BamE_after (5′-TCACCATACCTTTGCGATCA-3′) for ΔbamE. The ΔfimD null mutant of E. coli UT5600 was obtained with oligonucleotides described previously (30).
To obtain ΔbamB ΔbamC and ΔbamC ΔbamE double null mutants, the Kmr gene cassette in the ΔbamC mutant strain was deleted by transformation with pCP20, encoding FLP recombinase (9). The Km-sensitive ΔbamC mutant obtained, harboring plasmid pKD46 (9), was electroporated with PCR products that included the Kmr gene cassette and flanking DNA of bamB and bamE genes obtained using the primers described above. Km-resistant colonies were selected, and insertion of the Kmr gene into bamB, bamC, and bamE was confirmed by PCR using the oligonucleotides previously mentioned. The E. coli conditional mutant strain UTPBAD::surA (Table 1), in which the natural promoter of surA was replaced by the araC-PBAD promoter cassette, was also obtained by λred-driven homologous recombination after transformation with a PCR product containing the zeoRExBAD cassette (40) and flanking DNA homologous sequences of the surA promoter regions. The zeoRExBAD (zeor araC-PBAD) cassette was amplified from the chromosome of E. coli strain TG1zeoRExBAD (40) using specific oligonucleotides hybridizing with the upstream promoter region of the surA gene (5′ primer) and the beginning of their coding sequences (3′ primer), named ZEOBAD surA1 (5′-CGC AAG AGA TGC TGC GTT CGA ACA TTC TGC CGT ATC AAA ACA CTT TGT GAa gca atg ctt gca taa tgt gcc tgt c-3′) and ZEOBAD surA2 (5′-CTG GTA TTC GCG ATC ATG GCG ATA CCG AGA AGC AGC GTT TTC CAG TTC TTC CAT cgt ttc act cca tcc aaa aaa acg ggt-3′). The uppercase letters correspond to the sequence hybridizing to surA upstream or coding sequences. The lowercase letters correspond to the sequence hybridizing to the zeoRExBAD cassette. The PCR product was electroporated into E. coli UT5601 carrying pKD46. Zeor transformant colonies were selected in low-salt LB medium containing 0.4% (wt/vol) arabinose and Zeo (40 μg/ml). The insertion of the zeoRExBAD cassette in the promoter region of surA was tested by PCR with oligonucleotides Zeo1 (5′-CAC TGG TCA ACT TGG CCA TGG TTT AG-3′) and SurA2 (5′-CAT TAA TCC ATC AAC GTC GCT TTC CAG CAC-3′).
Plasmid constructions.
Cloning and DNA manipulation were done following standard methods. All DNA constructs were sequenced using an automated DNA sequencer (Perkin Elmer). Oligonucleotides were synthesized by Sigma Genosys. All the PCRs were done with Vent DNA polymerase (New England BioLabs). Plasmid pBamB is a derivative of pBAD30 (14), which contains bamB under the control of the PBAD promoter. The coding sequence of bamB was amplified with primers BamB-SacI (5′-ACT CGC GAG CTC CAT CTG AGA GGG ACC CGA TGC-3′) and BamB-XbaI (5′-CCG GGT TCT AGA GAG ACG ATT AAC GTG TAA TAG-3′) from the chromosomal DNA of E. coli UT5600. The resulting ∼1.3-kb DNA fragment was digested with XbaI and ScaI and was cloned into same sites of pBAD30.
FimD purification for immunization and production of polyclonal anti-FimD serum.
His6-tagged FimD (FimD-His6) was purified from the E. coli BL21(DE3) strain omp8 carrying the l-arabinose-inducible pETS7 (Cmr) plasmid (42). Bacteria were grown at 30°C in 10 ml of LB medium containing Cm with shaking at 250 rpm for 16 h, diluted into 500 ml of fresh medium to a final OD600 of 0.05 and grown under the same conditions until the OD600 reached 0.5. Then, the bacterial culture was induced with l-arabinose (0.1%, wt/vol) and grown for an additional 2 h. Afterwards, bacteria were harvested by centrifugation, resuspended in 50 ml of lysis buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl) and disrupted in a French press (SIM-Aminco Spectronic Instruments) at 1,000 lb/in2. Nonlysed bacteria were separated by centrifugation at 4,000 × g, and the supernatant that contained the bacterial envelope membranes was treated with 0.5% (wt/vol) Sarkosyl (Sigma) for 20 min at 22°C to solubilize the inner membrane. Bacteria outer membrane was recovered by centrifugation at 100,000 × g for 1 h and was resuspended in 4 ml of urea buffer (20 mM HEPES [pH 7.5], 0.1 M NaCl, 8 M urea). OM proteins were solubilized for 1 h at 4°C and loaded onto a Talon metal affinity (Clontech) column equilibrated with the same buffer. Subsequently, FimD-His6 was eluted in the same buffer containing 200 mM imidazole (pH 7.5). Fractions containing FimD-His6 were pooled and dialyzed against storage buffer (20 mM HEPES [pH 7.5], 0.1 M NaCl, and 10% [wt/vol] glycerol). Polyclonal antiserum against purified FimD-His6 was raised by subcutaneous inoculation of New Zealand White rabbits (Charles River Laboratories) with 50 μg of purified protein (in 0.5 ml of storage buffer) mixed with 0.5 ml of Freund's Complete Adjuvant (Sigma). The initial immunization was followed by four boosters of 50 μg of FimD-His6 (in 0.5 ml of storage buffer) mixed with 0.5 ml of Freund's Incomplete Adjuvant (Sigma) at days 20, 42, 60, and 75. The rabbits were bled, and sera were collected 15 days after the final boost. Serum specificity and titers were analyzed by Western blotting using purified FimD-His6 and whole-cells extracts of E. coli UT5600 and its isogenic ΔfimD strains (Table 1).
Protein extracts and Western blotting.
To prepare whole-cell protein extracts, samples (0.25 OD600 units) were collected from each culture at mid-exponential growth phase. Harvested bacteria after centrifugation were resuspended in 25 μl mixed with the same volume of 2× SDS sample buffer. SDS sample buffer (1×) contains 60 mM Tris-HCl, pH 6.8, 1% (wt/vol) SDS, 5% (vol/vol) glycerol, 0.005% (wt/vol) bromophenol blue, and 1% (vol/vol) 2-mercaptoethanol (2-ME). Controls of unfolded FimD (c+ on the figures) were boiled for 10 min in 1× urea-SDS sample buffer (60 mM Tris-HCl, pH 6.8, 2% [wt/vol] SDS, 4 M urea, 5 mM EDTA, 5% [vol/vol] glycerol, 0.005% [wt/vol] bromophenol blue, and 1% [vol/vol] 2-ME). All samples were sonicated in a water bath (Transsonic 310; Elma) for two pulses of 1 min each. Samples were boiled or not as indicated in the figures. All samples were centrifuged for 1 min (20,000 × g) to eliminate insoluble material before loading. SDS-PAGE was performed using a Miniprotean III electrophoresis system (Bio-Rad). For immunoblotting, the proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Immobilon-P; Millipore) using a semidry electrophoresis transfer apparatus (Bio-Rad). For immunodetection, PVDF membranes were incubated for 1 h at room temperature with the following reagents: anti-GroEL monoclonal antibody-peroxidase (POD) conjugate (1:5,000; Sigma) and rabbit polyclonal serum anti-FimD (1:4,000), anti-OmpA (1:20,000; a gift of Hiroshi Nikaido), anti-Skp (1:1,000; a gift of Matthias Mueller), anti-MBP-DegP (1:5,000; a gift of Michael Ehrmann), anti-SurA (1:10,000; a gift of Roberto Kolter), anti-BamA (1:5,000), anti-BamB (1:8,000), anti-BamC (1:20,000), and anti-BamE (1:10,000; a gift of Thomas Silhavy). Bound rabbit antibodies were detected with protein A-POD conjugate (1:8,000; Zymed). Membranes were blocked, washed, and developed as previously described (20). Intensity signals from protein bands were quantified by densitometry with Quantity One software (Bio-Rad).
RESULTS
Role of periplasmic chaperones in FimD folding and OM insertion.
Expression and folding of FimD were initially analyzed in E. coli K-12 strain UT5600 (wild type [WT]) and isogenic single null mutant strains lacking SurA, Skp, DegP, and FkpA periplasmic chaperones (Table 1). In addition, an isogenic fimD null mutant strain (Table 1) was included to control the specific detection of FimD in these experiments. Cultures of WT and mutant strains were incubated at 37°C in rich BHI medium under static conditions to induce type 1 fimbriae (and FimD) expression from the chromosomal fim operon. Similar growth rates of the WT and all mutant strains were observed (data not shown). The steady-state levels and the folding pattern of FimD were analyzed using bacterial samples from these cultures that were lysed in SDS-containing buffer and directly loaded, without boiling, for SDS-PAGE and Western blotting with an anti-FimD serum (Fig. 1, lanes 1 to 6). Similar to some other β-barrel OMPs, the compact β-barrel of folded FimD is resistant to SDS denaturation at low temperatures and migrates faster in SDS-PAGE than its unfolded form (31). The distinct mobility of the folded and unfolded forms of FimD was monitored in samples from the WT strain either boiled or not (Fig. 1, lanes c+ and c−). In the nonboiled samples, along with an intense high-mobility band corresponding to folded FimD, a faint band of unfolded FimD can be detected, likely caused by partial denaturation of FimD in SDS buffer (31). In addition to anti-FimD serum, all samples were probed by Western blotting with specific antibodies against OmpA, SurA, DegP, Skp, and GroEL (a cytoplasmic chaperone used as an internal loading control). As seen in Fig. 1 (FimD panel), FimD levels are similar in the WT, fkpA, degP, and skp mutants but are dramatically reduced in the surA null mutant (∼30-fold). These data are in agreement with the reported expression of FimD in surA and fkpA mutants (21). In contrast, the levels of folded OmpA are reduced ∼4-fold in the surA mutant and ∼2-fold in both skp and degP mutants (Fig. 1, OmpA panel). Importantly, the steady-state levels of DegP are strongly upregulated in the surA mutant (∼10-fold) and, to a lesser extent, in the skp mutant (∼3-fold) (Fig. 1, DegP panel). A small upregulation of Skp (∼1.3-fold) is also observed in the surA null mutant (Fig. 1, Skp panel). Finally, the level of SurA, DegP, or Skp is not altered in the fkpA null mutant. GroEL levels were similar in all samples (Fig. 1, GroEL panel). Therefore, under the growth conditions for type 1 fimbria expression, envelope stress responses are clearly induced in the surA null mutant, leading to a strong DegP upregulation, which makes it difficult to conclude that there is a direct effect of SurA on FimD.
Fig. 1.
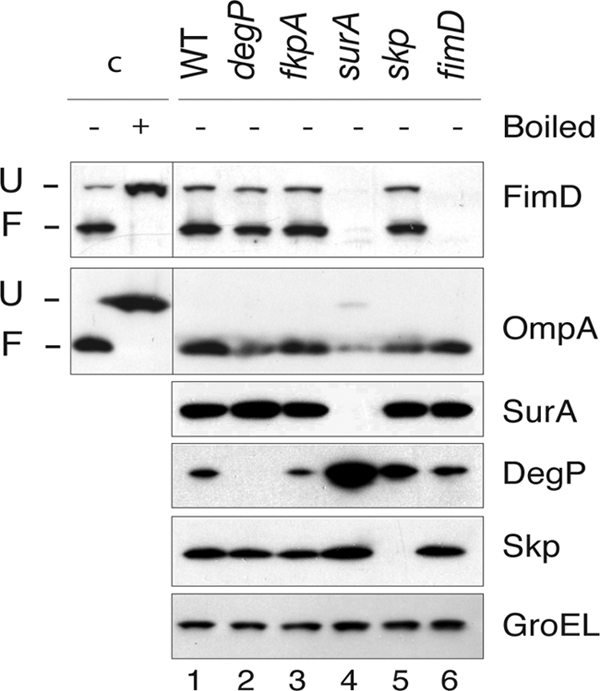
FimD folding in E. coli null mutants in periplasmic chaperones. Western blot analysis of whole-cell protein extracts from E. coli UT5600 (WT) strain and degP, fkpA, surA, skp, and fimD null mutants developed with anti-FimD, anti-OmpA, anti-SurA, anti-DegP, and anti-Skp antibodies, as indicated on the right of each panel. Detection of cytoplasmic GroEL with specific antibodies is used as a loading control (lower panel). Whole-cell extracts were prepared in SDS sample buffer and not boiled for detection of the folded forms of FimD and OmpA. Control sample c+ was boiled in urea-SDS sample buffer before loading in order to fully denature FimD and OmpA. The mobility of the protein bands corresponding to folded (F) and unfolded (U) FimD and OmpA are labeled on the left. Control samples are obtained from a culture of the WT strain.
To actually determine whether FimD depends on SurA for its assembly in the OM, a conditional surA mutant strain was generated. This mutant strain, named UTPBAD::surA (Table 1), was obtained by replacing the native promoter of chromosomal surA by an arabinose-inducible araC-PBAD genetic cassette (Fig. 2A) (40). In this conditional mutant strain, the level of surA can be controlled by the presence in the growth medium of the inducer l-arabinose or the repressor d-glucose. A culture of this strain was grown under static conditions in rich medium with 0.4% l-arabinose and used to inoculate fresh medium with either 0.4% l-arabinose or 0.4% d-glucose (in order to deplete SurA from bacteria). Both cultures were maintained in exponential growth by repeated dilutions in fresh medium (with l-arabinose or d-glucose, respectively) with similar growth rates (Fig. 2A). Aliquots of both cultures were taken at the time of culture dilutions (indicated by roman numbers I, II, and III in Fig. 2A). Harvested bacteria were processed for Western blotting as described previously to detect the levels of folded and unfolded FimD and OmpA; the periplasmic chaperones SurA, Skp, and DegP; and cytoplasmic GroEL as an internal loading control. SurA was completely depleted from bacteria grown with d-glucose but not in medium with l-arabinose. Importantly, in parallel to SurA, FimD was depleted during growth in d-glucose but not in medium containing l-arabinose (Fig. 2A, FimD panel). OmpA levels also decreased in medium with d-glucose, although not as dramatically as FimD, since folded OmpA was clearly detectable when SurA was depleted from bacteria. In contrast to the situation of the surA null mutant, DegP (and Skp) levels were not significantly upregulated in the conditional surA mutant under depletion conditions (Fig. 2A, DegP and Skp panels). Quantification of DegP bands reveals identical levels of DegP at time II in medium with l-arabinose or d-glucose and only a slight upregulation of DegP (1.3-fold) at time III in d-glucose medium. A small reduction in the level of SurA was observed in bacteria grown with l-arabinose, likely due to a moderate accumulation of SurA in this strain in the stationary-phase culture used as a preinoculum. Nevertheless, the steady-state level of SurA during continuous exponential growth with l-arabinose is clearly sufficient for proper folding of FimD and OmpA (Fig. 2A). Thus, under conditions in which DegP is not significantly upregulated, FimD is depleted from cells in parallel to SurA, indicating that SurA is required for FimD folding and insertion in the OM of E. coli.
Fig. 2.

FimD folding in conditional E. coli surA mutants. (A and B) Growth curves of static cultures of UTPBAD::surA and UTdegP-PBAD::surA strains grown in rich BHI medium containing d-glucose (Glu) or l-arabinose (Ara) and maintained in exponential phase by repeated dilutions with the same medium. Samples from these cultures were taken at the indicated times (I, II, and III) for Western blot analysis. A schematic drawing of the relevant genetic structure of these strains is depicted at the top of the growth curves. (C and D) Whole-cell protein extracts from bacteria harvested at the indicated times (I, II, and III) from cultures shown in panel A or B were analyzed by Western blotting with r anti-FimD, anti-OmpA, anti-SurA, anti-Skp, anti-DegP, and anti-GroEL antibodies. Samples from UTPBAD::surA are shown in panel C. Samples from UTdegP-PBAD::surA are shown in panel D. Whole-cell protein samples were prepared as indicated in the legend of Fig. 1. Control samples were obtained from cultures of the corresponding strain (UTPBAD::surA in panel C; UTdegP-PBAD::surA in panel D) grown in medium with l-arabinose.
Unassembled β-barrel OMPs are frequently eliminated from the periplasm by the protease activity of DegP (3, 26, 28). To assess whether unfolded FimD in SurA-depleted cells is degraded by DegP, FimD levels were analyzed in a double conditional mutant strain, UTdegP-PBAD::surA (Table 1), which carries a deletion of degP and the surA gene under the control of the PBAD promoter. Static cultures of this strain were grown in medium containing l-arabinose (control) or d-glucose (for depletion of SurA), as described above. Bacterial samples were taken at the indicated times (time points I, II, and III in Fig. 2B), and the folding and protein levels of FimD, OmpA, SurA, Skp, DegP, and GroEL were analyzed by Western blotting (Fig. 2B). In contrast to the UTPBAD::surA single conditional mutant strain, the double mutant UTdegP-PBAD::surA strain stopped its growth after 8 h in medium with d-glucose, as expected from the inactivation of both the SurA and DegP/Skp pathways (38). Higher levels of unfolded FimD are detected in this strain than in the WT or UTPBAD::surA strains (Fig. 2A and B), likely due to the constitutive absence of DegP. Depletion of SurA in the conditional double mutant strain caused the parallel disappearance of folded FimD and the increased accumulation of its unfolded form, with a maximum at 6 h of growth in d-glucose, when this conditional double mutant strain is still actively growing and when SurA is already depleted from bacteria (Fig. 2B). At later incubation times (8 h) only the unfolded form of FimD is detected but at lower levels than at 6 h, probably because of cell death and/or leakiness of the OM barrier, which may be responsible of the reduction of periplasmic Skp observed at this time (Fig. 2B, Skp panel). Taken together, these data indicate that the protease activity of DegP is involved in the degradation of the unfolded form of FimD that accumulates in the periplasm.
Role of individual components of the BAM complex in the assembly of FimD into the OM.
We tested initially whether a functional BAM complex was required for folding and insertion of FimD in the OM, as could be expected from its β-barrel structure. In E. coli, depletion of the essential BamA leads to a nonfunctional BAM complex and a reduction in the levels of integral β-barrel OMPs, such as OmpA, OmpC/OmpF, LamB, and TolC (52). A conditional BamA depletion strain of E. coli, named UTPBAD::bamA (Table 1), carrying the chromosomal bamA gene under the control of PBAD, was grown under static conditions in rich medium with either l-arabinose or d-glucose. As shown in Fig. 3A, growth rates of these cultures were similar for ∼7 h of continuous exponential growth by repeated dilutions with fresh medium. After that time, the culture with d-glucose stopped its growth, as expected from the lethal phenotype of BamA depletion (52). Bacteria from these cultures were taken at the time of dilutions (I, II, and III, indicated in Fig. 3A), lysed in SDS buffer, and subjected to Western blotting to detect FimD, OmpA, BamA, and GroEL. FimD was mainly present in the folded conformation when bacteria were grown in l-arabinose (Fig. 3B, FimD panel). However, already at 5 h of growth in medium with d-glucose, BamA was completely depleted (Fig. 3B, BamA panel), and levels of FimD and OmpA were reduced, becoming undetectable after 8 h of growth in d-glucose (Fig. 3B, FimD and OmpA panels). This experiment demonstrates that the BAM complex is necessary for the OM insertion of FimD. Next, we investigated the role of the nonessential lipoprotein components of the BAM complex (BamB, BamC, and BamE) for the biogenesis of FimD in the OM. To this end, FimD levels and folding status were evaluated in the WT (UT5601) and isogenic single null mutant strains lacking BamB (UTbamB), BamC (UTbamC), and BamE (UTbamE) (Table 1). No significant differences were observed in the growth rates among these strains grown under static conditions in rich medium (data not shown). Whole-cell extracts of bacteria from these cultures at exponential growth phase were used for Western blotting developed with antibodies against FimD, OmpA, BamB, BamC, BamE, BamA, DegP, and GroEL (Fig. 4A). This experiment revealed that the levels of folded FimD were strongly reduced (∼5-fold) in the mutant strain lacking BamB (lane 2), whereas in the single mutants lacking BamC or BamE, the levels of folded FimD were similar to the level in the WT (bamC mutant, lane 3) or reduced 1.6-fold (bamE mutant, lane 4). In contrast, the unfolded form of FimD accumulates in both the bamC and bamE single mutants (∼2-fold) but not in the bamB mutant. It has been previously reported that a moderate activation of the σE stress response takes place in bamB null mutants (33). A small increase of DegP (∼1.6-fold) was found in the bamB mutant (Fig. 4A, DegP panel, lane 2), which may help to explain the absence of unfolded FimD accumulated in this strain. However, DegP levels were found upregulated to the same extent in the bamE mutant and not in the bamC mutant (lanes 3 and 4), which both accumulate unfolded FimD. Therefore, the different levels of unfolded FimD in these mutants cannot be simply correlated to changes in DegP levels, and other mechanisms should be involved (see Discussion). In addition, we found that the levels of other components of BAM complex were unaffected in these mutants (Fig. 4A, panels BamB, BamC, BamE, and BamA, lanes 1 to 4). In contrast to the changes observed in FimD, the levels of folded OmpA were reduced only ∼1.4-fold in the bamB, bamC, and bamE single mutants, whereas only a small accumulation of unfolded OmpA is detected in the bamB mutant (Fig. 4A, OmpA panel, lanes 1 to 4). Thus, the requirement of accessory components of the BAM complex and the phenotypes of their single mutants are clearly different for FimD and OmpA. The role of BamB in the folding of FimD was confirmed by complementation of the bamB null mutant strain with a plasmid carrying bamB under the control of the PBAD promoter (pBAD-bamB) (Fig. 4B). Cultures of the E. coli bamB mutant strain grown in the presence of l-arabinose and carrying pBAD-bamB, but not carrying an empty vector (pBAD30), showed wild-type levels of folded FimD, as determined by Western blot analysis with anti-FimD serum, along with antibodies against BamB, DegP, and GroEL (Fig. 4B, lanes 1 and 2). In addition, when a culture of E. coli bamB carrying pBAD-bamB and grown in l-arabinose was used to inoculate rich medium with d-glucose and was maintained in exponential growth with d-glucose to deplete BamB, the level of folded FimD diminished in parallel to BamB without upregulation of DegP (Fig. 4B, lanes 3 to 5). Collectively, these data demonstrate that the BamB lipoprotein plays a direct and major role for proper OM assembly of FimD.
Fig. 3.
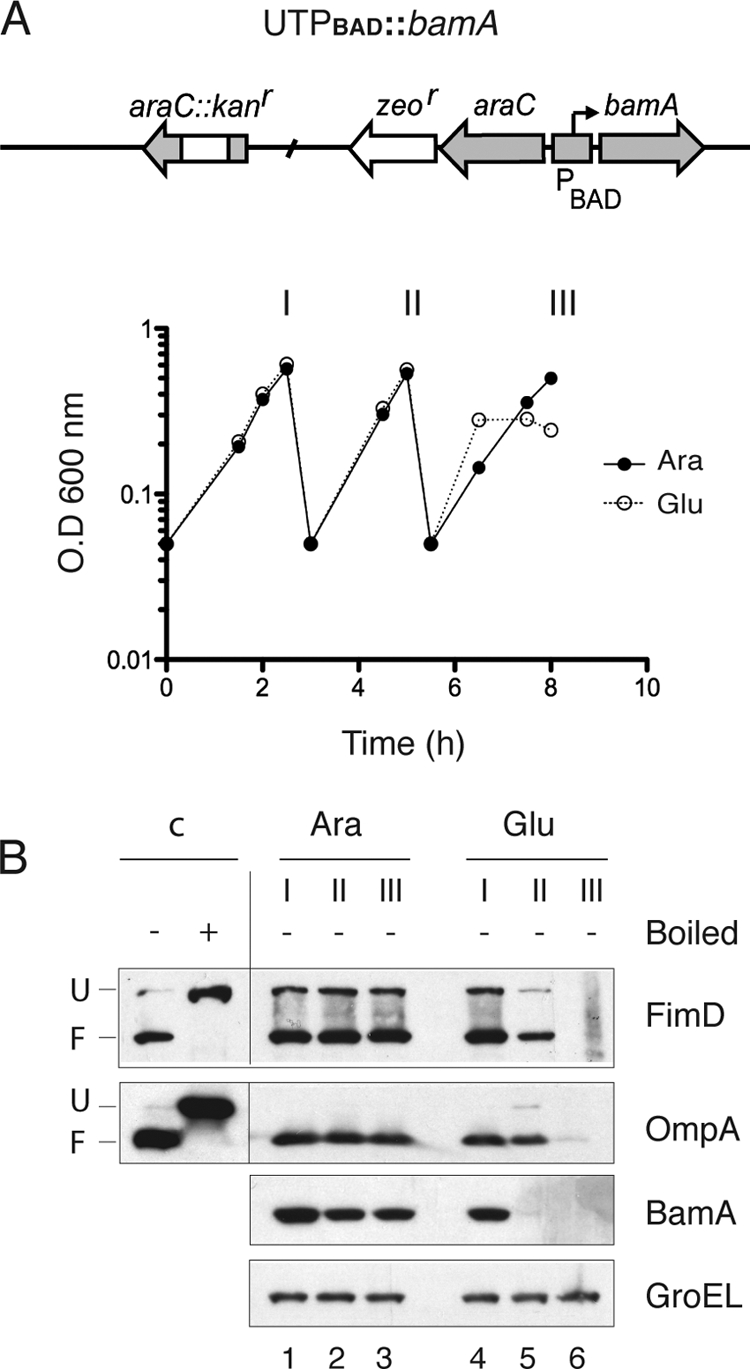
FimD folding in a conditional E. coli bamA mutant. (A) Growth curves of static cultures of the UTPBAD::bamA strain grown in rich BHI medium containing d-glucose (Glu) or l-arabinose (Ara) and maintained in exponential phase by repeated dilutions with the same medium. Samples from these cultures were taken at the indicated times (I, II, and III) for Western blot analysis. A schematic drawing of the relevant genetic structure of this strain is shown at the top. (B) Western blot analysis with anti-FimD, anti-OmpA, anti-BamA, and anti-GroEL antibodies of whole-cell protein extracts from bacteria harvested at the indicated times (I, II, and III) from cultures in panel A. Whole-cell protein samples were prepared as indicated in the legend of Fig. 1. Samples loaded in lanes 1 to 6 were not boiled except for detection of BamA. Control samples were obtained from a culture of the UTPBAD::bamA strain grown in medium with l-arabinose.
Fig. 4.

FimD folding in E. coli null mutants of nonessential lipoproteins of the BAM complex. (A) Western blot analysis of whole-cell protein extracts from the E. coli UT5601 (WT) strain and isogenic ΔbamB, ΔbamC, ΔbamE, ΔbamB ΔbamC, and ΔbamC ΔbamE null mutants grown statically in BHI medium. Western blots were developed with anti-FimD, anti-OmpA, anti-BamB, anti-BamC, anti-BamE, anti-BamA, anti-DegP, and anti-GroEL antibodies, as indicated on the right of each panel. The mobilities of the protein bands corresponding to folded (F) and unfolded (U) FimD and OmpA are labeled on the left. A nonspecific cross-reactive band against anti-FimD serum is also indicated with an asterisk. Samples loaded in lanes 1 to 6 were not boiled except for detection of BamA. (B) Complementation of the E. coli ΔbamB null mutant with plasmid pBamB (bearing bamB under control of the PBAD promoter). Western blot analysis of whole-cell protein extracts from ΔbamB null mutant bacteria carrying plasmid vector pBAD30 (lane 1) or pBamB (lanes 2 to 5) developed with anti-FimD, anti-BamB, anti-DegP, and anti-GroEL antibodies. Bacteria were cultured in medium with l-arabinose (Ara) (lanes 1 and 2) to induce the PBAD promoter or with d-glucose (Glu) (lanes 3 to 5) to deplete BamB bacteria previously grown with l-arabinose. For depletion in the induced E. coli ΔbamB (pBamB) strain, bacteria were grown with d-glucose and maintained in exponential growth for ∼8 h by repeated dilutions in fresh medium when the culture reached an OD600 of ∼0.5 (indicated as I, II, and III). In both panels, control samples (c) of FimD were obtained from a culture of the WT strain.
Folding of FimD was also analyzed in bamC bamB and bamC bamE double mutant strains. The bamB bamE double mutant was not generated due to its lethal phenotype (43). Double mutant strains UTbamC bamE and UTbamC bamE (Table 1) were grown in static cultures, and harvested bacteria were analyzed by Western blotting to determine the levels and folding status of FimD and OmpA, along with the levels of BamB, BamC, BamE, BamA, DegP, and GroEL (Fig. 4A, lanes 5 and 6). FimD levels were very low in both double mutants (>15-fold). However, whereas in the case of the bamC bamB mutant, no folded FimD was detectable, in the bamC bamE mutant a faint band of folded FimD was found. In contrast, OmpA exhibits a 2- to 3-fold reduction in bamC bamB and bamC bamE mutants, respectively, in agreement with previous results (44), and most OmpA was found in its folded form. Thus, when both BamC and BamE lipoproteins are absent, the overall FimD levels are clearly compromised, despite normal expression of BamB (Fig. 4A, BamB panel, lane 6). Because the lack of both BamC and BamE provokes a severe defect in the assembly of OMPs, such as LamB (44) and FimD and OmpA (this work), an increase in DegP levels could be expected in order to eliminate the unassembled β-barrel proteins. We found that steady-state levels of DegP were elevated ∼4-fold in bamC bamB and bamC bamE double mutants (Fig. 4A, DegP panel, lanes 5 and 6), reflecting the envelope stress caused by defective assembly of most OMPs. Taken together, these results indicate that BamC and BamE play an additive role in the folding of FimD and other OMP.
DISCUSSION
In this work we have studied the role of periplasmic chaperones and individual components of the BAM complex for the insertion and folding of FimD in E. coli under conditions for type 1 fimbria expression from the endogenous single-copy chromosomal fim locus. Our results demonstrate that OM assembly of the FimD usher is highly dependent on not only the essential BamA but also the nonessential SurA and BamB. In addition, we found that at least one of the other accessory proteins of the BAM complex (i.e., BamC and BamE) is needed for efficient assembly of FimD, supporting their role as enhancers of the activity of the BAM complex. Previous studies had demonstrated that the constitutive absence of SurA in E. coli K-12 and UPEC strains dramatically reduces the levels of FimD and type 1 fimbriae (21, 51). In agreement with these reports, we found that FimD levels are strongly diminished in an E. coli surA null mutant strain, whereas no change is seen in isogenic degP, skp, and fkpA null mutant strains (Fig. 1). However, DegP is highly upregulated in the surA null mutant under conditions of type 1 fimbria expression (Fig. 1). When FimD was analyzed in a conditional surA mutant strain, depletion of SurA from bacteria elicited a concomitant and parallel decrease of folded and unfolded FimD levels, without significant upregulation of DegP (Fig. 2A). Further, depletion of SurA in a conditional degP surA double mutant strain diminished the level of folded FimD and simultaneously generated the accumulation of unfolded FimD (Fig. 2B), indicating that DegP is involved in the proteolysis of unfolded FimD that accumulates when SurA is depleted. Collectively, these results are consistent with a direct involvement of SurA in FimD folding. The dependency on SurA is clearly higher for FimD than for other integral β-barrel OMPs, such as OmpA from E. coli K-12 or intimin from enteropathogenic E. coli (3, 27). In the case of OmpA and intimin, the use of the DegP/Skp pathway seems responsible for the protein levels found in the absence of SurA (3, 44). According to our data, FimD mainly uses the SurA pathway for its assembly although the reason for this selection is unclear.
FimD was found to require a functional BAM complex for its assembly in the OM (Fig. 3). Several examples have been reported of integral β-barrel OMPs of E. coli that fail to insert in the OM when BamA is depleted, including OmpA, OmpC/OmpF, LamB, PhoE, TolC, autotransporters, and intimins (3, 10, 19, 52, 53). In contrast, a special class of OMPs lacking integral β-barrels does not depend on BamA for OM insertion (7, 8). Hence, a functional BAM complex is needed for the OM assembly of FimD, most likely due to the presence of a large central β-barrel domain in its structure (37). In contrast to BamA, the nonessential lipoproteins BamB, BamC, and BamE seem to be required at different stages of the insertion of OMPs in the OM and/or for delivery of OMPs to the BAM complex. Mutants lacking BamB exhibit a strong decrease in FimD levels (∼5-fold), whereas the levels of OmpA were reduced less than 2-fold (Fig. 4). This observation agrees with previous works reporting variable decreasing levels of OMPs in bamB null mutants, with LamB (18 β-strands; 3-fold reduction) more sensitive than OmpA (8 β-strands; 1.4-fold reduction) (5, 33). Our results show that FimD (24 β-strands) is strongly dependent on BamB for insertion in the OM of E. coli. The precise role of BamB in OMP biogenesis has not been established, but its recent crystallographic structure revealed an eight-bladed β-propeller fold that may interact with the β-strands of OMPs by β-augmentation (17, 23). BamB and SurA have been suggested to define a distinct pathway for assembly of certain OMPs in E. coli, given the similar phenotypes of their single null mutants and the synthetic lethality associated with bamB degP, surA degP, and surA bamC double mutants (2, 5, 33, 38). Current models of OMP biogenesis suggest that SurA captures unfolded OMPs in the periplasm and transport them to BamA, where they are taken by the periplasmic polypeptide transport-associated (POTRA) domains through β-augmentation (17). BamB would increase the capacity of BamA for interaction with β-strands of OMPs by β-augmentation (17). Therefore, a hypothetical model to explain the strong dependency of FimD on BamB could be that folding of its large central β-barrel, with 24 β-strands, may require an increased β-strand binding capacity that is provided by BamB. This model could also explain the high dependency of LamB (18 β-strands) and OmpF (16 β-strands) on BamB. However, in addition to the complexity of the β-barrel, other factors may contribute to the requirement of BamB in OMP folding. Interestingly, FimD, LamB, and OmpF are also more dependent on SurA than OmpA (2, 5, 33). Thus, the observed BamB dependency of FimD could also reflect a preferential use of BamB lipoprotein for folding of OMPs delivered to the BAM complex by SurA. Both BamB and SurA directly interact with BamA although the interaction of SurA with BamA is independent on the presence of BamB (2, 44, 48).
Unlike mutants lacking BamB, E. coli cells lacking BamC or BamE show no significant reduction in OMPs such as OmpA and LamB (25, 43). Our results show that overall FimD levels are only weakly affected in bamC or bamE single null mutants although the level of unfolded FimD clearly increases in these mutants (Fig. 4). This situation contrasts with the strong decrease in both folded and unfolded FimD in bamB mutants. Interestingly, DegP was found slightly upregulated in bamE and bamB single mutants but not in the bamC mutant, indicating that the different levels of accumulation of unfolded FimD in these mutants do not seem to be related only to changes in DegP levels. Alternatively, BamC and BamE may enhance the capacity of the BAM complex for the insertion into the OM of FimD, already bound to BamA-BamB in its unfolded form. Lack of only one of these accessory lipoproteins appears to cause a small reduction in the efficiency of FimD insertion, resulting in some accumulation of unfolded FimD in the BAM complex but not an amount sufficient to significantly activate envelope stress responses that upregulate DegP. In contrast, the absence of both accessory lipoproteins (in the bamC bamE double mutant) appears to dramatically reduce the capacity of the BAM complex to insert FimD and other OMPs in the OM and strongly upregulates DegP (Fig. 4), perhaps due to the accumulation of misfolded OMPs associated to both the BAM complex and the periplasm. The recent determinations of the structures of all the individual BAM lipoproteins (1, 17, 22, 23, 41, 50) will help to unveil the specific activities of these proteins for the folding and insertion of FimD and other OMPs in E. coli.
In conclusion, our work demonstrates that the fimbrial usher FimD is highly dependent on SurA and BamB for its folding and OM assembly in E. coli, supporting the existence of a SurA-BamB pathway in E. coli that is followed by FimD, and which might be preferentially used by OMPs with large β-barrels. Other accessory lipoproteins of the BAM complex, BamC and BamE, appear to enhance FimD folding in the BAM complex, with at least one of them being needed for efficient folding and OM insertion of FimD.
ACKNOWLEDGMENTS
We thank Hiroshi Nikaido, Matthias Mueller, Michael Ehrmann, Roberto Kolter, and Thomas Silhavy for their kind gift of polyclonal sera.
This work was supported by grants to L.A.F. from the Spanish Ministry of Science and Innovation (BIO2008-05201), the Autonomous Community of Madrid (S-BIO-236-2006), and the VI Framework Program from the European Union (FP6-LSHB-CT-2005-512061 NoE, EuroPathogenomics). E.M. is supported by contract Apoyo a la Investigación of the Autonomous Community of Madrid.
We declare that there is no conflict of interest.
Footnotes
Published ahead of print on 22 July 2011.
REFERENCES
- 1. Albrecht R., Zeth K. 2011. Structural basis of the outer membrane protein biogenesis in bacteria. J. Biol. Chem. 286:27792–27803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bennion D., Charlson E. S., Coon E., Misra R. 2010. Dissection of beta-barrel outer membrane protein assembly pathways through characterizing BamA POTRA 1 mutants of Escherichia coli. Mol. Microbiol. 77:1153–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bodelón G., Marín E., Fernández L. A. 2009. Role of periplasmic chaperones and BamA (YaeT/Omp85) in folding and secretion of intimin from enteropathogenic Escherichia coli Strains. J. Bacteriol. 191:5169–5179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bos M. P., Robert V., Tommassen J. 2007. Biogenesis of the gram-negative bacterial outer membrane. Annu. Rev. Microbiol. 61:191–214 [DOI] [PubMed] [Google Scholar]
- 5. Charlson E. S., Werner J. N., Misra R. 2006. Differential effects of yfgL mutation on Escherichia coli outer membrane proteins and lipopolysaccharide. J. Bacteriol. 188:7186–7194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen R., Henning U. 1996. A periplasmic protein (Skp) of Escherichia coli selectively binds a class of outer membrane proteins. Mol. Microbiol. 19:1287–1294 [DOI] [PubMed] [Google Scholar]
- 7. Collin S., Guilvout I., Chami M., Pugsley A. P. 2007. YaeT-independent multimerization and outer membrane association of secretin PulD. Mol. Microbiol. 64:1350–1357 [DOI] [PubMed] [Google Scholar]
- 8. Collins R. F., Derrick J. P. 2007. Wza: a new structural paradigm for outer membrane secretory proteins? Trends Microbiol. 15:96–100 [DOI] [PubMed] [Google Scholar]
- 9. Datsenko K. A., Wanner B. L. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Doerrler W. T., Raetz C. R. 2005. Loss of outer membrane proteins without inhibition of lipid export in an Escherichia coli YaeT mutant. J. Biol. Chem. 280:27679–27687 [DOI] [PubMed] [Google Scholar]
- 11. Ford B., et al. 2010. Structural homology between the C-terminal domain of the PapC usher and its plug. J. Bacteriol. 192:1824–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gerlach R. G., Hensel M. 2007. Protein secretion systems and adhesins: the molecular armory of Gram-negative pathogens. Int. J. Med. Microbiol. 297:401–415 [DOI] [PubMed] [Google Scholar]
- 13. Grodberg J., Dunn J. J. 1988. OmpT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification. J. Bacteriol. 170:1245–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guzman L. M., Belin D., Carson M. J., Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hagan C. L., Silhavy T. J., Kahne D. E. 2011. b-Barrel Membrane Protein Assembly by the Bam Complex. Annu. Rev. Biochem. [DOI] [PubMed] [Google Scholar]
- 16. Henderson N. S., Ng T. W., Talukder I., Thanassi D. G. 2011. Function of the usher N terminus in catalysing pilus assembly. Mol. Microbiol. 79:954–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heuck A., Schleiffer A., Clausen T. 2011. Augmenting β-augmentation: structural basis of how BamB binds BamA and may support folding of outer membrane proteins. J. Mol. Biol. 406:659–666 [DOI] [PubMed] [Google Scholar]
- 18. Huang Y., Smith B. S., Chen L. X., Baxter R. H. G., Deisenhofer J. 2009. Insights into pilus assembly and secretion from the structure and functional characterization of usher PapC. Proc. Natl. Acad. Sci. 106:7403–7407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jain S., Goldberg M. B. 2007. Requirement for YaeT in the outer membrane assembly of autotransporter proteins. J. Bacteriol. 189:5393–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jurado P., Ritz D., Beckwith J., de Lorenzo V., Fernández L. A. 2002. Production of functional single-chain Fv antibodies in the cytoplasm of Escherichia coli. J. Mol. Biol. 320:1–10 [DOI] [PubMed] [Google Scholar]
- 21. Justice S. S., et al. 2005. Periplasmic peptidyl prolyl cis-trans isomerases are not essential for viability, but SurA is required for pilus biogenesis in Escherichia coli. J. Bacteriol. 187:7680–7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim K. H., et al. 2011. Structural characterization of Escherichia coli BamE, a lipoprotein component of the beta-barrel assembly machinery complex. Biochemistry 50:1081–1090 [DOI] [PubMed] [Google Scholar]
- 23. Kim K. H., Paetzel M. 2011. Crystal structure of Escherichia coli BamB, a lipoprotein component of the β-barrel assembly machinery complex. J. Mol. Biol. 406:667–678 [DOI] [PubMed] [Google Scholar]
- 24. Kline K. A., Falker S., Dahlberg S., Normark S., Henriques-Normark B. 2009. Bacterial adhesins in host-microbe interactions. Cell Host Microbe 5:580–592 [DOI] [PubMed] [Google Scholar]
- 25. Knowles T. J., Scott-Tucker A., Overduin M., Henderson I. R. 2009. Membrane protein architects: the role of the BAM complex in outer membrane protein assembly. Nat. Rev. Microbiol. 7:206–214 [DOI] [PubMed] [Google Scholar]
- 26. Krojer T., et al. 2008. Structural basis for the regulated protease and chaperone function of DegP. Nature 453:885–890 [DOI] [PubMed] [Google Scholar]
- 27. Lazar S. W., Kolter R. 1996. SurA assists the folding of Escherichia coli outer membrane proteins. J. Bacteriol. 178:1770–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lehr U., et al. 2010. C-terminal amino acid residues of the trimeric autotransporter adhesin YadA of Yersinia enterocolitica are decisive for its recognition and assembly by BamA. Mol. Microbiol. 78:932–946 [DOI] [PubMed] [Google Scholar]
- 29. Munera D., Hultgren S., Fernández L. A. 2007. Recognition of the N-terminal lectin domain of FimH adhesin by the usher FimD is required for type 1 pilus biogenesis. Mol. Microbiol. 64:333–346 [DOI] [PubMed] [Google Scholar]
- 30. Munera D., Palomino C., Fernández L. A. 2008. Specific residues in the N-terminal domain of FimH stimulate type 1 fimbriae assembly in Escherichia coli following the initial binding of the adhesin to FimD usher. Mol. Microbiol. 69:911–925 [DOI] [PubMed] [Google Scholar]
- 31. Ng T. W., Akman L., Osisami M., Thanassi D. G. 2004. The usher N terminus is the initial targeting site for chaperone-subunit complexes and participates in subsequent pilus biogenesis events. J. Bacteriol. 186:5321–5331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nishiyama M., Ishikawa T., Rechsteiner H., Glockshuber R. 2008. Reconstitution of pilus assembly reveals a bacterial outer membrane catalyst. Science 320:376–379 [DOI] [PubMed] [Google Scholar]
- 33. Onufryk C., Crouch M. L., Fang F. C., Gross C. A. 2005. Characterization of six lipoproteins in the σE regulon. J. Bacteriol. 187:4552–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Phan G., et al. 2011. Crystal structure of the FimD usher bound to its cognate FimC-FimH substrate. Nature 474:49–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Prilipov A., Phale P. S., Van Gelder P., Rosenbusch J. P., Koebnik R. 1998. Coupling site-directed mutagenesis with high-level expression: large scale production of mutant porins from E. coli. FEMS Microbiol. Lett. 163:65–72 [DOI] [PubMed] [Google Scholar]
- 36. Rego A. T., Chandran V., Waksman G. 2010. Two-step and one-step secretion mechanisms in Gram-negative bacteria: contrasting the type IV secretion system and the chaperone-usher pathway of pilus biogenesis. Biochem. J. 425:475–488 [DOI] [PubMed] [Google Scholar]
- 37. Remaut H., et al. 2008. Fiber formation across the bacterial outer membrane by the chaperone/usher pathway. Cell 133:640–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rizzitello A. E., Harper J. R., Silhavy T. J. 2001. Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J. Bacteriol. 183:6794–6800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rouvière P. E., Gross C. A. 1996. SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev. 10:3170–3182 [DOI] [PubMed] [Google Scholar]
- 40. Roux A., Beloin C., Ghigo J. M. 2005. Combined inactivation and expression strategy to study gene function under physiological conditions: application to identification of new Escherichia coli adhesins. J. Bacteriol. 187:1001–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sandoval C. M., Baker S. L., Jansen K., Metzner S. I., Sousa M. C. 2011. Crystal structure of BamD: an essential component of the β-barrel assembly machinery of gram-negative bacteria. J. Mol. Biol. 409:348–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saulino E. T., Bullitt E., Hultgren S. J. 2000. Snapshots of usher-mediated protein secretion and ordered pilus assembly. Proc. Natl. Acad. Sci. U. S. A. 97:9240–9245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sklar J. G., et al. 2007. Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 104:6400–6405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sklar J. G., Wu T., Kahne D., Silhavy T. J. 2007. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev. 21:2473–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Spiess C., Beil A., Ehrmann M. 1999. A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell 97:339–347 [DOI] [PubMed] [Google Scholar]
- 46. Veiga E., De Lorenzo V., Fernández L. A. 2004. Structural tolerance of bacterial autotransporters for folded passenger protein domains. Mol. Microbiol. 52:1069–1080 [DOI] [PubMed] [Google Scholar]
- 47. Vertommen D., Ruiz N., Leverrier P., Silhavy T. J., Collet J. F. 2009. Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics 9:2432–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vuong P., Bennion D., Mantei J., Frost D., Misra R. 2008. Analysis of YfgL and YaeT interactions through bioinformatics, mutagenesis, and biochemistry. J. Bacteriol. 190:1507–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Waksman G., Hultgren S. J. 2009. Structural biology of the chaperone-usher pathway of pilus biogenesis. Nat. Rev. Microbiol. 7:765–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Warner L. R., et al. 2011. Structure of the BamC two-domain protein obtained by Rosetta with a limited NMR data set. J. Mol. Biol. 411:83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Watts K. M., Hunstad D. A. 2008. Components of SurA required for outer membrane biogenesis in uropathogenic Escherichia coli. PLoS One 3:e3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Werner J., Misra R. 2005. YaeT (Omp85) affects the assembly of lipid-dependent and lipid-independent outer membrane proteins of Escherichia coli. Mol. Microbiol. 57:1450–1459 [DOI] [PubMed] [Google Scholar]
- 53. Wu T., et al. 2005. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121:235–245 [DOI] [PubMed] [Google Scholar]