

Abstract
A maximum likelihood estimator based on the coalescent for unequal migration rates and different subpopulation sizes is developed. The method uses a Markov chain Monte Carlo approach to investigate possible genealogies with branch lengths and with migration events. Properties of the new method are shown by using simulated data from a four-population n-island model and a source–sink population model. Our estimation method as coded in migrate is tested against genetree; both programs deliver a very similar likelihood surface. The algorithm converges to the estimates fairly quickly, even when the Markov chain is started from unfavorable parameters. The method was used to estimate gene flow in the Nile valley by using mtDNA data from three human populations.
Keywords: coalescence theory, population genetics
Estimation of migration rates from genetic data has a long history. As soon as the first analyses of population samples by using enzyme electrophoresis were available, migration rates estimated from Wright's FST (1) were used to infer patterns of gene flow. With the advent of other types of genetic data, such as restriction fragment length polymorphism data, DNA sequences, and microsatellite loci, migration rates have been routinely estimated by using modified versions of FST (2–6). Translation of these FST equivalents into migration rate estimates most often assumes that all subpopulations have the same size, or that there are infinitely many subpopulations, and that the migration rates are all symmetric. If the true migration pattern has been asymmetric, or the subpopulation sizes are unequal, FST-based methods will deliver wrong estimates (7).
Recently, it became possible to estimate migration rates and population sizes without the assumption that subpopulation sizes are all equal (8) or that the migration rates between the subpopulations are symmetric (9–12).
We describe here an extension of our two-population method (10) that calculates maximum likelihood estimates of migration rates and subpopulation sizes by using coalescence theory (13, 14). Our method allows us to analyze more than two subpopulations, to specify arbitrary migration scenarios, and to test a hierarchy of different migration scenarios. Success of estimating migration pattern is assessed with simulated sequence data in an n-island population model and a source–sink population scenario. Convergence to the correct result is investigated through simulation. The performance of our method, implemented in the program migrate, is compared with that of genetree (11). Finally, we analyze a human mtDNA hypervariable region I data set from three populations in the Nile valley, a total of 225 individuals.
Materials and Methods
Model.
We infer the population parameters by using a maximum likelihood approach based on coalescence theory (13, 14). This likelihood is the probability of the data given the parameters 𝒫
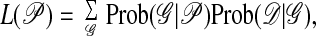 |
1 |
where Prob(𝒢|𝒫) is the probability of a genealogy 𝒢 given the population parameters 𝒫, such as population size (15), exponential population growth rate (16), migration rates (10), and recombination rate (17, 18).
Prob(𝒟|𝒢) is the likelihood of the data given the genealogy; this quantity is widely used in phylogenetic inference (19, 20). In this paper, we focus on estimation of migration rates and population sizes while assuming a molecular clock at each locus. For a system with n populations, we use the following set of parameters:
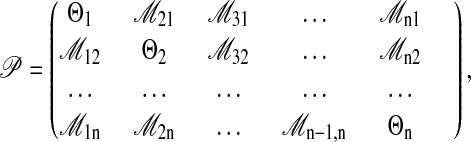 |
2 |
where ℳji is
mji/μ, where
mji is the immigration rate from population
j into i, and μ is the mutation rate
per generation. For sequence data, μ is the mutation rate
per site and for allelic data, such as allozyme or microsatellite
markers, and μ is the mutation rate per locus.
Θi is 4N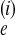 μ,
where N
μ,
where N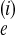 is the effective population size
of population i in a Wright–Fisher population model.
Sometimes we use γji, which is
Θiℳji =
4N
is the effective population size
of population i in a Wright–Fisher population model.
Sometimes we use γji, which is
Θiℳji =
4N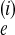 mji.
mji.
Kingman's coalescent can be extended to include migration (21). Instead of just one type of event, the coalescence of lineages, we need to record n2 different events: coalescences in different subpopulations and migration events that switch lineages from one population to another. This migration–coalescence prior is a product over all time intervals T on the genealogy (21). Going backwards in time and using a time scale in which the units of time is the ratio of the generation time and the expectation of the mutation rate, we have
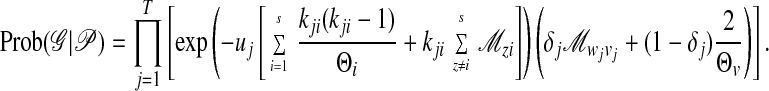 |
3 |
The exponential term is the probability that in the jth time interval with length uj neither a migration nor a coalescent event happens; uj is scaled by generations and mutation rate. The remaining term is the point probability density of the actual event that happens. The events in genealogy 𝒢 are either migrations from subpopulation wj to vj or coalescences in subpopulation vj. The indicator variable δj is 1 when the event at the bottom of interval j is a migration event and is 0 otherwise, and kji is the number of lineages in subpopulation i during time interval j. No modification of the genealogy likelihood Prob(𝒟|𝒢) is necessary to accommodate migration events, as they occur independently of mutation. The modified coalescent probability (3) and the genealogy likelihood are rather easy to calculate.
Unfortunately, the sum of the probabilities of all possible genealogies with different topologies and branch lengths cannot be calculated because there are infinitely many of them and no analytical solution for the integral over branch lengths or topologies is available. But it can be approximated by using the Metropolis–Hastings Markov chain Monte Carlo approach (22, 23). This approach (which we denote MH) concentrates the sampling of genealogies 𝒢 in those regions of the genealogy space that contribute most to the final likelihood. With MH importance sampling, we compute instead of the likelihood L(𝒫) the ratio L(𝒫)/L(𝒫0), where 𝒫0 are the parameters that were used to sample the genealogies. This is
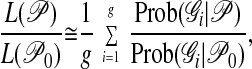 |
4 |
where g is the number of sampled genealogies. Our derivation of formula 4 from formula 1 was described earlier (10) and is essentially a standard MH scheme (24). The denominator L(𝒫0) is unknown, so that we cannot estimate the absolute likelihood, but this likelihood ratio is proportional to the absolute likelihood function. The parameter estimates obtained by maximizing this approximate likelihood ratio will approach the maximum likelihood estimates as the number of sampled genealogies g becomes infinite (22, 23). We find that very good estimates are achieved with a moderate number of genealogies (10, 15–17).
Our MH approach needs a set of starting parameters 𝒫0 and an initial genealogy. These starting parameters can often be found by using a simpler method, such as methods based on FST (25). The first genealogy is created by using a UPGMA (Unweighted Pair Group Method using arithmetic averages) method (see ref. 19) to construct the topology, and by using Sankoff's parsimony method (26) to reconstruct the minimal number of migrations on that topology. The branch lengths of this initial genealogy are drawn randomly from a coalescent density for that topology given 𝒫0. Our MH method (10) moves through genealogy space by making small rearrangements of branches of the current genealogy. A new genealogy is accepted with probability equal to the ratio of the likelihood of the old genealogy and the likelihood of the new [(for details, see ref. 10) r = min(1, Prob(D|Gnew)/Prob(D|Gold)].
With a random or otherwise inappropriate starting genealogy and an inappropriate 𝒫0, the program can spend much time in regions with highly improbable genealogies. To overcome these starting conditions, we use an adaptive scheme that samples 10 short chains, in each of which several thousand genealogies are sampled, followed by two or three long chains, each of which samples many tens or hundreds of thousands of genealogies. After each chain, we reestimate the parameters 𝒫 by maximizing the likelihood ratio (4). These 𝒫 are taken as the 𝒫0 of the next chain. The last long chain is used for the final estimates of 𝒫; the earlier chains are used only to obtain good starting parameters.
One would like to know not only the maximum likelihood values of the population parameters, but also confidence intervals for these parameters. Approximate confidence intervals can be generated in a maximum likelihood framework by using either the curvature of the likelihood at its maximum or profile likelihoods (27). The latter are more appropriate for our purposes, as curvature-based estimates can be unreliable with many parameters unless there are many loci. For an approximative confidence interval for a single parameter, we compare twice the logarithm of the ratio of its profile likelihoods to the quantiles of the χ2-distribution with one degree of freedom (27) for the desired level of confidence. If a researcher needs multiparameter confidence intervals, she would need to use a Bonferroni correction or a likelihood ratio test with the correct number of degrees of freedom. The latter method is implemented in migrate. This likelihood-ratio-based approach may be inappropriate for data that in theory cannot be extended, such as mtDNA, because the χ2 approximation becomes exact only as we add a large number of loci.
Results
Simulation Study.
Data sets were created by using an approach first described by Hudson (28). For some given set of true parameters Θi and 4N(i)mji, a coalescent genealogy is created. This genealogy is then used to evolve sites according to the Kimura two-parameter substitution model (29), starting at the root of that genealogy. The sites resulting at the tips are taken as the data. We used 500 sites for each locus in all simulations, except for the comparison of our own results with those from genetree (11).
n-island model.
We simulated a 4-island model and were generating 100 10-locus data sets with 25 individuals sampled from each of 4 subpopulations (Fig. 1).
Figure 1.
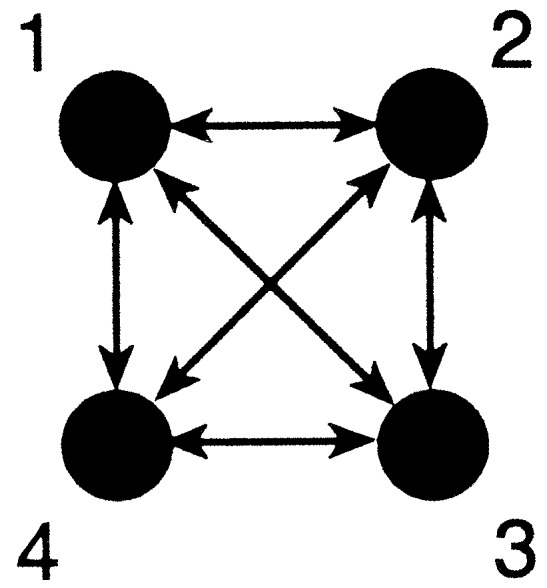
n-island model with four populations of equal size, exchanging migrants with equal rates.
The values for the Θi were taken to be equal and set to 0.01, a value that is moderately close to the estimate of Θ = 0.039 from mtDNA control region domain I sequences from the Nuu-Chah-Nulth people (15). The migration parameters 4Nm were all set to 1.0. Data sets were analyzed twice, once under the assumption that this is a symmetric n-island model with two parameters Θ and 4Nm, which are the same in all populations, and once under the assumption that we have n2 parameters, n different Θi and n(n − 1) different 4Nimji.
The averages for the two parameters of the n-island
model were Θ̄ = 0.00999 with an SE of 0.00007 and
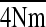 = 0.96327 with an SE of 0.01351. Averages
of the limits of the one-parameter 95% profile confidence intervals
were (0.00888, 0.01089) and (0.86656, 1.12981), respectively. The
estimates for the full migration matrix model with all 16 parameters is
shown in Table 1. The estimates for the
Θi are surprisingly precise given the
parameter-rich model, but most of the
4Nimji are lower than the true value
parameter, although the averages of the individual 95% profile
confidence intervals include the true parameter values.
= 0.96327 with an SE of 0.01351. Averages
of the limits of the one-parameter 95% profile confidence intervals
were (0.00888, 0.01089) and (0.86656, 1.12981), respectively. The
estimates for the full migration matrix model with all 16 parameters is
shown in Table 1. The estimates for the
Θi are surprisingly precise given the
parameter-rich model, but most of the
4Nimji are lower than the true value
parameter, although the averages of the individual 95% profile
confidence intervals include the true parameter values.
Table 1.
Averages of population parameters of a n-island population (see Fig. 1) based on 100 simulated data sets, each with 10 unlinked loci
| Population i | Θ | 4Nm
|
||||
|---|---|---|---|---|---|---|
| 1→i | 2→i | 3→i | 4→i | |||
| 1 | 2.5% | 0.0085 | — | 0.7394 | 0.6716 | 0.6586 |
| ML | 0.0104 | — | 1.0043 | 0.9280 | 0.9136 | |
| 97.5% | 0.0121 | — | 1.4329 | 1.3361 | 1.3161 | |
| 2 | 2.5% | 0.0083 | 0.6688 | — | 0.6745 | 0.6316 |
| ML | 0.0102 | 0.9236 | — | 0.9201 | 0.8752 | |
| 97.5% | 0.0118 | 1.3254 | — | 1.3197 | 1.2626 | |
| 3 | 2.5% | 0.0083 | 0.6882 | 0.6311 | — | 0.5933 |
| ML | 0.0101 | 0.9471 | 0.8840 | — | 0.8343 | |
| 97.5% | 0.0117 | 1.3605 | 1.2782 | — | 1.2155 | |
| 4 | 2.5% | 0.0084 | 0.6616 | 0.6740 | 0.6255 | — |
| ML | 0.0103 | 0.9149 | 0.9354 | 0.8757 | — | |
| 97.5% | 0.0119 | 1.3219 | 1.3536 | 1.2711 | — | |
All sequences were 500 bp long. The values shown are the 2.5% percentile, the maximum likelihood estimates, and the 97.5% percentile of the population sizes Θ and 4Nm, where m are the immigration rates. The true Θ was 0.01 and the true 4Nm was 1.0. The populations in the rows receive immigrants from those in the columns. ML, maximum likelihood.
Source–sink model.
The full model with n population sizes and n(n − 1) migration rates is able to detect asymmetric gene flow and differences in population sizes. But for some analyses, this freedom is undesirable because the researcher already has some idea of the pattern of gene flow or the population sizes and may want to fix population sizes, force migration rates to be symmetric, use equal migration rates, or set some migration rates to zero. This goal can be achieved in migrate by using a migration connection matrix (http://evolution.genetics.washington.edu/lamarc/migratedoc/migratedoc.html), such as the one shown in Fig. 2.
Figure 2.

A source–sink population complex. (A) Arrows mark directions of migration, and disk sizes are proportional to population sizes. (B) The corresponding population connection matrix used in migrate. The matrix contains specifications for Θ on the diagonal and for the ℳ off-diagonal. * indicates this parameter is estimated without restriction, and 0 indicates it is held to 0 so there is no direct gene flow between these subpopulations.
One hundred simulated 10-locus data sets from the populations shown in Fig. 2 were analyzed by using the full set of 16 parameters, and 50 data sets were analyzed by using only the 7 parameters implied by the migration-connection matrix.
Some of the simulations in Table 2 do not recover the true values of the migration parameters very well: all parameters with true parameter values of 0.0 are overestimated. Most disturbing are the values for migrations from population 2 to 1, from 3 to 2, and from 4 to 1. But this fact is not surprising, as all parameters are bounded by zero but have no upper limit. Our estimates must deliver a value greater than zero, so that the result must be an upwards bias. In addition, if we do not know the directionality of gene flow, finding the same haplotype in two or more populations will force the program to estimate at least a small migration rate in the wrong direction. Only a few mutations will arise in the small population and be visible in the sample, and only those unique mutations would contribute to inferring that that gene flow from the small population to the big population is very small. Thus, we would need many loci to establish this directional pattern.
Table 2.
Averages of population parameters of a source-sink population (see Fig. 2)
| Population i | Θ | 4Nm
|
||||
|---|---|---|---|---|---|---|
| 1→i | 2→i | 3→i | 4→i | |||
| 1 | True | 0.05 | — | 0.0 | 0.0 | 0.0 |
| 2.5% | 0.0408 | — | 0.1655 | 0.0358 | 0.0338 | |
| ML | 0.0465 | — | 0.2734 | 0.0892 | 0.0817 | |
| 97.5% | 0.0547 | — | 0.4465 | 0.1730 | 0.1623 | |
| 2 | True | 0.05 | 1.0 | — | 0.0 | 0.0 |
| 2.5% | 0.0411 | 0.6676 | — | 0.5783 | 0.0157 | |
| ML | 0.0471 | 0.8917 | — | 0.7775 | 0.0533 | |
| 97.5% | 0.0560 | 1.3088 | — | 1.1648 | 0.1215 | |
| 3 | True | 0.01 | 0.0 | 1.0 | — | 0.0 |
| 2.5% | 0.0097 | 0.0425 | 0.5600 | — | 0.0030 | |
| ML | 0.0113 | 0.0949 | 0.7238 | — | 0.0156 | |
| 97.5% | 0.0136 | 0.1807 | 1.0451 | — | 0.0403 | |
| 4 | True | 0.01 | 0.1 | 0.0 | 0.0 | — |
| 2.5% | 0.0086 | 0.0356 | 0.0076 | 0.0046 | — | |
| ML | 0.0101 | 0.0699 | 0.0252 | 0.018 | — | |
| 97.5% | 0.0121 | 0.1309 | 0.0579 | 0.0441 | — | |
Each of the 100 simulated data sets had 10 unlinked loci, each of which was 500 bp long. The values shown are the true values, the 2.5% percentile, the maximum likelihood estimates, and the 97.5% percentile estimates of Θ and 4Nm. ML, maximum likelihood.
If we know the migration model and need to estimate only 7 instead of 16 parameters, the maximum likelihood estimates are almost identical to those for the parameter-rich model shown in Table 2, but the profile confidence intervals are slightly smaller: the coefficients of variation (CV) of the parameters are about 25% smaller than with the full model. For example, with 7 parameters, the CV for γ12 is 0.299, whereas for the full model, the CV for γ12 is 0.392.
Convergence of Our Metropolis–Hastings Sampling Method.
Our Metropolis–Hastings Markov chain algorithm is irreducible, as it can reach any possible genealogy from any other. However, it may take a very long time until the proper regions of the parameter space are found, because the algorithm is sensitive to the start parameters. This problem is specific not to our algorithm but to any MH algorithms that draw correlated samples. There is no simple criterion to judge when the program has converged to the best possible answer. Several convergence measures have been suggested, but there is no guarantee that convergence occurs in a given run (30). We have used a simple graphical method to explore convergence of the two-parameter model (Fig. 3). A single-locus data set of DNA sequences with 500 bp with 100 individuals sampled from a symmetric model with 4 populations was generated with Θ = 0.01 and ℳ = 100. We ran four cases each with starting parameters (Θ0, ℳ0) equal to (0.0001, 2), (0.001, 200), (0.1, 200), and (0.1, 2) (see Fig. 3). Each run had 10 short chains, each with a total of 20,000 genealogies, and 3 long chains each with a total of 110,000 genealogies. The first 10,000 genealogies in each chain were discarded. At the end of each chain, the parameters were estimated and recorded, and the next chain was then started with these new parameters. These four cases were then compared with a very long run (five times longer) that started from estimates Θ0 and ℳ0, which were based on FST estimates.
Figure 3.

Convergence of parameter estimates in migrate. Four runs by using different initial parameter settings in migrate: (A) Θ0 = 0.1 and ℳ0 = 2, (B) 0.001 and 2, (C) 0.1 and 200, (D) 0.001 and 200. E shows run that was started with values from an FST-based method with Θ0 = 0.0097 and M0 = 97. F compares the average surface of A–D with E. In A–E, the line marks the trajectory of the parameter estimates over successive chains to the final estimate (black disk). Solid contour lines depict approximate 50 and 95% likelihood-based confidence regions. Gray-scale contour areas are from dark to light, 50, 95, 99% confidence regions of the average of A–D. The data set was generated by using Θ = 0.01 and M = 100 (dashed lines).
The starting points 𝒫0 chosen for this convergence study are fairly far from the true parameter values (see ref. 31), but the adaptive improvement of the 𝒫0 moves gradually toward parameters of highest likelihood for this data set, as can be seen in Fig. 3: in A, B, D, and F, the trajectories are moving toward values close to the true parameters, namely toward the maximum likelihood estimates for this specific data set, 𝒫data. In Fig. 3C, the trajectory first moves toward high ℳ values (256.4, outside of the shown frame) while staying at low Θ (0.0066) and then returns toward 𝒫data.
Comparison with genetree.
genetree (11) can use only sequence data that evolve according to an infinitely-many-sites model. To approximate this model, we simulated data according to the Kimura two-parameter model, but when more than one mutation occurred on the genealogy for a given site, we split the site into two or more new sites, so that each of these new sites would have mutated only once or not at all. We simulated sequence data for 100 loci with 500 bp each that evolved according to this infinite-sites model with 2 populations with 2 sampled individuals each. The subpopulations had the same size (Θi = 0.002) and had a symmetric migration model with rate 4Nm = 0.1. This data set was analyzed with genetree after removal of the invariant sites and also analyzed with migrate for the full set of sites. We chose to evaluate a data set with few individuals and these parameters so that we can compare the outcomes of both programs independent of their ability to search the genealogy space. With genetree, we sampled 1,000 genealogies per locus and used the true parameters to run the sampling process. Our true parameters defined the driving parameters for genetree to be θ0 = 4, and 4Nm0 = 0.2. The θ0 and 4Nm0 used in genetree are computed from the mutation rate per locus, and N is the size of the whole population. migrate was run at its default values, except for the following settings: the starting 𝒫0 were set to the true values, and the lengths of sampling were set to 10 short chains each of 400 genealogies, of which the first 100 were discarded and then every third genealogy used for the parameter estimation, and then two long chains with 1,600 genealogies each, of which the first 100 were discarded and then every third one was used for parameter estimation. This was a total of 7,200 genealogies visited per locus.
With a large number of loci, one expects that the results should converge to the values used to generate the data. With 100 loci and only 4 sampled individuals, there is much uncertainty about the parameters, but both programs include the true parameter values in their 50% confidence regions (Fig. 4).
Figure 4.

Comparison between results from migrate and genetree. The logarithm likelihood contours inferred by migrate are drawn with solid lines, and those inferred by genetree with broken lines. Contour lines enclose approximate 50, 95, and 99% likelihood-based confidence regions. The dashed lines mark the true parameter values used to generate the data set.
migrate spent 45 min for the whole 100-locus data set on a computer with a 166-MHz Pentium processor, running linux. It was sampling ≈271 genealogies with 4 tips per second; genetree spent roughly 350 min on the same computer, and it evaluated about 5 genealogies per second. However, migrate's genealogies are autocorrelated, whereas genetree's are independently sampled.
Example Data Set.
For a real-world example, we used aligned mtDNA control region domain I sequences of populations from the Nile valley, described by Krings et al. (32). The aligned sequences were taken from the compilation of Handt et al. (33). We chose the following three groups: Egypt (79 sequences), Nubia (69 sequences), and Sudan (79 sequences). These 225 sequences certainly violate several of the assumptions migrate is based on: the “populations” are assemblages of local populations, some or all of the population sizes were not constant, and migration rates between the populations were most likely not constant over time.
In our analysis, we ignored the existence of unsampled populations but took into account that mutation rates of the mtDNA control region Domain I data are heterogenous among sites by using a Hidden Markov Model with F84 mutation model with 4 rates (0.025, 0.239, 0.787, 2.354) with equal probability (34). This is an approximation to a Gamma distribution with α = 0.3 (see ref. 35). We used a transition–transversion ratio of 15 and the empirical base frequencies (A: 0.3302, C: 0.3313, G: 0.1161, T: 0.2225). We analyzed the data by using starting Θi of 0.5 and ℳji of 5.0. Because of the size of the problem, we used a Metropolis-coupled MH algorithm (36) with four independent chains that accept at different rates. The chain that was used for the estimates uses an unmodified acceptance ratio, whereas the others accept more often. Switching between neighboring chains followed the approach of Kuhner and Felsenstein (37).
The results of the analysis are shown in Table 3. The gene flow among the populations seems to be moderate, except that there is considerable gene flow from Egypt into Nubia and from Sudan into Nubia.
Table 3.
Gene flow between three human populations (Egypt, Nubia, Sudan) in the Nile valley (32)
| Population i | Θ | 2Nfm
|
||
|---|---|---|---|---|
| Sudan → i | Nubia → i | Egypt → i | ||
| Sudan | 0.122 | — | 3.19 | 3.70 |
| 0.094–0.158 | 1.70–5.61 | 2.06–6.29 | ||
| Nubia | 0.107 | 37.50 | — | 28.41 |
| 0.072–0.162 | 27.04–54.97 | 19.11–43.16 | ||
| Egypt | 0.108 | 5.14 | 4.45 | — |
| 0.169–0.372 | 2.83–8.78 | 2.38–7.78 | ||
A total of 225 mtDNA control region Domain I sequences from the database of (ref. 33) were analyzed. The maximum likelihood estimate and the 95% profile confidence intervals of population sizes Θ = 2Nfμ, where Nf is the effective population size of females and μ is the mutation rate per generation and per site, and 2Nfm, the number of immigrant females per generation, are shown. The receiving populations are in the rows.
Discussion
The present method allows us to analyze a wide range of different population models. It allows us to estimate as many as n population sizes Θi and n(n − 1) immigration rates ℳij or as few as two parameters, a Θ that is equal for all subpopulations and an ℳ that is the same between all pairs of subpopulation. With the migration connection matrix, one can analyze arbitrary migration models where some migration routes are not allowed. This versatility allows us to consider biologically relevant migration scenarios and to put some of the complication under the control of the user.
The MH technique for this method is identical to that described in ref. 10. Our method wanders through the sample space by proposing local changes on a genealogy and rejecting or accepting such a changed genealogy according to their likelihood. These changes in genealogy are reversible: we showed (10) that this branch insertion and removal process allows us to connect any two genealogies with a modest number of rearrangements, and that genealogies are sampled in proportion to Prob(𝒢|𝒫0) Prob(𝒢|𝒟). This MH sampler will converge to the correct answer when run for an infinitely long time, but of course our hope is that convergence will be achieved much earlier. For simple population scenarios, such as in our simulations of the n-island model, we can get similar parameter estimates even from bad starting parameters (Fig. 3). Our adaptive scheme using many short chains and a few long chains helps move the sampler into regions with genealogies that have high probabilities. As a result, estimates are more accurate than if they came from a single long chain run at an arbitrary 𝒫0. For practical purposes, this self-targeting process for finding the appropriate distribution is important, as it seems less desirable to rely on the user to find good starting parameters or to ask the user to restart the estimation process many times.
For large data sets, the researcher may need to run migrate several times with different chain lengths to see whether the length and the number of the chains are influencing the result. Alternative strategies are the use of Metropolis coupling (36) (e.g., our real-world example) or summarizing over different chains (15, 38) or even over different runs. These extensions will improve results but will increase the time for analysis considerably. In genetree (11), which is currently the only competing coalescent likelihood program, no such adaptive scheme is used, and the researcher needs to find appropriate starting parameters by doing the iterations by hand. Our and Bahlo and Griffiths' schemes will produce strange estimates when the starting parameters are far from the truth (Fig. 3; ref. 31). Bayesian approaches, although they vary the parameters of interest during the sampling process, will have similar problems if they are based on MH: with increasing numbers of parameters, the search space gets larger and much more sampling needs to be done to produce a proper posterior distribution. So far, there is no Bayesian method for analyzing migration models by using the coalescent, except for the two-population method developed by Nielsen and Slatkin (39).
The comparison with genetree shows that for the cases chosen, both methods deliver similarly shaped likelihood surfaces, as they should, because both are approximating the same likelihood criterion (17). For this data set, genetree has slightly wider confidence intervals in the Θ direction than migrate (Fig. 4). When using only a single population, both programs deliver almost identical likelihood curves and therefore confidence intervals (data not shown). The small differences might be caused by the different assumptions about the mutation model or by the different distributions from which the programs sample their genealogies.
Advantages of migrate over the current version of genetree are that the researcher can take into account different mutation models, such as the infinite allele model, a stepwise mutation model for microsatellite data, and sequence evolution models with rate heterogeneity among sites (34). All models can be combined with a model of rate heterogeneity among loci (10).
In the simulations tests of an n-island migration model, the averages of the two-parameter model are rather close to the values used to generate the data sets but are most often slightly smaller. This is in stark contrast to theoretical results showing that expectations of parameters over all simulations do not exist or at least are highly biased upwards (41). There exists a very small but nonzero probability that the data are compatible with a genealogy of infinite length. If such a data set is encountered in a simulation study, the program will return very large parameter estimates. The distributions of the 100 10-locus estimates from the simulation have heavy right tails (skewness S for Θ is 0.063 and for 4Nm is 0.11). The skewness is more pronounced if we look at the distribution of the 1,000 single locus estimates (SΘ = 0.195, S4Nm = 0.896). In fact, there is an upwards bias that is reflected in the skewness but not much in the averages because in these 100 simulation runs, we have not yet encountered a very high parameter value. On the other hand, there is a “fatal attraction” to zero: once a parameter becomes very small, it is unlikely that our adaptive MH procedure will succeed in reaching higher parameter values, because the events that are under the control of that parameter are not proposed and therefore subsequent parameter estimates tend to stay small.
For some population structures, such as a hidden source–sink scenario with large gene flow from a large population to a small population, results are not very enlightening without additional information about the migration structure. It remains to be shown that other methods are superior for these kinds of data. We expect that when we do not know the migration structure, many loci and many individuals will be needed to detect the few new mutations in the small sink population. Only then would we be able to see whether any of these rare mutations migrate back into the source population.
The difficulty of retrieving the true parameters increases with the number of parameters. Estimates for two parameters show smaller profile confidence intervals than the estimates with 16 parameters, but if the true population structure is complicated, the two-parameter model also delivers much less information than a more parameter-rich model.
Krings et al. (32) infer gene flow in the Nile valley by using analyses of molecular variance (AMOVA) (40). Their findings coincide with ours, in that Nubia seems to have received a considerable number of genes from Egypt and Sudan. The population sizes are most likely inflated, because we did not take into account that the individual populations are substructured, and because all populations exchange migrants with their other neighbors, who contribute genetic variation which we do not account for in our analysis.
The rather large range of possible values for the migration parameters in the example of possible migration directions in the Nile valley makes it evident that single locus data, even if it is highly variable, does not help much in clarifying current discussions in anthropology. Additional unlinked loci, each with its own coalescent history, can reduce this uncertainty greatly (10, 17).
Conclusion
We have presented three lines of evidence that our method works even for rather complicated migration models: derivation of the MH sampling strategy (10, 15, 16), simulation to test convergence to true parameters, and simulations to assess biases and to see whether the method achieves results that are reproducible and can be found in a reasonable amount of time.
Maximum likelihood methods for the estimation of population parameters, as implemented in migrate or genetree, will make the current FST-based estimators obsolete, because these do not take into account the genealogical relationship of the sample and the possibility of asymmetry in gene flow. However, the practical use of these new programs is limited to few subpopulations or few parameters because of current lack of computation power. Incorporation of the machinery of migrate into a program that can handle additional forces such as recombination and population growth is under way in our laboratory. The program migrate is available at http://evolution.genetics.washington.edu/lamarc.html.
Acknowledgments
We thank M. K. Kuhner and J. Yamato for many discussions and comments on the manuscript, and R. C. Griffiths and J. Wakeley for helpful comments on the manuscript. This work was funded by National Science Foundation grant no. BIR 9527687 and National Institutes of Health grants nos. R01 GM 51929, R01 GM 01989, all to J.F.
Abbreviation
- MH
Metropolis–Hastings Markov chain Monte Carlo approach
References
- 1.Wright S. Genetics. 1952;37:312–321. doi: 10.1093/genetics/37.3.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hudson R R, Slatkin M, Maddison W P. Genetics. 1992;132:583–589. doi: 10.1093/genetics/132.2.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slatkin M. Genetics. 1993;139:457–462. doi: 10.1093/genetics/139.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Excoffier L, Smouse P E. Genetics. 1994;136:343–359. doi: 10.1093/genetics/136.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weir B S. Genetic Data Analysis II. Sunderland, MA: Sinauer; 1996. [Google Scholar]
- 6.Rousset F. Genetics. 1996;142:1357–1362. doi: 10.1093/genetics/142.4.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beerli P. In: Advances in Molecular Ecology, NATO Science Series A: Life Sciences. Carvalho G, editor. Vol. 306. Amsterdam: IOS Press; 1998. pp. 39–53. [Google Scholar]
- 8.Gaggiotti O E, Excoffier L. Proc R Soc London Ser B. 2000;267:81–87. doi: 10.1098/rspb.2000.0970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tufto J, Engen S, Hindar K. Genetics. 1996;144:1911–1921. doi: 10.1093/genetics/144.4.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beerli P, Felsenstein J. Genetics. 1999;152:763–773. doi: 10.1093/genetics/152.2.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bahlo M, Griffiths R C. Theor Popul Biol. 2000;57:79–95. doi: 10.1006/tpbi.1999.1447. [DOI] [PubMed] [Google Scholar]
- 12.Wakeley, J. (2000) Theor. Popul. Biol., in press.
- 13.Kingman J. In: Essays in Statistical Science. Gani J, Hannan E, editors. London: Applied Probability Trust; 1982. pp. 27–43. [Google Scholar]
- 14.Kingman J. Stochastic Processes and Their Applications. 1982;13:235–248. [Google Scholar]
- 15.Kuhner M K, Yamato J, Felsenstein J. Genetics. 1995;140:1421–1430. doi: 10.1093/genetics/140.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuhner M K, Yamato J, Felsenstein J. Genetics. 1998;149:429–434. doi: 10.1093/genetics/149.1.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felsenstein J, Kuhner M K, Yamato J, Beerli P. Lecture Notes—Monograph Series. Hayward, CA: IMS; 1999. 33, 163–185. [Google Scholar]
- 18.Kuhner M K, Yamato J, Felsenstein J. Genetics. 2000;156:1393–1401. doi: 10.1093/genetics/156.3.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swofford D, Olsen G, Waddell P, Hillis D. In: Molecular Systematics. Hillis D, Moritz C, Mable B, editors. Sunderland, MA: Sinauer; 1996. pp. 407–514. [Google Scholar]
- 20.Felsenstein J. Annu Rev Genet. 1988;22:521–565. doi: 10.1146/annurev.ge.22.120188.002513. [DOI] [PubMed] [Google Scholar]
- 21.Hudson R R. Oxford Surv Evol Biol. 1990;7:1–44. [Google Scholar]
- 22.Hammersley J M, Handscomb D C. Monte Carlo Methods. London: Methuen; 1964. [Google Scholar]
- 23.Chib S, Greenberg E. Am Stat. 1995;49:327–335. [Google Scholar]
- 24.Gilks W R, Richardson S, Spiegelhalter D J. Markov Chain Monte Carlo in Practice. Boca Raton, FL: Chapman & Hall/CRC; 1996. [Google Scholar]
- 25.Wright S. Genetics. 1931;16:97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sankoff D. SIAM J Appl Math. 1975;28:35–42. [Google Scholar]
- 27.Meeker Q, Escobar L A. Am Stat. 1995;49:48–53. [Google Scholar]
- 28.Hudson R R. Theor Popul Biol. 1983;23:183–201. doi: 10.1016/0040-5809(83)90013-8. [DOI] [PubMed] [Google Scholar]
- 29.Kimura M. J Mol Biol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 30.Kass R E, Carlin B P, Gelman A, Neal R M. Am Stat. 1998;52:93–100. [Google Scholar]
- 31.Stephens M, Donnelly P. Philos Trans R Soc London B. 2000;354:1–31. [Google Scholar]
- 32.Krings M, el Halim Salem A, Bauer K, Geisert H, Malek A K, Chaix L, Simon C, Welsby D, Di Rienzo A, Utermann G, et al. Am J Hum Genet. 1999;64:1166–1176. doi: 10.1086/302314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Handt O, Meyer S, von Haeseler A. Nucleic Acids Res. 1998;26:126–129. doi: 10.1093/nar/26.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Felsenstein J, Churchill G A. Mol Biol Evol. 1996;13:93–104. doi: 10.1093/oxfordjournals.molbev.a025575. [DOI] [PubMed] [Google Scholar]
- 35.Excoffier L, Yang Z. Mol Biol Evol. 1999;16:1357–1368. doi: 10.1093/oxfordjournals.molbev.a026046. [DOI] [PubMed] [Google Scholar]
- 36.Geyer C. In: Computing Science and Statistics. Keramidas E M, editor. Fairfax Station, VA: Interface Foundation; 1991. pp. 156–163. [Google Scholar]
- 37.Kuhner M K, Felsenstein J. Genetic Epidemiology. 2000;19,(Suppl. 1):S15–S21. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI3>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 38.Geyer C. Technical Report 568 R(4) Twin Cities, MN: Univ. of Minnesota; 1994. [Google Scholar]
- 39.Nielsen R, Slatkin M. Evolution (Lawrence, KS) 2000;54:44–50. doi: 10.1111/j.0014-3820.2000.tb00006.x. [DOI] [PubMed] [Google Scholar]
- 40.Excoffier L, Smouse P E, Quattro J M. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuhner M K, Beerli P, Yamato J, Felsenstein J. Genetics. 2000;156:439–447. doi: 10.1093/genetics/156.1.439. [DOI] [PMC free article] [PubMed] [Google Scholar]