

Abstract
The genome annotations of all sequenced Dehalococcoides strains lack a citrate synthase, although physiological experiments have indicated that such an activity should be encoded. We here report that a Re face-specific citrate synthase is synthesized by Dehalococcoides strain CBDB1 and that this function is encoded by the gene cbdbA1708 (NCBI accession number CAI83711), previously annotated as encoding homocitrate synthase. Gene cbdbA1708 was heterologously expressed in Escherichia coli, and the recombinant enzyme was purified. The enzyme catalyzed the condensation of oxaloacetate and acetyl coenzyme A (acetyl-CoA) to citrate. The protein did not have homocitrate synthase activity and was inhibited by citrate, and Mn2+ was needed for full activity. The stereospecificity of the heterologously expressed citrate synthase was determined by electrospray ionization liquid chromatography-mass spectrometry (ESI LC/MS). Citrate was synthesized from [2-13C]acetyl-CoA and oxaloacetate by the Dehalococcoides recombinant citrate synthase and then converted to acetate and malate by commercial citrate lyase plus malate dehydrogenase. The formation of unlabeled acetate and 13C-labeled malate proved the Re face-specific activity of the enzyme. Shotgun proteome analyses of cell extracts of strain CBDB1 demonstrated that cbdbA1708 is expressed in strain CBDB1.
INTRODUCTION
Dehalococcoides species are known for their ability to use a wide range of persistent halogenated compounds as terminal electron acceptors in an anaerobic respiration with hydrogen as the electron donor. Apart from this organohalide respiration, no other mode of energy conservation has been described for Dehalococcoides species, although several strains have been isolated and physiologically characterized (1, 4, 12, 13, 23). Also, genome analyses of several Dehalococcoides strains have not indicated further energy fixation capabilities (20, 24, 29).
The anabolism of Dehalococcoides species relies on acetate as a carbon source (1, 23). In our model organism, Dehalococcoides strain CBDB1, six acyl coenzyme A (acyl-CoA) (EC 6.2.1.3) or acetyl-CoA (EC 6.2.1.1) synthetases are annotated in the genome (locus tags are shown in Fig. 1), and the encoded enzymes are candidates for the activation of acetate to acetyl-CoA. Acetyl-CoA is used to generate biocomponents via different pathways (20). One of these pathways is the reversible reductive carboxylation of acetyl-CoA to pyruvate and is catalyzed by pyruvate:ferredoxin oxidoreductase (EC 1.2.7.1), all subunits of which are encoded in strain CBDB1. Together with oxaloacetate, acetyl-CoA should also be used to synthesize citrate via citrate synthase (CS) (EC 2.3.3.1); however, this key enzyme was not identified in the genome of strain CBDB1 or any other Dehalococcoides strain (2, 20, 24, 29, 32). In contrast to citrate synthase, several other enzymes of the citric acid cycle have been annotated: aconitase, isocitrate dehydrogenase, malate dehydrogenase, and fumarase (Fig. 1). According to the actual annotations, the genome of strain CBDB1 is lacking the genes for oxoglutarate dehydrogenase and succinate dehydrogenase, indicating that an incomplete citric acid cycle is operating, with an oxidative half-cycle to 2-oxoglutarate and a reductive half-cycle to fumarate. Such a “horseshoe-type” citric acid cycle has been reported for many anaerobic bacteria (see, e.g., references 14, 15, 18, and 21), along with aerobic bacteria such as Mycobacterium tuberculosis (33). These horseshoe-type reactions have no energetic function but serve solely anabolic purposes. Because ATP citrate lyase (EC 2.3.3.8), 2-oxoglutarate:ferredoxin oxidoreductase (EC 1.2.7.3), and fumarate reductase (EC 1.3.99.1) are missing in the annotation, CBDB1 seems also to be unable to catalyze a reductive citric acid cycle as shown for other anaerobic bacteria (16). Oxaloacetate is thought to be synthesized by pyruvate carboxylase (EC 6.4.1.2). A gene for pyruvate-water dikinase (EC 2.7.9.2), irreversibly forming phosphoenolpyruvate from pyruvate, is present in the genome of strain CBDB1 (cbdbA529) (20). Genes for phosphoenolpyruvate carboxylase or phosphoenolpyruvate carboxykinase are not annotated.
Fig. 1.

Metabolic pathways of acetate fixation and glutamine biosynthesis in strain CBDB1 inferred from annotation of the genome. Resulting isotopomer distributions for Re-type and Si-type citrate synthase reactions are shown. The carbon positions derived from [1-13C]acetate are marked in green, those from [2-13C]acetate in red, and carbons from hydrogen [13C]carbonate in blue. The measured l-glutamine data were in accordance with l-glutamine resulting from a Re-citrate synthase activity.
As described above, physiological and genomic studies together indicate that catabolic and anabolic reactions are strictly separated from each other in the metabolism of Dehalococcoides species, which is in contrast to the case for most other organisms, where catabolic and anabolic reactions are intertwined. Two key reactions at which such an interference of catabolic and anabolic functions can be normally seen are the phosphofructokinase/fructose-1,6-bisphosphatase and the citrate synthase steps, and both steps involve unusual enzymes in strain CBDB1 (28). In aerobic organisms, citrate synthase initiates both catabolic reactions of the citric acid cycle and anabolic reactions leading to the biosynthesis of, e.g., glutamate and glutamine via 2-oxoglutarate. Such citrate synthases have to be controlled in a complex way to balance catabolic and anabolic needs.
In most organisms, and in all aerobic organisms, citrate synthase is Si stereospecific with respect to the C-2 of oxaloacetate (Si-CS) (EC 2.3.3.1). However, some bacteria do not encode such a Si-CS but still show biochemical citrate synthase activity. For example, extracts of the anaerobic archaeon Ignicoccus hospitalis catalyzed the formation of citrate from oxaloacetate and acetyl-CoA, but a putative gene for citrate synthase was not detected (18). From their detailed analysis, the authors concluded that a Re-type citrate synthase (Re-CS) (EC 2.3.3.3) was present. Similar studies were recently done for several other strictly anaerobic bacteria (3, 5, 8, 32). Such atypical citrate synthases of the Re type were first observed in the strictly anaerobic firmicute Clostridium kluyveri (11) but were later also found in other strictly anaerobic bacteria (9, 10, 21). In contrast to Si-CS, the Re-CS was found to be oxygen sensitive and therefore appears to function only in anaerobic bacteria (21). Recently, Tang et al. (32) found biochemical evidence for a Re-CS also in Dehalococcoides strains by analyzing isotopically labeled protein hydrolysates.
In the present study we investigated the hypothesis that gene cbdbA1708 (NCBI accession number CAI83711, UniProt identifier Q3ZW76), annotated as encoding homocitrate synthase in the genome of strain CBDB1, encodes a Re-CS, fulfilling a central role in the metabolism of CBDB1. The hypothesis for our experiments was generated based on the facts that (i) a citrate synthase is missing in the annotation of strain CBDB1, (ii) a Re-CS activity was proposed by Tang et al. (32), (iii) a homocitrate synthase appears to be redundant in strain CBDB1 because the lysine biosynthesis seems to be catalyzed via the α-aminoadipate pathway (Discussion), (iv) some described homocitrate synthases showed low citrate synthase activity in in vitro assays (35), (v) homocitrate and Re-CS share similarity both in gene sequence and in biochemical mechanisms (21), and (vi) other possible candidates such as cbdbA803 and cbdbA808, annotated as encoding isopropylmalate synthases, appear to be necessary for leucine biosynthesis and are both located in the leucine biosynthesis operon of strain CBDB1.
MATERIALS AND METHODS
Chemicals.
Sodium acetate labeled with 99 atom% 13C at the first ([1-13C]acetate) or second ([2-13C]acetate) position and labeled sodium hydrogen carbonate (hydrogen [13C]carbonate, 98 atom% 13C) were acquired from Sigma-Aldrich-Isotec (Munich, Germany). Si-citrate lyase (EC 4.1.3.6) from Aerobacter aerogenes was purchased from Roche (Grenzach-Wyhlen, Germany). Si-citrate synthase (EC 2.3.3.1) from porcine heart, malic dehydrogenase (EC 1.1.1.37) from recombinant Escherichia coli, acetyl-CoA synthetase (EC 6.2.1.1), and amino acid standard solution were purchased from Sigma-Aldrich (Munich, Germany). CoA, acetyl-CoA, Tris-HCl, MnCl2, oxaloacetate, 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), malate dehydrogenase, NADH, and other chemicals used were of the highest available purity and were purchased from Sigma-Aldrich (Munich, Germany).
Heterologous expression of the gene cbdbA1708.
The 1,248 bp of the cbdbA1708 gene were amplified from chromosomal DNA of Dehalococcoides. For PCR, Pfu polymerase was used according to the instructions of the manufacturer (Promega, Mannheim, Germany). Clones were constructed using the vector pASK-IBA7plus (IBA, Göttingen, Germany). The digested PCR product was ligated into the BsaI site of the expression vector, resulting in plasmid pAeW15, which contains the Strep tag sequence at the N terminus of the enzyme.
E. coli BL21(DE3) harboring pAeW15 was cultivated in 10 ml LB supplemented with 100 μg ml−1 ampicillin (LB-Amp) overnight. The overnight culture was transferred into 100 ml LB-Amp and cultivated at 37°C until the optical density (OD) reached about 0.5. Half of this preculture was transferred in 500 ml LB-Amp, and the culture was further incubated at 37°C. At an OD of about 0.5, the expression of the cloned gene was induced by adding anhydrotetracycline (AHTc) in ethanol to a final concentration of 0.2 μg/ml. The culture was incubated for an additional 3 h, after which cells were collected by centrifugation (6,000 × g, 4°C, 10 min).
Purification of the cbdbA1708 gene product.
Cells collected from a 500-ml culture were suspended in 9 ml 0.1 M Tris-HCl (pH 8.0) containing 150 mM NaCl and disrupted by French press treatment (2 passages, 96.5 MPa). The cell homogenate was centrifuged at 16,000 × g for 20 min, and the supernatant was applied to a 1-ml Strep tag affinity column (IBA, Göttingen, Germany). Chromatography was performed according to the instructions of the supplier. However, a buffer without EDTA was used, since EDTA interferes with the following activity assay. Purification was performed in an anaerobic tent. The eluate was used for activity tests and enzymatic characterization.
Determination of citrate synthase activity.
Citrate synthase activity was determined photometrically at 412 nm using a microplate reader (SynergyHT; BioTek Instruments, Winooski, VT) in an anaerobic tent. DTNB reacts with CoA, and the enzyme activity was assayed by monitoring the formation of the chromophoric thionitrobenzoate. The molar absorption coefficient is 13.6 × 103 M−1 cm−1. Assays modified from those described by Danson and Hough (6) were performed in 0.1 M Tris-HCl (pH 8.0) containing 0.1 mM acetyl-CoA, 0.2 mM oxaloacetate, 0.2 mM DTNB, and 0.2 mM MnCl2 in a final volume of 0.2 ml. The reaction was started by the addition of about 50 μg protein. Because of a slow spontaneous hydrolysis of acetyl-CoA, controls were set up containing protein and acetyl-CoA but no 2-oxo substrate. Using the same sample, standard deviations obtained with this test were calculated as ±29% (n = 8). The detection limit of this test was about 0.25 nkat ml−1 (15 mU ml−1).
The Michaelis-Menten constant (Km) and the maximum velocity of the enzyme (Vmax) were graphically determined. To determine the Km for acetyl-CoA, oxaloacetate concentrations were held constant at 0.2 mM and acetyl-CoA was varied between 0.01 and 0.3 mM. To determine the Km for oxaloacetate, acetyl-CoA concentrations were held constant at 0.1 mM and oxaloacetate was varied between 0.005 and 0.25 mM. We used mean values from triplicate determinations. For the analysis of the substrate specificity, oxoglutarate (0.5, 1, or 10 mM) and pyruvate (1 or 10 mM) also were used.
Protein concentrations were determined using the method of Bradford with serum albumin as the standard.
In vivo stereospecificity of citrate synthase in cultures of Dehalococcoides strain CBDB1.
The stereospecificity of citrate synthase in strain CBDB1 was determined by studying the glutamine labeling pattern when the bacterium was grown with 13C-labeled carbon sources. Dehalococcoides strain CBDB1 was cultured under strictly anaerobic conditions in 60-ml glass serum bottles containing 30 ml medium and 30 ml gas phase (N2-CO2, 80:20 [vol/vol]) (1). A synthetic mineral medium containing vitamins, trace elements, and 5 mM sodium acetate as a carbon source was reduced with 0.8 mM Ti(III) citrate, as previously described (1). Perchloroethylene (PCE) was used as an electron acceptor and added in doses of 50 μM, and all the cultures were fed with H2 (+0.3 bar) as an electron donor (22). All cultures were set up in triplicate. Three different experiments were performed to study carbon fluxes to glutamine: (i) cultures containing 5 mM [1-13C]acetate and 30 mM unlabeled carbonate as pH buffer, (ii) cultures containing 5 mM [2-13C]acetate and 30 mM unlabeled carbonate, and (iii) cultures containing 5 mM unlabeled acetate and 30 mM hydrogen [13C]carbonate. Unlike standard cultures, cultures with labeled carbonate were not gassed with N2-CO2 to avoid contamination with unlabeled carbonate. However, some unlabeled carbonate was still present in the medium as a component in the reducing agent Ti(III) that was neutralized with carbonate during preparation. This unlabeled carbonate accounted for about 10% of the total carbonate. To remove unlabeled acetate traces from the inoculum, strain CBDB1 was repeatedly transferred (5% [vol/vol] inoculum, at least four times) to fresh medium containing the corresponding labeled substrate before being collected for glutamine analysis.
Glutamine was determined mainly following the method of Silfer et al. (30). Cells of strain CBDB1 were collected by passing 200 ml of culture medium containing 5 × 107 to 8 × 107 cells ml−1 through a 0.2-μm cellulose acetate filter. The retained cells were lysed, and proteins were fully hydrolyzed by backflushing the filter with 2 ml of 6 M HCl and incubating eluates at 110°C for 22 h. Hydrolysates were then dried under a nitrogen stream. For derivatization, samples were incubated with 1 ml of water-free isopropanol and 250 μl of acetyl chloride overnight at 70°C to obtain the corresponding isopropylesters. The esterified samples were dried under nitrogen and then acetylated by applying a mixture of 500 μl of dichloromethane and 500 μl of trifluoroacetic anhydride for 1 h at 60°C. After removal of residual dichloromethane and trifluoroacetic anhydride by evaporation under nitrogen, the derivatives were dissolved in 100 μl dichloromethane. An external standard was prepared by dissolving 500 μl of a commercial amino acid standard solution. In addition, an internal standard was included by adding 200 μl of 0.5-mg ml−1 trans-4-(aminomethyl)-cyclohexanecarboxylic acid to each sample. The derivatized sample was analyzed by using an Agilent 7890A gas chromatograph coupled to an Agilent 5975C mass spectrometer with triple-axis detector (Agilent, Palo Alto, CA). The derivatized amino acids were separated on a DB5 column (30 m by 0.25 mm by 0.25 μm; Agilent) by injection of a 3-μl sample with a split ratio of 1:1 using the following temperature program: initial temperature of 40°C held for 2 min; then temperature increased from 40°C to 160°C at 10°C min−1, then from 160°C to 190°C at 4°C min−1, and finally from 190°C to 300°C at 10°C min−1; and temperature held for 10 min. Glutamine was identified by comparison of the retention time and mass spectrum with those of the glutamine derivatized from the amino acid standard solution.
In vitro stereospecificity of the purified cbdbA1708 gene product.
Citrate was synthesized from oxaloacetate and [2-13C]acetyl-CoA with the heterologously expressed and purified cbdbA1708 gene product. A parallel experiment under the same conditions but using a commercially available Si-citrate synthase instead of the heterologously expressed cbdbA1708 gene product was included for comparison. The resulting labeled [13C]citrate was subsequently cleaved to oxaloacetate plus acetate with Si-citrate lyase in the presence of NADH and malate dehydrogenase so that the oxaloacetate formed was converted to malate. Therefore, the expected products formed from Si-citrate synthase were malate plus [2-13C]acetate, whereas in the case of Re-citrate synthase the products were [2-13C]malate plus acetate (21). The stereospecificity of the purified enzyme was corroborated by detecting 13C-labeled and unlabeled malic acid produced during the citrate cleavage both in the Si-citrate synthase and in the recombinant enzyme assays. The details of the reactions were as follows.
(i) Synthesis of [13C]citrate.
The 10-ml reaction mixture contained 0.1 M Tris-HCl (pH 8.0), 0.2 mM MnCl2, purified Re- or Si-citrate synthase (0.5 and 0.15 U, respectively), 2.5 mM ATP, 1 mM oxaloacetic acid, 1 mM [2-13C]acetate, 1 mM CoA, and 5 U acetyl-CoA synthetase. The reaction was performed under anaerobic conditions in an anaerobic tent. After 2 h of incubation at 25°C, the reaction mixture was heated to 95°C for 10 min and denatured proteins were removed by centrifugation.
(ii) Cleavage of [13C]citrate.
The sample was concentrated to dryness by flash evaporation and redissolved in a 1-ml reaction mixture containing 0.1 M Tris-HCl (pH 7.3), 0.2 mM MgCl2, 18 U malate dehydrogenase, 0.6 mM NADH, and 0.25 U Si-citrate lyase. After 2 h of incubation at 25°C, the reaction mixture was heated to 95°C for 10 min and denatured proteins were removed by centrifugation.
(iii) Analysis of labeled and unlabeled malic acid by electrospray ionization liquid chromatography-mass spectrometry (ESI LC-MS).
Chromatographic analysis of the supernatant was carried out on a Shimadzu model 2010A liquid chromatograph and mass spectrometer (Shimadzu, Kyoto, Japan) with a UV detector at 210 nm. The isocratic mobile phase contained 0.03% (vol/vol) formic acid in water and was delivered at a flow rate of 0.6 ml min−1. The column temperature was 25°C, and a sample volume of 20 μl was injected from an autosampler. Chromatographic separation of malic acid was achieved on an ICSep COREGEL-87H3 column (250 mm by 4 mm) (Transgenomic, Glasgow, United Kingdom). The nebulizer gas flow was set at 0.5 ml min−1, the detector voltage was set to 1.65 kV, and the instrument operated in negative mode. All MS data were collected in a full-scan m/z range of 10 to 500 Da. Malic acid was identified by comparison of the retention time and mass spectrum with that of a standard solution containing 20 mM malic acid.
Protein extraction, peptide preparation, and nano-LC-MS Orbitrap analyses.
Cells from 30-ml cultures of Dehalococcoides strain CBDB1 grown under strictly anaerobic conditions with PCE were harvested by filtration through a 0.2-μm cellulose acetate filter, backflushed with anaerobic water, and subsequently centrifuged for protein extraction. The collected cells were suspended in 50 mM ammonium bicarbonate, and cell lysis was performed by a freeze/thaw step and a 30-s ultrasonic bath treatment. Prevention of methionine oxidation and carbamidomethylation of cysteines were performed by a treatment with 50 mM (final concentration) dithiothreitol at 30°C for 1 h and 100 mM (final concentration) iodacetamide at ambient temperature for 1 h in the dark. Trypsin (0.6 μg) was added, and the mixture was incubated at 37°C for 16 h. The reaction was stopped by adding 0.1% (final concentration) formic acid. Peptides were purified by using C18 Zip Tip columns (Millipore).
Peptides were reconstituted in 0.1% formic acid, injected by use of an autosampler, and concentrated on a trapping column (nanoAcquity UPLC column, C18, 180 μm by 2 cm, 5 μm; Waters, Eschborn, Germany) with water containing 0.1% formic acid at a flow rate of 15 μl min−1. After 6 min, the peptides were eluted on a separation column (nanoAcquity UPLC column, C18, 75 μm by 100 mm, 1.7 μm; Waters, Eschborn, Germany). Chromatography was performed by using 0.1% formic acid in solvents A (100% water) and B (100% acetonitrile), with peptides eluted over 90 min with a 6 to 40% solvent B gradient using a high-pressure liquid chromatography (nano-HPLC) system (nanoAcquity; Waters) coupled to an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Continuous scanning of eluted peptide ions was carried out between 400 and 1,400 m/z, automatically switching to MS/MS collision-induced dissociation (CID) mode for ions exceeding an intensity of 3,000.
Raw data were processed for database search using Thermo Proteome Discoverer software (v. 1.0, build 43; Thermo Fisher Scientific). The search was performed with tandem mass spectrometry ion search algorithms from the Mascot house server (v2.2.1) (27). The following parameters were selected: Taxonomy ID 255470 of NCBInr (National Center for Biotechnology Information, Rockville, MD) as criterion for taxonomy, tryptic cleavage, and maximum of two missed cleavage sites. A peptide tolerance threshold of ±10 ppm and an MS/MS tolerance threshold of ±0.2 Da were chosen. Carbamidomethylation at cysteines was given as static and oxidation of methionines as variable modification. Peptides were considered to be identified by Mascot when a false-positive probability of <0.05 (probability-based ion score threshold of >40) was achieved.
Phylogenetic analyses.
Local sequence similarities were found by using Blastp implemented in UniProt (http://www.uniprot.org), and selected amino acid sequences were retrieved, including those for which biochemical data were available. Amino acid sequences were aligned with ClustalW implemented in MEGA4 (31). Trees were constructed with MEGA4 using the neighbor-joining or minimum-evolution method, with Poisson correction and 500 bootstrap replications.
RESULTS
Evidence of a Re-CS in strain CBDB1.
The labeling pattern of glutamine in protein hydrolysates of strain CBDB1 grown with either unlabeled carbon sources, [1-13C]acetate, [2-13C]acetate, or 13C-labeled hydrogen carbonate was examined in parallel experiments, each in triplicate. After harvesting of the cells, hydrolysis of proteins, and derivatization, glutamine isotopomers were analyzed by gas chromatography (GC)-MS. Two types of positively charged derivatized glutamine fragments were identified by mass spectrometry (see Fig. S1 in the supplemental material): a fragment that contained all the glutamine carbons but had released a 59-Da fragment from the ester bond ([M − 59]+) and another fragment that had released an 87-Da group containing the first carbon (α-carboxyl group) of the amino acid ([M − 87]+).
Strain CBDB1 incorporated two labeled carbons into the glutamine carbon skeleton when it was grown with either [1-13C]acetate or [2-13C]acetate, as observed from the difference in the molecular weights of fragment [M − 59]+ in cultures growing with unlabeled substrates (see Fig. S2A in the supplemental material, m/z = 226) and [1-13C]acetate (see Fig. S2B, m/z = 228) or [2-13C]acetate (see Fig. S2C, m/z = 228), respectively. In contrast, only one label was incorporated if the cells were grown with 13C-labeled hydrogen carbonate (see Fig. S2D, m/z = 227). The analysis of the [M − 87]+ fragment provided additional information about the origin of the first carbon atom of glutamine, because the [M − 87]+ fragment does not contain this atom. The gain of only 1 Da in fragment [M − 87]+ in cultures growing with [1-13C]acetate (see Fig. S2B, m/z = 199) in comparison with those growing with unlabeled substrates (see Fig. S2A, m/z = 198) showed that the α-carboxyl group of glutamine originated from the carboxyl carbon of acetate.
Heterologous expression and purification of the cbdbA1708 gene product.
From genome analyses it was hypothesized that gene cbdbA1708 was misannotated in strain CBDB1 and in fact encodes the Re-type citrate synthase. To biochemically study the gene product, gene cbdbA1708 was heterologously expressed in E. coli together with a N-terminal Strep tag, and the recombinant enzyme was purified by affinity chromatography. The gene cbdbA1708 encodes a 415-amino-acid polypeptide that was calculated to have a mass of 45.6 kDa without the Strep tag and a calculated isoelectric point of 5.5. Heterologous expression of the gene yielded a protein with the expected molecular mass as determined by SDS-PAGE.
Biochemical characteristics of the cbdbA1708 gene product.
The citrate, homocitrate, citramalate, and isopropylmalate synthases belong to a group of transferases that all transfer an acetyl group from acetyl-CoA to a 2-oxo-acid and release CoA (Table 1). We therefore analyzed the substrate spectrum of the expressed cbdbA1708 gene product. In all assays, acetyl-CoA was given as a substrate together with different 2-oxo-acids, and the release of CoA was monitored photometrically using DTNB. With the cbdbA1708 gene product, neither 2-oxoglutarate nor pyruvate resulted in CoA release above the detection limit. Only with oxaloacetate did the overexpressed protein show a clear reaction in the assay. The purified cbdbA1708 gene product (about 0.05 mg protein per assay) catalyzed the synthesis of citrate from oxaloacetate and acetyl-CoA with a specific activity of 18.3 nkat mg−1 (1.1 U mg−1) protein.
Table 1.
Acetyl-CoA-condensing enzymes and their annotation in strain CBDB1a
| Enzyme | EC no. | Substrateb | Productc | Annotation in CBDB1 genome |
|---|---|---|---|---|
| Si-citrate synthase | 2.3.3.1 | Oxaloacetate | Citrate | Not annotated |
| Re-citrate synthase | 2.3.3.3 | Oxaloacetate | Citrate | Not annotated |
| Homocitrate synthase | 2.3.3.14 | 2-Oxoglutarate | Homocitrate | cbdbA1708 |
| Isopropylmalate synthase | 2.3.3.13 | 2-Oxoisovalerated | 2-Isopropylmalate | cbdbA803, cbdbA808 |
| Citramalate synthase | 2.3.1.182 | Pyruvate | Citramalate | Not annotated |
| Malate synthase | 2.3.3.9 | Glyoxylate | Malate | Not annotated |
Our present study shows that gene cbdbA1708 is misannotated and represents the Re-citrate synthase in strain CBDB1.
Substrate to which acetyl-CoA is bound. In addition, water is needed to hydrolyze CoA.
In addition, CoA is released.
Minor activity with pyruvate to citramalate (7).
The effects of different additions on the activity of the cbdbA1708 gene product with oxaloacetate and acetyl-CoA were tested (Fig. 2). Under standard conditions, 0.2 mM Mn2+ was present. Such controls showed only slightly higher activity than in assays in which Mn2+ was omitted. The enzyme was inhibited by citrate. The activity dropped by 54% in 10 mM citrate and by 73% in 20 mM citrate. Lysine, 2-oxoglutarate, and AMP did not have an apparent influence on the activity. An inhibitory effect of 5 mM ATP was reversed by the addition of 5 mM MnCl2. The activity responded to changing substrate concentrations in a Michaelis-Menten-type behavior. The Km values for acetyl-CoA and for oxaloacetate were determined to be 75 μM and 50 μM, respectively. Graphically determined Vmax values were 9.1 μmol l−1 s−1 and 11.2 μmol l−1 s−1 for acetyl-CoA and oxaloacetate, respectively.
Fig. 2.
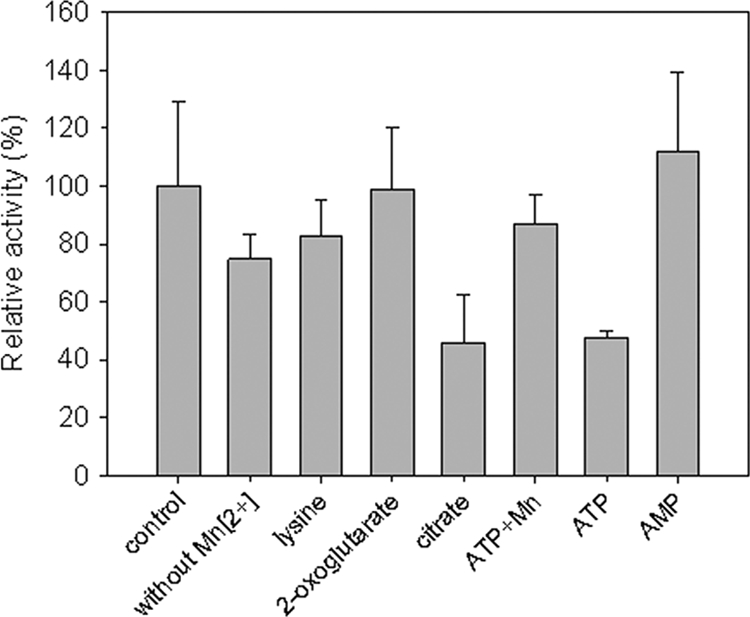
Effectors of enzyme activity of citrate synthase. Enzyme reactions were performed as described in Materials and Methods and in the presence of activators/inhibitors at the following concentrations: lysine, 0.5 mM; 2-oxoglutarate, 10 mM; citrate, 10 mM; ATP, 5 mM; and AMP, 0.15 mM. All assays mixtures except that for the sample designated “without Mn2+” contained 0.2 mM Mn2+, as described in Materials and Methods. Shown are means for triplicate assays ± standard deviations (SD). The mean of the control activity was set to 100%.
The activity was oxygen sensitive. Incubation at ambient oxygen pressure resulted in a loss of 25% of the activity after 1 h, 40% after 4 h, and 85% after 24 h.
Re-citrate stereospecificity of the recombinant enzyme.
We tested whether the heterologously expressed enzyme also had Re-type stereospecificity in vitro. The recombinant enzyme was used to synthesize citrate from oxaloacetate and [2-13C]acetyl-CoA. For comparison, the same reaction was set up with commercial Si-type citrate synthase. To analyze the 13C distribution, the produced citrate was cleaved to oxaloacetate and acetate by Si-citrate lyase and the formed oxaloacetate was further converted to malate by malate dehydrogenase. The final products of these reactions were analyzed by ESI LC-MS in full-acquisition mode, and malic acid was identified at a retention time of about 9.13 min by comparing the chromatograms with the retention time of a standard solution of malic acid and by analyzing the mass spectra. The deprotonated molecular ion [M − H]− of non-isotopically labeled malic acid showed an m/z value of 133 (see Fig. S3A in the supplemental material). When the recombinant citrate synthase was used for the experiment, the resulting malate showed an m/z value of 134, revealing that this ion fragment contained one 13C atom (see Fig. S3C). This demonstrated that the expressed citrate synthase was Re stereospecific. In contrast to these results, malic acid with the predicted m/z of 133 was formed when commercial Si-citrate synthase was used, corroborating the Si stereospecificity of the commercial enzyme (see Fig. S3B) (21).
Identification of cbdbA1708 expression by shotgun proteomics.
To evaluate the in vivo expression of cbdbA1708, seven independent protein shotgun analyses were run, each from a single CBDB1 culture grown with PCE as an electron acceptor. In all seven runs, peptides of the cbdbA1708 gene product were identified. Numbers varied between 3 and 8 peptides per analysis, and protein coverage varied between 10 and 27%. Figure S4 in the supplemental material provides an overview of the peptides found. The corresponding exponentially modified protein abundance index (emPAI) values were between 0.23 and 1.03. emPAI values give an estimate of protein abundance (17). Ranking all proteins in an analysis according to the emPAI value revealed that the cbdbA1708 gene product fell between rank 59 and 149 out of about 150 to 200 identified proteins in the different analyses. This calculation indicates that the protein CbdbA1708 is abundantly expressed in strain CBDB1.
Phylogenetic analysis of CbdbA1708.
Comparison of the amino acid sequence of CbdbA1708 with other published sequences reveals that CbdbA1708 is not related to Si-citrate synthases. In contrast, CbdbA1708 is a member of a large cluster of sequences annotated, e.g., as 2-isopropylmalate, citramalate, homocitrate, or Re-citrate synthase. However, only very few of these proteins are biochemically characterized, and no substantiated functional prediction is possible from the alignments and trees (Fig. 3). CbdbA1708 forms a tight cluster with the Dehalococcoides gene products DehalGT_1408, DehaBAV1_1360, DET1614, and DhcVS_1496 and gene products from Dehalogenimonas lykanthroporepellens (Dehly_1331), Thermodesulfovibrio yellowstonii (THEYE_A1277), and Elusimicrobium minutum (Emin_0401). The gene products CbdbA803 and CbdbA808 form different clusters together with respective gene products from other Dehalococcoides strains. The previously biochemically described Re-CS from Clostridium kluyveri (21) is only distantly related to CbdbA1708. Whereas the putative Re-CS of Clostridium acetobutylicum (3, 5) clusters with the Clostridium kluyveri Re-CS, the putative Re-CS of Thermoanaerobacter sp. strain X514 (8) is more closely related to the Re-CS identified here from Dehalococcoides strain CBDB1. The amino acid sequence of CbdbA1708 contains all the conserved residues involved in metal binding and substrate binding and most other conserved amino acids without apparent function as proposed by Li et al. (21).
Fig. 3.

Minimum-evolution tree of CbdbA1708 and related sequences based on deduced amino acid sequences. Numbers on branches give bootstrap values. C. kluyveri Si-CS was used as an outgroup. The topology was stable as tested by neighbor-joining analysis. Given are the bacterium or strain name, UniProt number (http://www.uniprot.org), locus tag for Dehalococcoides gene products, and an annotation if biochemical data were available for the gene product (marked with a black dot). Abbreviations: IPMS, 2-isopropylmalate synthase; CMS, citramalate synthase; HCS, homocitrate synthase; Re-CS, Re-type citrate synthase; Si-CS, Si-type citrate synthase.
DISCUSSION
Citrate synthase represents a vital enzymatic function in the central metabolism of most organisms. It is involved in anabolism by initiating reactions leading to crucial metabolic precursors such as 2-oxoglutarate but also in catabolism, mainly for acetyl-CoA oxidation via the citrate cycle. In Dehalococcoides species the citrate cycle is not closed as described for other anaerobic bacteria; however, the citrate synthase activity is still needed for anabolic purposes (2, 29, 32). The missing citrate synthase gene in the annotations of fully sequenced Dehalococcoides genomes was the major gap in the understanding of the metabolism of Dehalococcoides species (20, 24, 29), and the goal of our study was to fill this gap by identifying the responsible gene in the genome.
With the analysis of the labeling pattern of glutamine in protein hydrolysates, we first verified in our model organism strain CBDB1 previous results with strain 195 that suggested the presence of a Re-CS in Dehalococcoides species (32). As deduced from glutamine fragments in cultures growing with [1-13C]acetate, the α-carboxyl group of glutamine was derived from the carboxyl carbon of acetate. A Si-CS would instead result in the formation of glutamine with an α-carboxyl group originating from carbonate (Fig. 1). The α-carboxyl group of glutamine was not labeled in the [2-13C]acetate (see Fig. S2C in the supplemental material, m/z = 200) and hydrogen [13C]carbonate (see Fig. S2D, m/z = 199) experiments, showing that the first carbon of glutamine is labeled solely by [1-13C]acetate. The described genes coding for Re-CS have little sequence similarity with Si-CS genes but high sequence similarity with isopropylmalate synthase (EC 2.3.3.13) (18, 21) and homocitrate synthase (EC 2.3.3.14) genes (21). The catalyzed reaction types also are similar, as all these enzymes catalyze the transfer of an acetyl group from acetyl-CoA to a 2-oxo-acid. In some bacteria homocitrate synthase catalyzes the initial reaction of lysine biosynthesis through the α-aminoadipate pathway, whereby 2-oxoglutarate and acetyl-CoA are condensed to homocitrate (19). However, while gene cbdbA1708 was annotated as encoding homocitrate synthase, no other enzymes of the α-aminoadipate pathway are encoded in the genome of strain CBDB1 (20), making this pathway unlikely to occur in vivo. In contrast, all genes needed for the synthesis of lysine through the diaminopimelate pathway are carried in the genome of strain CBDB1. These observations suggested that the cbdbA1708 gene product is redundant for lysine biosynthesis and that its annotation as homocitrate synthase is erroneous. Isopropylmalate synthase catalyzes the initial reaction of the leucine biosynthetic pathway from 2-oxoisovalerate. Two such genes were annotated in strain CBDB1 (cbdbA803 and cbdbA808 [leuA]), and all other genes of this leucine biosynthesis pathway are present, indicating that these genes fulfill their annotated functions. Therefore, gene cbdbA1708 was heterologously expressed in E. coli to elucidate whether it encodes a Re-CS rather than a homocitrate synthase as annotated in the genome. The substrate screening of the heterologously expressed cbdbA1708 gene product supported this hypothesis and showed that the gene product has only citrate synthase activity. Homocitrate and citramalate synthase activities were below the detection threshold. Omitting Mn2+ from the activity tests did not have a significant effect on the activity; however, the enzymes might have been loaded with Mn2+ in the E. coli host, and traces of Mn2+ might have been remained in the buffer. Addition of ATP, however, significantly reduced the activity. Because this inhibition was neutralized by the addition of more Mn2+, the effect of ATP was attributed to the complexation of Mn2+ ions rather than to an effect by ATP directly, indicating a dependence of the enzyme on Mn2+ ions. A dependence on Mn2+ for activity has been found for Re-CS activity before (21). Notably, lysine did not have inhibitory effects, but it had a strong allosteric inhibitory effect on homocitrate synthase in other bacteria (35), corroborating that the cbdbA1708 gene product is not involved in lysine biosynthesis.
In the strictly anaerobic bacterium Clostridium kluyveri the synthesis of citrate is catalyzed by a Re-CS, which was previously annotated as isopropylmalate synthase. The heterologously expressed and purified gene product catalyzed the synthesis of citrate from oxaloacetate (Km = 40 μM) and acetyl-CoA (Km = 50 μM) with a specific activity of 21.7 nkat mg−1 protein (1.3 U mg−1) (21), values that were very similar to those now found for the Re-CS of strain CBDB1 (50 μM, 75 μM, and 18.3 nkat mg−1, respectively). The Si-CS of Clostridium kluyveri showed a similar specific activity of 15 nkat mg−1 (0.9 U mg−1) (21). Like Dehalococcoides spp., the strictly anaerobic archaeon Ignicoccus hospitalis encodes an incomplete citric acid cycle that serves solely anabolic purposes. Isoleucine is synthesized via the citramalate pathway and lysine via the α-aminoadipate pathway. Although a putative gene for CS was not detected, an enzyme activity could clearly be detected in cell extracts, and from labeling experiments the authors assumed that a Re-CS must be present (18). In fact, a gene annotated as encoding isopropylmalate/citramalate/homocitrate synthase with high sequence similarity to cbdbA1708 is present in I. hospitalis (GI:156937774; E value, 4 × 10−33).
The Thermus thermophilus homocitrate synthase catalyzes the initial reaction of lysine biosynthesis through the α-aminoadipic acid pathway, forming homocitrate from 2-oxoglutarate and acetyl-CoA. Homocitrate synthase is strongly regulated by the end product lysine via feedback inhibition. Homocitrate synthase also catalyzes the reaction using oxaloacetate in place of 2-oxoglutarate as a substrate. Kinetic analysis of the heterologously expressed enzyme yielded Km values of 44 μM, 32 μM, and 255 μM for 2-oxoglutarate, acetyl-CoA, and oxaloacetate, respectively (35). Homocitrate synthase from Azotobacter vinelandii was heterologously expressed in E. coli. The enzyme catalyzed the condensation of acetyl-CoA and 2-oxoglutarate but also other 2-oxo-acids. Km values of 60 μM for acetyl-CoA, 2.24 mM for 2-oxoglutarate, and 2.83 mM for oxaloacetate were determined (36).
The results from this study showed an in vitro Re-CS activity of the heterologously expressed enzyme, which supports the conclusion that the cbdbA1708 gene product is responsible for the in vivo Re-CS activity found by us and others (32) by the analysis of protein hydrolysates. In addition, the identification as Re-CS excludes interference of the Si-CS activity of the E. coli host on our results. Additional evidence for the in vivo involvement of the CbdbA1708 protein was obtained by analyzing the proteome of strain CBDB1. With this approach we demonstrated that gene cbdbA1708 is expressed in large amounts during the growth phase of strain CBDB1. However, we cannot presently exclude that other enzymes might also contribute to in vivo citrate synthase activity.
In summary, we demonstrate that the product of gene cbdbA1708 is a Re-CS and present evidence that this gene is the missing link in the metabolism of Dehalococcoides species catalyzing the citrate synthase reaction in vivo. To our knowledge, this study is only the second report on the functional heterologous expression and biochemical characterization of a Re-CS. Genes with very high sequence similarity to cbdbA1708 (96 to 100% amino acid identity) are conserved in all other Dehalococcoides genomes and other strictly anaerobic bacteria. The gene product was also found in one (34) out of three proteomic studies with Dehalococcoides strain 195 (25, 26, 34). Many other strictly anaerobic microorganisms also carry a gene with sequence similarity to cbdbA1708, and biochemical studies on more of these genes might demonstrate a wider distribution of Re-CS activities in the future than yet anticipated.
Supplementary Material
ACKNOWLEDGMENTS
We thank Petra Bombach for help in learning the protocols for extraction and analysis of glutamine.
E.M.-U. was supported by a Marie Curie Intra-European Fellowship within the Seventh Framework Program of the European Commission (PIEF-GA-2009-235049). L.A. was supported by the European Research Council (ERC) (project no. 202903-2, Microflex) and the DFG (FOR1530).
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
Published ahead of print on 22 July 2011.
REFERENCES
- 1. Adrian L., Szewzyk U., Wecke J., Görisch H. 2000. Bacterial dehalorespiration with chlorinated benzenes. Nature 408:580–583 [DOI] [PubMed] [Google Scholar]
- 2. Ahsanul Islam M., Edwards E. A., Mahadevan R. 2010. Characterizing the metabolism of Dehalococcoides with a constraint-based model. PLoS Comput. Biol. 6:e1000887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Amador-Noguez D., et al. 2010. Systems-level metabolic flux profiling elucidates a complete, bifurcated tricarboxylic acid cycle in Clostridium acetobutylicum. J. Bacteriol. 192:4452–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheng D., He J. 2009. Isolation and characterization of “Dehalococcoides” sp. strain MB, which dechlorinates tetrachloroethene to trans-1,2-dichloroethene. Appl. Environ. Microbiol. 75:5910–5918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crown S. B., et al. 2011. Resolving the TCA cycle and pentose-phosphate pathway of Clostridium acetobutylicum ATCC 824: isotopomer analysis, in vitro activities and expression analysis. Biotechnol. J. 6:300–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Danson M. J., Hough D. W. 2001. Citrate synthase from hyperthermophilic archaea. Methods Enzymol. 331:3–12 [DOI] [PubMed] [Google Scholar]
- 7. de Carvalho L. P. S., Blanchard J. S. 2006. Kinetic and chemical mechanism of α-isopropylmalate synthase from Mycobacterium tuberculosis. Biochemistry 45:8988–8999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feng X., et al. 2009. Characterization of the central metabolic pathways in Thermoanaerobacter sp. strain X514 via isotopomer-assisted metabolite analysis. Appl. Environ. Microbiol. 75:5001–5008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gottschalk G. 1968. The stereospecificity of the citrate synthase in sulfate-reducing and photosynthetic bacteria. Eur. J. Biochem. 5:346–351 [DOI] [PubMed] [Google Scholar]
- 10. Gottschalk G., Barker H. A. 1967. Presence and stereospecificity of citrate synthase in anaerobic bacteria. Biochemistry 6:1027–1034 [DOI] [PubMed] [Google Scholar]
- 11. Gottschalk G., Barker H. A. 1966. Synthesis of glutamate and citrate by Clostridium kluyveri, a new type of citrate synthase. Biochemistry 5:1125–1133 [DOI] [PubMed] [Google Scholar]
- 12. He J., Ritalahti K. M., Yang K. L., Koenigsberg S. S., Löffler F. E. 2003. Detoxification of vinyl chloride to ethene coupled to growth of an anaerobic bacterium. Nature 424:62–65 [DOI] [PubMed] [Google Scholar]
- 13. He J., Sung Y., Krajmalnik-Brown R., Ritalahti K. M., Löffler F. E. 2005. Isolation and characterization of Dehalococcoides sp. strain FL2, a trichloroethene (TCE)- and 1,2-dichloroethene-respiring anaerobe. Environ. Microbiol. 7:1442–1450 [DOI] [PubMed] [Google Scholar]
- 14. Herlemann D. P. R., et al. 2009. Genomic analysis of “Elusimicrobium minutum,” the first cultivated representative of the phylum “Elusimicrobia” (formerly termite group 1). Appl. Environ. Microbiol. 75:2841–2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huber H., et al. 2008. A dicarboxylate/4-hydroxybutyrate autotrophic carbon assimilation cycle in the hyperthermophilic archaeum Ignicoccus hospitalis. Proc. Natl. Acad. Sci. U. S. A. 105:7851–7856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hügler M., Huber H., Molyneaux S. J., Vetriani C., Sievert S. M. 2007. Autotrophic CO2 fixation via the reductive tricarboxylic acid cycle in different lineages within the phylum Aquificae: evidence for two ways of citrate cleavage. Environ. Microbiol. 9:81–92 [DOI] [PubMed] [Google Scholar]
- 17. Ishihama Y., et al. 2005. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 4:1265–1272 [DOI] [PubMed] [Google Scholar]
- 18. Jahn U., Huber H., Eisenreich W., Hügler M., Fuchs G. 2007. Insights into the autotrophic CO2 fixation pathway of the archaeon Ignicoccus hospitalis: comprehensive analysis of the central carbon metabolism. J. Bacteriol. 189:4108–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kosuge T., Hoshino T. 1998. Lysine is synthesized through the α-aminoadipate pathway in Thermus thermophilus. FEMS Microbiol. Lett. 169:361–367 [DOI] [PubMed] [Google Scholar]
- 20. Kube M., et al. 2005. Genome sequence of the chlorinated compound-respiring bacterium Dehalococcoides species strain CBDB1. Nat. Biotechnol. 23:1269–1273 [DOI] [PubMed] [Google Scholar]
- 21. Li F., Hagemeier C. H., Seedorf H., Gottschalk G., Thauer R. K. 2007. Re-citrate synthase from Clostridium kluyveri is phylogenetically related to homocitrate synthase and isopropylmalate synthase rather than to Si-citrate synthase. J. Bacteriol. 189:4299–4304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marco-Urrea E., Nijenhuis I., Adrian L. 2011. Transformation and carbon isotope fractionation of tetra- and trichloroethene to trans-dichloroethene by Dehalococcoides sp. strain CBDB1. Environ. Sci. Technol. 45:1555–1562 [DOI] [PubMed] [Google Scholar]
- 23. Maymó-Gatell X., Chien Y. T., Gossett J. M., Zinder S. H. 1997. Isolation of a bacterium that reductively dechlorinates tetrachloroethene to ethene. Science 276:1568–1571 [DOI] [PubMed] [Google Scholar]
- 24. McMurdie P. J., et al. 2009. Localized plasticity in the streamlined genomes of vinyl chloride respiring Dehalococcoides. PLoS Genet. 5:e1000714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morris R. M., et al. 2007. Comparative proteomics of Dehalococcoides spp. reveals strain-specific peptides associated with activity. Appl. Environ. Microbiol. 73:320–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morris R. M., Sowell S., Barofsky D., Zinder S., Richardson R. 2006. Transcription and mass-spectroscopic proteomic studies of electron transport oxidoreductases in Dehalococcoides ethenogenes. Environ. Microbiol. 8:1499–1509 [DOI] [PubMed] [Google Scholar]
- 27. Perkins D. N., Pappin D. J. C., Creasy D. M., Cottrell J. S. 1999. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20:3551–3567 [DOI] [PubMed] [Google Scholar]
- 28. Say R. F., Fuchs G. 2010. Fructose 1,6-bisphosphate aldolase/phosphatase may be an ancestral gluconeogenic enzyme. Nature 464:1077–1084 [DOI] [PubMed] [Google Scholar]
- 29. Seshadri R., et al. 2005. Genome sequence of the PCE-dechlorinating bacterium Dehalococcoides ethenogenes Science 307:105–108 [DOI] [PubMed] [Google Scholar]
- 30. Silfer J. A., Engel M. H., Macko S. A., Jumeau E. J. 1991. Stable carbon isotope analysis of amino acid enantiomers by conventional isotope ratio mass spectrometry and combined gas chromatography/isotope ratio mass spectrometry. Anal. Chem. 63:370–374 [DOI] [PubMed] [Google Scholar]
- 31. Tamura K., Dudley J., Nei M., Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599 [DOI] [PubMed] [Google Scholar]
- 32. Tang Y. J., et al. 2009. Investigation of carbon metabolism in “Dehalococcoides ethenogenes” strain 195 by use of isotopomer and transcriptomic analyses. J. Bacteriol. 191:5224–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tian J., Bryk R., Itoh M., Suematsu M., Nathan C. 2005. Variant tricarboxylic acid cycle in Mycobacterium tuberculosis: identification of α-ketoglutarate decarboxylase. Proc. Natl. Acad. Sci. U. S. A. 102:10670–10675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Werner J. J., Ptak A. C., Rahm B. G., Zhang S., Richardson R. E. 2009. Absolute quantification of Dehalococcoides proteins: enzyme bioindicators of chlorinated ethene dehalorespiration. Environ. Microbiol. 11:2687–2697 [DOI] [PubMed] [Google Scholar]
- 35. Wulandari A. P., et al. 2002. Characterization of bacterial homocitrate synthase involved in lysine biosynthesis. FEBS Lett. 522:35–40 [DOI] [PubMed] [Google Scholar]
- 36. Zheng L., White R. H., Dean D. R. 1997. Purification of the Azotobacter vinelandii nifV-encoded homocitrate synthase. J. Bacteriol. 179:5963–5966 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.