

Abstract
Findings from a number of studies suggest that the PilA pilin proteins may play an important role in the pathogenesis of disease caused by species within the genus Francisella. As such, a thorough understanding of PilA structure and chemistry is warranted. Here, we definitively identified the PglA protein-targeting oligosaccharyltransferase by virtue of its necessity for PilA glycosylation in Francisella tularensis and its sufficiency for PilA glycosylation in Escherichia coli. In addition, we used mass spectrometry to examine PilA affinity purified from Francisella tularensis subsp. tularensis and F. tularensis subsp. holarctica and demonstrated that the protein undergoes multisite, O-linked glycosylation with a pentasaccharide of the structure HexNac-Hex-Hex-HexNac-HexNac. Further analyses revealed microheterogeneity related to forms of the pentasaccharide carrying unusual moieties linked to the distal sugar via a phosphate bridge. Type A and type B strains of Francisella subspecies thus express an O-linked protein glycosylation system utilizing core biosynthetic and assembly pathways conserved in other members of the proteobacteria. As PglA appears to be highly conserved in Francisella species, O-linked protein glycosylation may be a feature common to members of this genus.
INTRODUCTION
Francisella tularensis is a facultative intracellular bacterium whose virulence is associated with resistance to killing by macrophages (15). Highly virulent strains of F. tularensis, exemplified by the highly pathogenic subspecies tularensis (designated type A strains), are the etiologic agent of tularemia, a severe and sometimes fatal zoonotic disease in humans (36). Two other subspecies of F. tularensis implicated in human disease have been identified and differentiated based on comparative genomics, geographic distribution, and reduced virulence, F. tularensis subsp. holarctica (also known as type B strains) and F. tularensis subsp. mediasiatica, which are associated with milder forms of the disease in humans but retain full virulence in rodents (25). A fourth subspecies, subspecies novicida, has been proposed, but currently Francisella novicida retains its own species designation, despite being genetically closely related to F. tularensis. F. novicida is pathogenic only in immunocompromised individuals but can cause disease in mice (39). Current data suggest that F. novicida is ancestral to the human-pathogenic species and that the emergence of strains with increased virulence may be associated with adaptation to increased survival and persistence in macrophages (18, 28). Such adaptation has been linked to a number of genomic rearrangements and gene losses, rather than the acquisition of virulence factors. Interest in the pathogenesis of disease caused by these agents and evolution within the genus has been fueled by the designation of F. tularensis as a category A biological agent and the current lack of any licensed vaccine against tularemia (10, 19).
Type IV pili (Tfp) are complex filamentous appendages expressed by many eubacteria of medical, environmental, and ecological importance. Expression of Tfp and associated phenotypes involve the concerted actions of multiple proteins that are highly conserved across taxonomic boundaries (34). The major pilin subunit of the pilus fiber is expressed as a membrane-associated prepilin that is proteolytically cleaved by a prepilin peptidase, PilD (35). In many cases, multiple proteins sharing structural similarities to the major pilin subunit, the so-called minor pilins, are required for Tfp expression and function (3–5, 40, 41, 43). However, the precise role they serve in Tfp biogenesis and function has yet to be identified. Furthermore, Tfp are dynamic structures that undergo retraction mediated by members of the PilT retraction ATPase family (23, 33). Sequential rounds of Tfp extension and retraction mediate twitching motility and susceptibility to pilus-specific bacteriophages.
Although the genomes of all F. tularensis subspecies contain a nearly complete repertoire of genes required for Tfp expression and function, mutations and rearrangements at such loci appear to impact on pathogenicity. For example, a spontaneous mutant of a type B strain carrying a deletion within a locus encompassing three tandemly arrayed genes encoding pilin-like proteins (pilA, pilE, and pilV, which are orthologous to those annotated as FTT0888c, FTT0889c, and FTT0890c, respectively, in the type A strain SCHU S4 genome) showed reduced virulence in a mouse model. In that strain, full virulence was restored by reintroduction of an intact copy of pilA (14). Similarly, another type B strain, LVS (named LVS for live vaccine strain), carries an analogous pilA-pilE deletion as well as a rearrangement at another locus termed RD18. In this case, full virulence was restored only by complementation with both pilA and the FTT_0918 gene that maps in RD18 (30). In F. novicida, the secretion of multiple proteins was abolished by mutational inactivation of genes predicted to encode Tfp and ancillary factors (16). This suggested that Tfp here might also be associated with type II secretion system (T2SS) activity as seen in Dichelobacter nodosus and Vibrio cholerae (17, 20). Interestingly, F. novicida mutants lacking either the secretion substrates or carrying insertions at Tfp-associated loci (including pilA) had increased virulence in a mouse infection model (16). These surprising results were reconciled by the finding that one of the secreted substrates (PepO) is a zinc protease that enhances vasoconstriction and possibly inhibits spread and dissemination of the organism.
Rearrangements and allelic variation of genes encoding components related to Tfp biology may also be of phylogenetic relevance. For example, type B strains have been reported to carry identical nonsense mutations in pilE, pilV, and pilT that would likely preclude the expression of functional products (14, 30). In addition, FTT_0861 (predicted to encode another pilin-like protein in the SCHU S4 nomenclature) appears to be highly polymorphic between species. In comparison, type A strains and F. novicida appear to be more similar to one another regarding Tfp-related gene repertoire and status with the exception of the polymorphisms described above in pilA and FTT_0861.
What remains unclear for these agents is whether the genotypes and phenotypes described above are related to the expression of Tfp. Despite many efforts and reports, the data remain equivocal. Tfp have yet to be purified and characterized from any F. tularensis subspecies, although morphologically analogous structures have been observed and correlated with genotype by forward genetics. In a recent study of F. novicida, it was concluded that Tfp expression and protein secretion could be dissociated and that PilE4 (orthologous to FTT0861) was the likely major subunit of Tfp (44). In contrast, a study in which pilin-like proteins from the type A strain SCHU S4 and F. novicida strain U112 were expressed in Neisseria gonorrhoeae, and their capacity to complement Tfp-related phenotypes was assessed concluded that PilA was most likely to represent a Tfp-forming pilus subunit in both the type A strain SCHU S4 and the F. novicida strain U112 (29).
Despite the apparent importance of PilA in the pathogenesis of disease caused by F. tularensis, our current understanding of PilA structure and chemistry is limited. Prior studies have noted aberrant mobility patterns of F. tularensis PilA expressed in an endogenous background, which were not seen when expressed in Pseudomonas aeruginosa PAK (14). Further, alterations in the mobility pattern of F. tularensis PilA expressed in N. gonorrhoeae correlated with the expression of distinct O-linked glycoforms, suggesting that they might undergo glycosylation in that background (29). Here, we report the identification of the cognate oligosaccharyltransferase (encoded by FTT_0905, SCHU S4 nomenclature), the activity of which was necessary for PilA glycosylation in the type B strain FSC200 and sufficient to glycosylate PilA in a reconstituted system in E. coli. We also purified PilA proteins from the type A strain SCHU S4 and the type B strain FSC200 and used mass spectrometry (MS) in order to demonstrate that they undergo O-linked glycosylation and to characterize the associated glycan.
MATERIALS AND METHODS
Bacterial strains, plasmids, growth conditions, and DNA methods.
The bacterial strains and plasmids used in this study are listed in Table 1. Francisella tularensis strains were grown on modified Thayer-Martin agar or blood cysteine glucose agar (BCGA) plates at 37°C in 5% CO2. E. coli strains were grown on blood agar base (BAB; Merck) plates or in Luria-Bertani broth (LB). The following antibiotics were used at the indicated concentrations: kanamycin, 50 μg/ml; chloramphenicol, 2.5 μg/ml (for F. tularensis) or 25 μg/ml (for E. coli); and tetracycline, 10 μg/ml. Preparation of plasmid DNA, restriction enzyme digestion, ligation, and transformation into E. coli were performed essentially as described previously (31). For studies of oligosaccharyltransferase activity of PglO and PglA in E. coli, we used the E. coli strain BL21(DE3) and the plasmids pACYC184, pJB658, and p2/16/1 to express the pgl locus (pglFBCD), the oligosaccharyltransferases (pglO and pglA) and the target substrate (pilA). Cloning behind the Pm promoter in pJB658 allows for inducible expression of the inserted sequence upon the addition of m-toluic acid.
Table 1.
Bacterial strains and plasmids used in this study
| Bacterial species and strain | Description, genotype, and/or phenotypea | Source or reference |
|---|---|---|
| F. tularensis strains | ||
| Subsp. tularensis SCHU S4 FSC237 | Human ulcer 1941, Ohio | |
| Subsp. holarctica FSC200 | Human ulcer 1998, Sweden | |
| PS1 | SCHU S4 expressing pEMS10 (pilAFLAG) in trans; Tcr | This study |
| FSC679 | FSC200 ΔpglA; in-frame deletion of codons 10 to 469 | This study |
| FSC749 | FSC200/pEMS10; FSC200 expressing pilAFLAG in trans; Tcr | This study |
| FSC703 | FSC679/pEMS10; FSC679 expressing pilAFLAG in trans; Tcr | This study |
| N. gonorrhoeae strains | ||
| KS101 | pilEind | 29 |
| KS67 | KS101 iga::pilAFt | 29 |
| KS180 | KS67 pglA | 29 |
| KS182 | KS67 pglC | 29 |
| KS186 | KS67 pglEon | 29 |
| KS190 | KS67 pglI | 29 |
| E. coli strains | ||
| Top10 | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 deoR araD139 (Δara-leu)7697 galU galK rpsL (Smr) endA1 nupG | Invitrogen |
| BL21(DE3) | E. coli B F−dcm ompT hsdS(rB− mB−) gal λ(DE3) | Invitrogen |
| HB101 | supE44 hsdS20(rB− mB−) recA13 ara-14 proA2 lacY1 galK2 rpsL20 xyl-5 mtl-1 | NEB |
| S17-1λpir | recA thi pro hsdR−M+ <RP4:2-Tc:Mu:Km:Tn7> Tpr Smr | 32 |
| Plasmids | ||
| pCR2.1 | TOPO cloning vector; Ampr Kmr | Invitrogen |
| pDM4 | Suicide plasmid; sacB mobRP4 oriR6K; Cmr | 22 |
| pKK214GFP | groELS promoter; Ft ori; p15A ori; GFP; Tcr | 2 |
| pAL19 | 2,148-bp fragment of approximately 1 kb upstream and 1 kb downstream of pglA (FTT_0905) cloned in XhoI and BglII sites of pDM4; Cmr | This study |
| pEMS10 | The pilA gene and FLAG epitope cloned into the NdeI and EcoRI sites of pKK214GFP; Tcr | 14 |
| pET24a(+) | Protein expression vector; T7 lac promoter; ColE1 ori; Kmr | Novagen |
| pCRII-TOPO | TOPO cloning vector; Ampr Kmr | Invitrogen |
| pACYC184 | Cloning vector; p15A ori; Cmr Tcr | NEB |
| pJB658 | Protein expression vector; Pm promoter; RK2 oriT; Ampr | 7 |
| pACYCpglFBCD | Gonococcal pglFBCD locus including about 250 bp of upstream DNA from strain VD300 (the parent of KS101) cloned into the BamHI and SphI sites in pACYC184; Cmr | This study |
| pJB658pglO | The pglO gene from N. gonorrhoeae N400 cloned into the NdeI and SmaI sites of pJB658 | This study |
| pJB658pglA | The pglA (FTT_0905) gene cloned into the NdeI and KpnI sites of pJB658 | This study |
| p2/16/1 | Cloning vector; ColE1 ori; Kmr | 42 |
| pEMS25 | pilAFt gene cloned into the Bsu36I and StuI sites in p2/16/1 | 29 |
pilEind, inducible pilE allele; pilAFt, pilA gene from F. tularensis; pglEon, pglE in a phase “on” configuration; Ft ori, F. tularensis origin; GFP, green fluorescent protein.
Construction of a deletion mutation of FTL_0425 in strain FSC200.
The Francisella pglA (FTH_0425) deletion construct was obtained by an overlap PCR using primer pairs PglA_A, PglA_B, PglA_C (Table 2), and PglA_D, where PglA_A and PglA_D have complementary sequences, and using genomic DNA from strain FSC155 as a template. Before subcloning the DNA fragment into the suicide mutagenesis vector pDM4, the construct were first introduced into a pCR-2.1 vector and sequenced (Eurofins MVG Operon) to ensure a correct sequence. The 2,148-bp fragment was digested with BamHI and XhoI (pCR-2.1 sites) and ligated into XhoI/BglII-digested pDM4, generating plasmid pAL19. The mutagenesis vector, pAL19, was introduced into the recipient strain FSC200 by conjugal mating as previously described (22) and verified by PCR and sensitivity to chloramphenicol.
Table 2.
Primers used in this study
| Primer | Primer sequence (5′–3′) | RE sitea |
|---|---|---|
| PglA_A | AGAGCCAGCAACAGCTACAA | XhoI |
| PglA_B | ATATAAGTGGTAATACAATATAATGCATAAAAGGCA | |
| PglA_C | CATTATATTGTATTACCACTTATATTTCCACCTTT | |
| PglA_D | TCAGATGATGAGCAAACTTTAC | BglII |
| pilA-comp_1F | GCATGTCATATGAAAAAGAAAATGCAAAAAGGT | NdeI |
| pilAFLAG_1R | GCATGTGAATTCTTATTTATCATCATCATCTTTATAATCGATAGCATTACAGTTAGATGGT | EcoRI |
| FE1175 | GTTTCATCGATAAGCGAAATCCTCGGACACG | |
| FE1170 | CGCCGATTAATAGCATATTGACGGGCTTGTCGC | |
| pglO5′ | CTTGGATCCATGTCCGCTGAAACGACCGTATCCG | BamHI |
| pglO3′ | GTTTACTCGAGTTTGCAGGGTTTTGTTTCCGGATGGC | XhoI |
| FE2108 | ATTTCATATGATAGAAGTGATAAATTACC | NdeI |
| FE2110 | CAAAGGTACCAAAGATGGCGAACAGAAAG | KpnI |
| RT-PCR primers | ||
| A | TACTCTAATGACGGCAACTTT | |
| B | GCAAGCTCTGAATTATCTGACT | |
| C | AAGGCGTCAGCGTAACATTT | |
| D | CCACTAAAACCACCTCTAACA | |
| E | GGCATTTAGGACACTCTTCTT | |
| F | GGCATCCATTTATTGAGAGAA |
RE site, restriction enzyme site.
Construction of C-terminal FLAG epitope-tagged PilA.
The Francisella pilin gene pilA, fused in frame to a C-terminal FLAG epitope, DYKDDDDK, was cloned under the groESL promoter of pKK214GFP (GFP stands for green fluorescent protein) by using PCR primers pilA-comp_1F and pilAFLAG_1R (Table 2), and strain FSC352 as template. The resulting DNA fragment was sequenced (Eurofins MVG Operon) to ensure a correct sequence and cloned into pKK214GFP (NdeI-EcoRI) downstream of the groELS promoter. The construct, denoted pEMS10 (14), was introduced into the recipient strains FSC200, FSC679 (FSC200ΔpglA), and SCHU S4 (FSC237) by cryotransformation (24). The resulting recombinant strains were verified by PCR and immunoblotting using antibodies specific for the FLAG epitope.
Affinity purification of epitope-tagged PilA.
Epitope-tagged PilA was purified from the type A strain SCHU S4 and the type B strains FSC200 and FSC697 (ΔpglA) by slightly differing protocols. In the case of the SCHU S4 derivative, the strain was cultured on BCGA supplemented with 10 ml of 10% (wt/vol) histidine per liter. Harvested cells were heat inactivated and lysed with CelLytic B plus kit (Sigma, United Kingdom) according to the manufacturer's instructions. Immunoprecipitation of tagged PilA from the soluble-protein fraction was performed using EZview red anti-FLAG M2 affinity gel (Sigma, United Kingdom) according to the manufacturer's instructions. FLAG-tagged PilA was eluted from the resin using 0.1 M glycine HCl at pH 3.5. Samples were lyophilized and resuspended in distilled H2O (dH2O) for mass spectrometric (MS) analysis. The type B strains FSC200 and FSC697 (ΔpglA) were grown on modified Thayer-Martin agar plates. Harvested cells were heat inactivated and lysed with CelLytic B plus kit (Sigma, United Kingdom) according to the manufacturer's instructions. Immunoprecipitation of tagged PilA from the soluble-protein fraction was performed using EZview red anti-FLAG M2 affinity gel (Sigma, United Kingdom) according to the manufacturer's instructions. Elution of FLAG-tagged PilA was carried out by competition with FLAG peptide as described by the manufacturer (Sigma). The samples were then concentrated by precipitation with 5 volumes of acetone, centrifuged, and air dried before resuspension in dH2O for MS analysis.
Cloning of the gonococcal pgl core locus (pglFBCD).
The gonococcal gene cluster containing the pglF, pglB, pglC, and pglD genes and upstream DNA were amplified by PCR from strain N400 using Expand Long Template PCR system kit (Roche) and the primers FE1175 and FE1170. The PCR product was cloned into TOPO vector pCRII-TOPO, released by the unique restriction sites BamHI and SphI, and then subcloned into the pACYC184 vector digested with BamHI and SphI. The resulting plasmid pACYCpglFBCD was transformed into E. coli BL21(DE3) cells.
Cloning of the PglO and PglA oligosaccharyltransferases.
The coding region of pglO was amplified by PCR from strain N400 using the primers pglO5′ and pglO3′. The PCR product was digested with BamHI/XhoI and cloned into BamHI/XhoI-digested pET24a(+) (Novagen), released by the unique restriction sites NdeI and SmaI, located in the pET24a(+) vector, and subcloned into the pJB658 plasmid digested with NdeI and SmaI. The resulting plasmid, pJB658pglO, was transformed into chemically competent E. coli BL21(DE3) cells containing the plasmid pACYCpglFBCD and selected on LB agar plates containing chloramphenicol and ampicillin. The sequences encompassing FTT_0905 were amplified by PCR from strain SCHU S4 using Advantage HD polymerase (Clontech) and the FE2108 and FE2110 primers. The resulting PCR product was digested with NdeI/KpnI and cloned into NdeI/KpnI-digested pJB658. The resulting plasmid, pJB658pilO2, was transformed into chemically competent E. coli BL21(DE3) cells containing the pACYCpglFBCD plasmid and selected on LB agar plates containing chloramphenicol and ampicillin.
Expression and detection of glycosylated PilA in E. coli.
E. coli BL21(DE3) cells containing the relevant plasmids were grown overnight in selective LB medium, diluted 100-fold in fresh selective LB medium, and then grown to an optical density at 600 nm (OD600) of 0.6 before induction for 2 h with 2 mM m-toluic acid to induce oligosaccharyltransferase expression (pglO or pglA). Whole-cell extracts were prepared from equivalent numbers of cells by heating cell suspensions to 100°C for 3 min in SDS sample loading buffer and analyzed by SDS-PAGE and immunoblotting. The primary antibody used to detect PilA was a polyclonal rabbit antibody (14) diluted 1:1,000, and the monoclonal antibodies (MAbs) npg1, npg2, and npg3 were used to detect glycan-dependent epitopes (8).
Protein gel electrophoresis and immunoblotting.
Samples containing SDS and β-mercaptoethanol were boiled for 5 min (denaturing conditions). All samples were analyzed by SDS-PAGE on a 12% gel as described previously (21). The antibodies used in immunoblotting were a polyclonal anti-PilA serum (14), anti-FLAG mouse monoclonal antibody (Sigma-Aldrich, Sweden) and the npg1, npg2, and npg3 rabbit monoclonal antibodies recognizing a defined neisserial protein glycan epitope (8). Proteins were transferred to Immobilon-P transfer membranes (Millipore) using Trans-Blot semidry transfer cell (Bio-Rad). Membranes were blocked in Tris-buffered saline (TBS) with 5% nonfat dry milk. When a horseradish peroxidase-conjugated secondary antibody system was used, filters were developed by using the ECL kit (Amersham Pharmacia Biotech). For the alkaline phosphatase-conjugated (Roche) secondary antibody system, visualization was accomplished by incubating the filters with 0.1% (wt/vol) Nitro Blue Tetrazolium (NBT; Sigma) and 0.05% (wt/vol) 5-bromo-4-chloro-3-indolylphosphate (BCIP; Sigma).
RNA isolation and RT-PCR.
Bacteria were grown for 18 h on Thayer-Martin agar plates, harvested, and suspended in TRIzol reagent (Life Technologies). Total RNA was extracted and treated with RNase-free DNase I (Roche), phenol extracted, and precipitated by ethanol. An aliquot of the RNA (3 μg) was used as a template to synthesize cDNA using random hexamers (final concentration, 25 ng/μl) and Superscript III reverse transcriptase (RT) as described by the manufacturer (Life Technologies). In control experiments, samples processed without the addition of RT enzyme were used.
In-gel protein digestion.
Coomassie blue-stained gel slices containing PilA protein were serially rehydrated and washed with 150 μl of high-performance liquid chromatography (HPLC)-grade water, 150 μl of acetonitrile-water (1:1 [vol/vol]), and 100% acetonitrile at room temperature. Protein reduction was carried out by the addition of 50 μl of 10 mM dithiothreitol, 0.05 mM NH4HCO3 (60 min, 56°C) to the dehydrated gel pieces. Excess reduction buffer was then removed, and thiol groups were alkylated by adding 50 mM iodoacetamide and 0.05 mM NH4HCO3 (45 min at room temperature in the dark). The gel pieces were washed twice with 150 μl of acetonitrile-water (1:1 [vol/vol]) and twice with 100% acetonitrile at room temperature. Digestion buffer (5 to 10 μl) containing 16 ng/μl trypsin (Sigma-Aldrich), 25 ng/μl chymotrypsin (Roche), or 40 ng/μl proteinase K (Sigma-Aldrich) in 0.05 mM NH4HCO3 (chymotrypsin/proteinase K, 0.05 M Tris-HCl, pH 8.0) was added, and the samples were kept on ice for 30 min to allow rehydration of the gel pieces. To limit autoproteolysis of proteases, the remaining solution was removed and replaced with 35 to 50 μl of 0.05 new buffer, and digestions were carried out overnight (trypsin, chymotrypsin) or over 1 h (proteinase K) at 37°C. Peptides were extracted successively with 5% formic acid and 5% formic acid, acetonitrile (1:1 [vol/vol]), and acetonitrile. The combined supernatants were dried by SpeedVac and then redissolved in 0.1% formic acid. Samples were subjected immediately to mass spectrometric analyses or frozen at −80°C.
MS analysis of proteolytic peptides.
Reverse-phase chip-based nanoflow liquid chromatographic (LC) MS and tandem MS (MS2) (collision-induced dissociation [CID] and electron transfer dissociation [ETD] mode) analyses (nano-LC-chip MS2) of proteolytic peptides were performed using an Agilent 1100 LC/MSD Trap XCT Ultra equipped with a Chip/MS Cube interface. In some cases, reverse-phase (C18) nanoflow on-line liquid chromatographic MS2 analyses of proteolytic peptides were performed using a HPLC system consisting of two Agilent 1200 HPLC binary pumps (nanoflow and capillary flow) with a corresponding autosampler, column heater, and integrated switching valve. This LC system was coupled via a nanoelectrospray ion source to a LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). For the analyses, 4 μl of peptide solution was injected onto the 5- by 0.3-mm extraction column filled with Zorbax 300 SB-C18 of 5-μm particle size (Agilent). The samples were washed with a mobile aqueous phase of 0.1% formic acid–3% acetonitrile. The flow rate was 4 μl/min provided by the capillary pump. After 7 min, the switching valve of the integrated switching valve was activated, and the peptides were eluted in the back-flush mode from the extraction column onto the 150- by 0.075-mm C18 column (GlycproSIL C18-80Å; Glycpromass, Stove, Germany) with 3-μm resin. The mobile phase consisted of acetonitrile and MS-grade water, both containing 0.1% formic acid. Chromatographic separation was achieved using a binary gradient from 5 to 55% of acetonitrile in 60 or 120 min. The nanoflow pump flow rate was 0.2 μl min−1.
Mass spectra were acquired in the positive-ion mode applying a data-dependent automatic switch between survey scan and MS2 acquisition. Peptide samples were analyzed either by higher-energy C-trap dissociation (HCD) or CID in the LTQ ion trap by acquiring one Orbitrap survey scan in the mass range of m/z 300 to 2,000 followed by HCD and/or CID of the three most intense ions in the Orbitrap. The target value in the LTQ Orbitrap was 1,000,000 for a survey scan at a resolution of 30,000 at m/z 400 using lock masses for recalibration to improve the mass accuracy of precursor ions. Fragmentation was performed with a target value of 5,000 ions. The ion selection threshold was 500 counts. Selected sequenced ions were dynamically excluded for 180 s.
MS data analysis.
Mass spectrometric data were analyzed with the in-house-maintained F. tularensis protein sequence database using SEQUEST. The mass tolerances of a fragment ion and a parent ion were set at 0.05 Da and 5 ppm, respectively. Methionine oxidation and cysteine carbamidomethylation were selected as a variable or fixed modification. A false discovery rate of 0.01 was required for proteins and peptides with a minimum length of 6 amino acids. Glycopeptide MS2 spectra were manually inspected by Qual Browser version 2.0.7.
To assign detected peptide backbone masses to corresponding peptide pieces, an in-house software program called Peptide Mass Tool was developed. Peptides cleaved by unspecific proteases were identified by this peptide mass tool, programmed with Perl. The tool accepts a monoisotopic mass M+H+, a list of possible modifications, and an allowed range of mass deviations (in ppm) and searches through a list of detected proteins (here PilA) by scanning each protein amino acid on the fly from the N terminus to the C terminus for matching peptides. All matched peptides were collected, sorted on mass deviation and/or protein mass, and written into an Excel spreadsheet file together with all search and program settings.
RESULTS
Identification and characterization of the endogenous protein-targeting oligosaccharyltransferase.
Studies of protein glycosylation in N. gonorrhoeae and pilin glycosylation in selected strains of P. aeruginosa have revealed related pathways in which oligosaccharides are synthesized on lipid-linked carriers in the cytoplasm, and these complexes are then translocated into the periplasm by the action of dedicated transporters or flippases (1, 11). The glycans are then transferred from the lipid-linked precursors onto serine residues of target proteins by the action of oligosaccharyltransferases structurally related to the WaaL family of O-antigen ligases (1, 12, 26, 27). The FTT_0905 open reading frame (ORF) is annotated in the F. tularensis SCHU S4 database as being a type IV pilin glycosylation protein based on its sequence similarity to the pilin-targeting P. aeruginosa PilO oligosaccharyltransferase (25% identity over a span of 274 residues). To test the potential role of FTT_0905 in PilA glycosylation, a null mutation encompassing a large in-frame deletion of the ORF was constructed and introduced into different F. tularensis strains. To facilitate characterization of PilA, a polypeptide in which the FLAG epitope domain was fused to the C terminus of the pilA ORF was expressed in the strains examined (see Fig. S1 in the supplemental material). Despite repeated attempts, mutants carrying the altered allele in the type A strain SCHU S4 could not be isolated. However, a mutant carrying a null allele of the FTL_0425 gene (the FTT_0905 homologue) was recovered in the type B strain FSC200 background. To determine whether the mutation had any major impact on transcription of neighboring genes, reverse transcriptase PCR (RT-PCR) analyses of RNAs extracted from the wild-type and mutant strains were performed. However, no alterations in the relative levels of RNA for the flanking genes were observed (data not shown).
The relative migrations of PilA and affinity-tagged derivatives on SDS-polyacrylamide gels were profoundly altered in the FTL0425 mutant with a single immunoreactive species of approximately 14 kDa being detected versus the predominant immunoreactive species of approximately 18 kDa seen in the wild-type backgrounds (Fig. 1). Thus, the absence of FTH0425 correlated with a substantial increase in the relative mobility of PilA. When examining PilA expression in strain SCHU S4, substantial differences in the relative migration of the wild-type and affinity-tagged proteins were observed that could not be simply attributed to differences in their primary structure. While wild-type PilA migrated as one major species of approximately 18 kDa, the affinity-tagged version was detected as a number of discrete species between 14 and 19 kDa. These findings suggested that the presence of the affinity tag on PilA may have differentially altered posttranslational modification in this background.
Fig. 1.

Characterization of PilA expression in different F. tularensis backgrounds using immunoblotting of whole-cell lysates with anti-PilA antibodies. From left to right, the lanes contain strains FSC200 (wild type [wt]), FSC679 (FSC200 ΔFTL_0425 [ΔpglA]), FSC749 (FSC200 pilAFLAG), FSC703 (FSC200 pilAFLAG ΔFTL_0425 [pglA]), SCHU S4 (wt), and PS1 (SCHU S4 pilAFLAG). Note that the presence of FLAG-tagged residues decreases the mobility of PilA and that in the SCHU S4 background (strain PS1 in the rightmost lane), it may alter posttranslational modification of PilA.
To gain more insight into the role served by FTL0425 and FTT0905, we attempted to functionally reconstitute PilA glycosylation in E. coli using an approach previously developed in studies of oligosaccharyltransferase from the Campylobacter jejuni N-linked and N. meningitidis O-linked glycosylation systems (12, 13). This entailed expressing the PilA and FTT0905 proteins together with genes encoding components required for the biosynthesis of a glycan donor in its lipid-linked carrier form. Here, a plasmid carrying the core glycosylation locus (pglF, -B, -C, and -D genes) from N. gonorrhoeae strain N400 was used to express the lipid-linked 2,4-diacetamido-2,4,6-trideoxyhexose (DATDH) monosaccharide precursor on the periplasmic side of the cytoplasmic membrane (1). The principal advantage of this approach was that proteins having undergone O-linked glycosylation with the DATDH monosaccharide could be detected by their reactivity with the monoclonal antibody npg1 that recognizes a DATDH-specific epitope (8). As shown in Fig. 2A, coexpression in E. coli of F. tularensis PilA, the N. gonorrhoeae targeting oligosaccharyltransferase PglO, and the N. gonorrhoeae core pgl locus was sufficient to establish PilA reactivity with the monoclonal antibody npg1, synonymous with DATDH modification. Thus, consistent with its established promiscuity for protein substrates in N. gonorrhoeae, PglO is capable of glycosylating type A SCHU S4 PilA. Similarly, coexpression of type A-derived PilA and the N. gonorrhoeae core pgl locus together with FTT0905 protein was sufficient to establish reactivity of PilA with the monoclonal antibody npg1 (Fig. 2B). All findings considered, we conclude that FTL0425 and FTT0905 are protein-targeting oligosaccharyltransferase-utilizing lipid-linked glycan donors. Accordingly, we recommend that the designation PglA (for protein glycosylation protein A) be used in reference to FTT0905 and its related orthologues in Francisella species.
Fig. 2.

Reconstitution of PglA-mediated PilA glycosylation in E. coli. Immunoblotting of whole-cell lysates from strains expressing PilA and gonococcal oligosaccharyltransferase PglO (A) and F. tularensis subsp. tularensis strain SCHU S4 PglA (B). The undecaprenylphosphate-linked 2,4-diacetamido-2,4,6-trideoxyhexose (DATDH) precursor was expressed by virtue of the pglFDCD core locus from N. gonorrhoeae. Immunoblotting utilized polyclonal anti-PilA serum and the monoclonal antibody npg1 that recognizes an epitope imparted by the presence of the DATDH sugar. Plus and minus signs denote the proteins expressed (+) and not expressed (−) in the backgrounds tested.
F. tularensis PilA undergoes glycosylation when expressed in N. gonorrhoeae.
Polyclonal antibodies generated against glycosylated gonococcal pili were previously shown to react with pilus-like appendages associated with F. tularensis PilA expression in gonococci (29). Taken together with the observations that the relative mobility of F. tularensis PilA varied in association with altered gonococcal pgl gene content, it was suggested that F. tularensis PilA was glycosylated when expressed in gonococci. To examine this possibility in more detail, immunoblotting using monoclonal antibodies recognizing epitopes specific to mono-, di-, and trisaccharide glycan forms (8) was performed using cell lysates of gonococci expressing PilA in defined pgl backgrounds. As shown in Fig. 3, the npg1 and npg3 MAbs (recognizing mono- and trisaccharide-associated epitopes, respectively) reacted specifically with F. tularensis PilA in the pglA and pglEon (phase “on”) backgrounds expressing the corresponding glycoforms. Likewise, npg2 (recognizing the disaccharide forms) reacted with F. tularensis PilA in the wild-type strain expressing the disaccharide glycan. PilA reactivity with npg1 was enhanced in the background lacking O-acetylation of the distal galactose (in the pglI background). The latter finding is consistent with the previously established association between glycan O-acetylation and diminished exposure or accessibility of the epitope recognized by the npg2 monoclonal antibody (8).
Fig. 3.
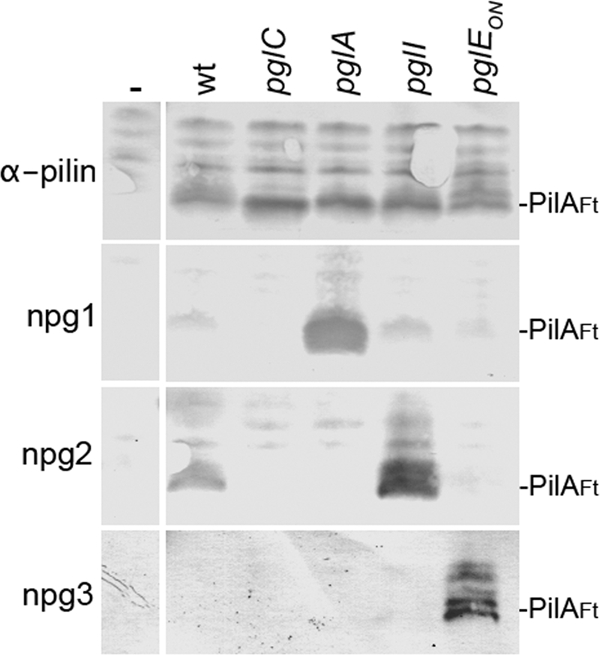
F. tularensis PilA is glycosylated by the endogenous O-linked system in N. gonorrhoeae. Immunoblotting of whole-cell lysates from defined backgrounds expressing PilA was performed. N. gonorrhoeae strains KS101 (lacking pilA) (-), KS67 (wt), KS182 (pglC), KS180 (pglA), KS190 (pglI), and KS186 (pglEon) were used. Immunoblotting utilized polyclonal anti-PilA serum (α-pilin) and the monoclonal antibodies npg1, npg2, and npg3 that recognize epitopes imparted by the presence of the DATDH, DATDH-Gal, and DATDH-Gal-Gal glycans, respectively. The positions of PilA from F. tularensis (PilAFt) are shown to the right of the blots.
Mass spectrometric analyses of F. tularensis subsp. tularensis PilA peptide digests.
As noted earlier, a polypeptide in which the FLAG epitope domain was fused to the C terminus of the pilA ORF was expressed in the type A strain SCHU S4 to facilitate characterization (see Fig. S1 in the supplemental material). Following purification, the fraction enriched in PilA was subjected to SDS-PAGE, and sequential gel slices were excised and processed by reduction, alkylation, and proteolytic digestion. It is not possible to digest PilA into peptides smaller than 3 kDa with trypsin. Therefore, we used a time-controlled digestion first with proteinase K and then with chymotrypsin in combination with trypsin proteolysis.
The proteinase K digest peptide mixture was analyzed using ion trap MS and manually inspected for typical fragment patterns related to glycosylation. Here, we used a loss of either 203 Da or 162 Da to identify glycopeptides with a glycan moiety containing N-acetylhexosamine (HexNAc) or hexose (Hex) residues, respectively. Figure 4 shows spectra of two peptides possessing related oligosaccharides based on the release of diagnostic reporter ions. Specifically, tandem MS (MS2) collision-induced dissociation (CID) of a species at 717.82+ generated two major daughter ions of m/z 934.2 and 501.2 along with a series of ions in the high-m/z region consistent with sequential losses of 203, 203, 162, 162, and 203 Da (Fig. 4A). Together, these data provided strong evidence for a peptide (signal at m/z 501.2) bearing a single pentasaccharide with the structure HexNAc-Hex-Hex-HexNAc-HexNAc (signal at m/z 934.2). In accordance with this, a dominant oxonium ion with a signal at m/z 407.2 corresponding to a HexNAcHexNAc moiety was also seen. Furthermore, the m/z 501.2 species and the m/z 331.1 species as a y4-fragment ion allowed the identification of the peptide as corresponding to residues 72 to 77 of PilA in its mature (prepilin peptidase cleaved) form. MS2 CID of a second species/signal at m/z 877.52+ generated a strikingly similar pattern of daughter ions at m/z 934.2 and m/z 407.2 together with a series of ions in the high-m/z region consistent with sequential losses of 203, 203, 162, 162, and 203 Da. In this case, the intact peptide (m/z 820.4) could be inferred to correspond to residues 101 to 108 of PilA in its mature form (Fig. 4B). Thus, two distinct PilA peptides carried a conserved pentasaccharide linked through a HexNAc residue (see Fig. S1 in the supplemental material).
Fig. 4.

Identification of glycopeptides and glycans derived from PilA of the type A strain SCHU S4. (A) MS2 CID spectrum generated using the ion trap mass spectrometer of the doubly charged peptide at m/z 717.82+ showing that the peptide 72GLPSGA77 derived from PilA is glycosylated. Abundance is shown as a percentage on the y axis. (B) MS2 CID spectrum generated using the ion trap mass spectrometer of the doubly charged peptide at m/z 877.52+ showing that the peptide 101VSSGAITW108 derived from PilA is glycosylated. Both spectra are dominated in the high-m/z region by the sequential losses of HexNAc and Hex carbohydrate moieties, allowing the identification of the glycan as a pentasaccharide consisting of HexNAc-Hex-Hex-HexNAc-HexNAc (gray squares, HexNAc; filled circles, Hex). pep, peptide.
Evidence for O-linked glycans at serine residues.
Locating the exact sites of glycosylation using conventional MS2 CID techniques is difficult owing to the relative lability of glycosidic bonds. We therefore used ion trap MS with electron transfer dissociation (ETD), a soft fragmentation technology that causes peptide backbone fragmentation due to rearrangement initiated by electron transfer to the peptide ion. Instead of fragmentation-induced generation of b- and y-ions, the major fragmentation process are cleavages of the N-Cα bonds along the peptide/protein backbone to give rise to complementary c- and z-type fragment ions. As shown for the 101VSSGAITW108 glycopeptide at m/z 877.5, the ETD spectrum provided the necessary peptide backbone fragmentation to indicate that serine residue 103 (S103) bears the glycan (Fig. 5). Its location to S103 is derived by detection of fragment ions c3+ (VSS) to c7+ (VSSGAIT) at m/z 1,224.4 to 1,566.5, respectively, representing five c-ions of the c-ion series c6+ to c11+ with a mass increase of 934.2 Da, which corresponds to the complete pentasaccharide glycan residue. However, the c2+ (VS) fragment ion at m/z 204.0 in its glycan-free form is of low abundance, and the corresponding glycan carrying C2+ ion at m/z 1,137.2 may be hidden in the noise of the MS2 spectrum. Therefore, the possibility that S102 may also carry the glycan cannot be completely excluded. Taken together with the observations that serine is the only hydroxyl-bearing residue within the peptide 72GLPSGA77 with an m/z of 501.2, these data strongly suggested that PilA protein glycosylation is attributable solely to linkage at serine residues.
Fig. 5.

Evidence for glycan O-linkage at serine residues. The ETD spectrum of the m/z 877.52+ precursor confirms the identity of the PilA peptide 101VSSGAITW108 and identifies serine 103 as the site of pentasaccharide occupancy. Abundance (in arbitrary units) is shown on the y axis. Its location to this residue is derived by detection of fragment ions c3+ (VSS) to c7+ (VSSGAIT) at m/z 1,224.4 to 1,566.5, respectively, representing five c-ions of the c-ion series c6+ to c11+ with a mass increase of 934.2 Da, which corresponds to the complete pentasaccharide glycan residue. Furthermore, the c2+ (VS) fragment ion at m/z 204.0 was detected only in its glycan-free form.
Mass spectrometric characterization of atypical glycan modifications.
A chymotrypsin/trypsin digest of PilA was further analyzed with the Orbitrap XL mass spectrometer using the higher-energy collisional fragmentation (HCD) mode. Those spectra within the ion chromatograms yielding fragment signals at m/z 204.086 (indicative of HexNAc residues) were then subjected to further evaluation. The HCD fragmentation spectrum of a doubly charged ion at m/z 1,068.5 confirmed the presence of a PilA glycopeptide carrying the earlier detected pentasaccharide but with a longer overlapping proteolytic backbone (97LAPTVSSGAITW108) (see Fig. S2 in the supplemental material). Surprisingly, additional signals were observed that were consistent with the presence of other glycopeptides with identical amino acid sequence but extended modifications of the already described pentasaccharide (see Fig. S4A in the supplemental material). The mass of one form observed corresponded to a peptide precursor signal at m/z 1,180.532+ and was consistent with the addition of a unknown moiety related to a mass increase of 242.078 Da to the pentasaccharide (Fig. 6A). Further information on the nature of m/z 242.078 species was derived from reporter ions in the low-mass-range pattern generated by HCD of the doubly charged ion at m/z 1,180.53. In addition to signals that could be attributed to HexNAc-associated fragment ions (at m/z 126.054, 138.053, 144.064, and 168.064), ions indicative of the presence of a phosphate moiety were observed (Fig. 6B). The latter included ions at m/z 144.101 and m/z 162.111 that could most plausibly be assigned to the neutral loss of phosphoric acid (97.977 Da) and metaphosphoric acid (79.966 Da) from the ion at m/z 242.078. MS3 of m/z 1,180.532+ confirmed this precursor-product relationship, as it gave a fragment ion at m/z 144.101 that could only be accounted for by the loss of phosphoric acid (data not shown). Moreover, an abundant ion at m/z 186.075 could represent the neutral loss of phosphoric acid from the species at m/z 284.05, suggesting that one HexNAc might be phosphorylated. This possibility was confirmed by examining a triple charge potassium adduct modified form of the 97LAPTVSSGAITW108 peptide at m/z 799.6713+ (Fig. 7). Here, the successive loss of the subunits of the pentasaccharide detected by the decreasing fragment ion signals at m/z 1,078.465, 985.930, 884.390, 722.337, and 620.797 revealed the presence of dehydrated HexNAc at the nonreducing end of the glycan that could be explained only by the neutral loss of phosphoric acid. Together, these data strongly suggest that the pentasaccharide is linked through its terminal HexNAc residue to a 162.111-Da moiety via a phosphate bridge.
Fig. 6.

Evidence for a glycan-associated phosphate and its association with the m/z 242.078 species. (A) MS2 spectrum of the doubly charged peptide at m/z 1,180.532+ (eluting at 70.76 min) confirms that the peptide 97LAPTVSSGAITW108 is modified with the pentasaccharide linked to a moiety of m/z 242.078. The identity of the peptide is confirmed by the fragment ions b8 to b11 at m/z 713.383 to 998.553, respectively, as well as the accurate peptide mass of 1202.641 Da (+2.5 ppm). P-compound X and P-HexNAC, phosphorylated compound X and HexNAC, respectively. (B) Fragment ions in the lower-m/z region generated by HCD MS2 analysis related to the m/z 242.067 species. The MS2 spectrum in the mass range from 70 to 250 m/z generated by analysis of doubly charged peptide at m/z 1,180.532+ is shown. Signals attributable to HexNAc-associated fragment ions are m/z 126.054, 138.053, 144.064, and 168.064. Ions indicative of a phosphate moiety were observed including those at 144.101 m/z and 162.111 m/z that could most plausibly assigned to the neutral loss of phosphoric acid (97.977 Da) and metaphosphoric acid (79.966 Da) from the ion at m/z 242.077. An abundant ion at m/z 186.075 could represent the loss of phosphoric acid from the species at m/z 284.05, suggesting that one HexNAc is phosphorylated.
Fig. 7.

Evidence that the glycan-associated phosphate is linked to the distal HexNAC residue. (A) MS2 (HCD) spectrum of the peptide 97LAPTVSSGAITW108 carrying the pentasaccharide with the extended version of 224.067 Da at m/z 799.6713+ (triply charged potassium adduct) using the Orbitrap XL mass spectrometer. The successive loss of the glycan moiety subunits detected by the decreasing fragment ion signals at m/z 1,078.465, 985.930, 884.390, 722.337, and 620.797 unambiguously determine that the pentasaccharide is attached in the orientation HexNAc-Hex-Hex-HexNAc-HexNAc to the peptide backbone. Furthermore, it suggests that the distal HexNAc is phosphorylated, indicated by its detected β-elimination (dehydro-HexNAc; mass difference, 185.069 instead of 203.086 Da).
The masses of the other two forms seen in the ion chromatograms corresponded to peptide precursor signals at m/z 1,166.512+ and 1,173.522+ (see Fig. S4B in the supplemental material) that could be attributed to the lack of one and two methyl groups, respectively, from the 162.111-Da moiety. The varied methylation status could be unambiguously defined by the exact mass differences of 14.016 Da between all three forms (Fig. S4B). Accordingly, two new reporter ions with m/z 228.062 and 214.046 appeared in the low-mass range in addition to m/z 204.086 species corresponding to HexNAc. In addition, each of these spectra contained a unique, abundant species with m/z 284.051 corresponding to a phosphorylated HexNAc. As each glycopeptide eluted at distinct retention times, these are distinct glycan modifications rather than experimental artifacts generated by in-source fragmentation.
Additional signals in the low-mass range at m/z 86.095, 100.111, 130.085, and 228.062 provided information relevant to the nature of the m/z 242.078 moiety (Fig. 6B). These signals are likely to be secondary fragment ions from further collision of the unknown ion signal. There is no direct evidence that those signals are related to further fragmentation of 242.078, 162.111, or 144.101, respectively. However, the exact mass difference of 43.990 Da between 100.111 to 144.101 can be explained only by the loss of CO2 and therefore suggests strongly that 100.111 is a further fragment of 144.101. In addition, the compound should have a carboxyl group, a core structure to allow dimethylation, and a hydroxyl group in order to be modified with a phosphate group. Furthermore, the nitrogen rule suggests the presence of an odd number of nitrogen atoms. Accurate mass measurements of the new fragment ions in the low-mass range were thus used to generate plausible elemental compositions, and the top-ranked formulae are shown in Table 3. The top-ranked elemental composition for the m/z 162.1112 moiety indicated this to be a compound with the formula C7H16NO3.
Table 3.
Accurate masses of the glycan oxonium- and glycan-related fragment ions and most plausible empirical formulae
| Observed m/z | Calculated m/z | Elemental composition | Δ m/z (mmu)a | Gross formula shift related to 242.078 |
|---|---|---|---|---|
| 86.0952 | 86.0963 | C5H12N | −1.764 | -HPO3, -H2O, -CO2, -CH2 |
| 100.1108 | 100.1120 | C6H14N | −1.794 | -HPO3, -H2O, -CO2 |
| 130.0849 | 130.0862 | C6H12O2N | −1.854 | -HPO3, -H2O, -CH2 |
| 144.1006 | 144.1018 | C7H14O2N | −1.824 | -HPO3, -H2O |
| 162.1112 | 162.1124 | C7H16O3N | −1.828 | -HPO3 |
| 228.0620 | 228.0631 | C6H15O6NP | −1.689 | -CH2 |
| 242.0776 | 242.0787 | C7H17O6NP | −1.759 | |
| 204.0855 (HexNAC control) | 204.0865 | C8H14O5N | −1.717 |
Difference between observed and predicted m/z values, in molecular mass units (mmu).
Direct evidence that PilA is nonglycosylated in a pglA background.
We sought to confirm that the altered migration of PilA in pglA null backgrounds was indicative of defective glycosylation. Accordingly, affinity-tagged forms were isolated from the wild-type type B strain FSC200 and its isogenic mutant and examined by MS protocols detailed previously (Fig. 8). Only analyses of digests from the wild-type background revealed the detection of a peak eluting at 37.25 min in the total ion chromatogram (TIC) related to the pentasaccharide-modified peptide 97LAPTVSSGAITW108. Corresponding MS2 analysis confirmed the detection of the earlier described glycopeptides (see Fig. S5 in the supplemental material). The peaks related to the unmodified peptide were detected in the TIC at 39.4 to 39.5 min in both backgrounds (Fig. 8, middle panels). Their relative abundance suggested that the concentration of unmodified peptide was approximately 10 times greater for the mutant than for the wild-type samples. Thus, the pglA null mutation was directly correlated with defective PilA glycosylation.
Fig. 8.

Characterization of PilA purified from type B strains FSC749 (wild type [left panels]) and FSC703 (ΔpglA background [right panels]). Base peak and selected ion chromatograms of chymotrypsin digests of PilA isolated from the two backgrounds were analyzed by nanoflow on-line LC-MS and manually inspected for glycan-related reporter ions and known precursor ions corresponding to modified PilA peptides. The top panel represents the base peak chromatograms (BPC), and the middle and bottom panels show the selected ion (SIC) chromatograms of precursor ions at m/z 1,202.6411+ (unmodified peptide 97LAPTVSSGAITW108) and it pentasaccharide modified form at m/z 1,129.0352+, respectively. Only the sample from the wild-type background showed a peak at 37.25 min related to the pentasaccharide modified peptide 97LAPTVSSGAITW108. Corresponding MS2 analysis confirmed the detection of the earlier described glycopeptides (see Fig. S2 in the supplemental material). The peak related to the unmodified peptide was detected at 39.4 to 39.5 min in both backgrounds (middle panels). Their abundance suggests that the concentration of the unmodified peptide in the sample from the ΔpglA background was approximately 10 times higher than that seen in the sample from the wild-type background (gray squares, HexNAc; filled circles, Hex).
DISCUSSION
Evidence from a number of studies indicate that the pilin-like PilA proteins of both type A and type B strains as well as that expressed by strains of F. novicida influence virulence-related phenotypes in animal models of disease (14, 16, 30). However, the manner by which PilA exerts its effects in these systems and its associated structure-function relationships remain poorly understood. In this study, we identified PglA as the endogenous PilA-targeting oligosaccharyltransferase in one F. tularensis subspecies background. Based on its structural similarities and functional activity, PglA represents the third member of an expanding family of bacterial oligosaccharyltransferases responsible for O-linked protein glycosylation utilizing lipid-linked glycan precursors.
In addition, we demonstrate that the PilA protein of two of the human-pathogenic subspecies of F. tularensis is subject to glycosylation with an unusually modified pentasaccharide that is targeted to a minimum of two defined serine attachment sites.
The identification of the PilA-associated phosphoglycan form and its linkage to compound X are particularly noteworthy. Although phosphoglycans are well-established constituents of cell surface- and cell wall-associated polysaccharides of many bacterial species, we are aware of only two other examples of phosphoglycans in protein-linked carbohydrate systems. These are the O-linked glycan attached to flagellin in some strains of Clostridium difficile (37) and Pseudomonas aeruginosa (37a). The phosphates there are proposed to bridge sugars to a methylated form of aspartic acid and an unknown terminal modification, respectively. The data on the Francisella phosphoglycan are consistent with a similar structural organization to these other systems. Further studies are needed to define the biosynthetic pathways operating in these protein-associated phosphoglycan systems and to confirm the identities of the moieties terminal to the phosphate groups. In this context, it should be clarified that the detection of the pentasaccharide modification in some cases and its further elaborated phosphoglycan form in others relates to the different MS methodologies employed. Specifically, this reflects the propensity for the phosphate-linked compound X to be lost due to fragmentation during the ion trap-based MS but not in Orbitrap HCD analysis.
Knowing the function of PglA in O-linked protein glycosylation provides further insights related to earlier observations made in the field. Specifically, an in vivo negative-selection screen using F. novicida mutants identified the orthologue of PglA as contributing to growth and survival in mice (38). Moreover, pglA expression in F. novicida has been shown to be controlled by MglA, a transcription factor that coordinately regulates expression of several genes necessary for replication in macrophages and for virulence in mice (9). Taken together, these data hint at an influential role of protein glycosylation in vivo.
It remains to be definitively determined whether the system described here is dedicated solely to PilA or whether other protein substrates are modified. In another recent study, putative glycoproteins in the type B strain FSC200 were identified through a combination of carbohydrate-related chemical detection, lectin affinity, and MS methodologies and included among these were PilA (6). It seems plausible then that broad-spectrum protein glycosylation occurs in both type A and type B strains within the F. tularensis complex. Similarly, as homologues of PilA and PglA are each readily identifiable within the genomes of less related Francisella species such as Francisella philomiragia, we hypothesize that O-linked pilin glycosylation is a general feature of strains within the genus Francisella.
In summary, we demonstrate that both type A and type B strains of Francisella subspecies express an PilA-targeting O-linked protein glycosylation system utilizing core biosynthetic and assembly pathways conserved in other members of the proteobacteria. Furthermore, a number of reports suggest that PilA may contribute to the virulence phenotypes of species within the genus Francisella. Thus, further studies of both PilA and protein glycosylation may significantly enhance our understanding of these intriguing and important pathogens.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported in part by Research Council of Norway grants 166931 and 183814, the Functional Genomics initiative (FUGE) directed through the Consortium of Advanced Microbial Sciences and Technologies (CAMST), and by funds and infrastructure from the Department of Molecular Biosciences, Centre for Molecular Biology and Neurosciences, and Mathematics and Natural Sciences Faculty at the University of Oslo.
We thank Stephen Michell and Helen Sharps for technical assistance and Einar Uggerud for analyses of the MS data relating to the oligosaccharide modifications.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
Published ahead of print on 29 July 2011.
REFERENCES
- 1. Aas F. E., Vik A., Vedde J., Koomey M., Egge-Jacobsen W. 2007. Neisseria gonorrhoeae O-linked pilin glycosylation: functional analyses define both the biosynthetic pathway and glycan structure. Mol. Microbiol. 65:607–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abd H., Johansson T., Golovliov I., Sandström G., Forsman M. 2003. Survival and growth of Francisella tularensis in Acanthamoeba castellanii. Appl. Environ. Microbiol. 69:600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alm R. A., Hallinan J. P., Watson A. A., Mattick J. S. 1996. Fimbrial biogenesis genes of Pseudomonas aeruginosa: pilW and pilX increase the similarity of type 4 fimbriae to the GSP protein-secretion systems and pilY1 encodes a gonococcal PilC homologue. Mol. Microbiol. 22:161–173 [DOI] [PubMed] [Google Scholar]
- 4. Alm R. A., Mattick J. S. 1995. Identification of a gene, pilV, required for type 4 fimbrial biogenesis in Pseudomonas aeruginosa, whose product possesses a pre-pilin-like leader sequence. Mol. Microbiol. 16:485–496 [DOI] [PubMed] [Google Scholar]
- 5. Alm R. A., Mattick J. S. 1996. Identification of two genes with prepilin-like leader sequences involved in type 4 fimbrial biogenesis in Pseudomonas aeruginosa. J. Bacteriol. 178:3809–3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Balonova L., et al. 2010. Multimethodological approach to identification of glycoproteins from the proteome of Francisella tularensis, an intracellular microorganism. J. Proteome Res. 9:1995–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blatny J. M., Brautaset T., Winther-Larsen H. C., Karunakaran P., Valla S. 1997. Improved broad-host-range RK2 vectors useful for high and low regulated gene expression levels in gram-negative bacteria. Plasmid 38:35–51 [DOI] [PubMed] [Google Scholar]
- 8. Borud B., et al. 2010. Genetic, structural, and antigenic analyses of glycan diversity in the O-linked protein glycosylation systems of human Neisseria species. J. Bacteriol. 192:2816–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brotcke A., et al. 2006. Identification of MglA-regulated genes reveals novel virulence factors in Francisella tularensis. Infect. Immun. 74:6642–6655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dennis D. T., et al. 2001. Tularemia as a biological weapon: medical and public health management. JAMA 285:2763–2773 [DOI] [PubMed] [Google Scholar]
- 11. DiGiandomenico A., et al. 2002. Glycosylation of Pseudomonas aeruginosa 1244 pilin: glycan substrate specificity. Mol. Microbiol. 46:519–530 [DOI] [PubMed] [Google Scholar]
- 12. Faridmoayer A., Fentabil M. A., Mills D. C., Klassen J. S., Feldman M. F. 2007. Functional characterization of bacterial oligosaccharyltransferases involved in O-linked protein glycosylation. J. Bacteriol. 189:8088–8098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feldman M. F., et al. 2005. Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 102:3016–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Forslund A. L., et al. 2006. Direct repeat-mediated deletion of a type IV pilin gene results in major virulence attenuation of Francisella tularensis. Mol. Microbiol. 59:1818–1830 [DOI] [PubMed] [Google Scholar]
- 15. Golovliov I., Baranov V., Krocova Z., Kovarova H., Sjöstedt A. 2003. An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect. Immun. 71:5940–5950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hager A. J., et al. 2006. Type IV pili-mediated secretion modulates Francisella virulence. Mol. Microbiol. 62:227–237 [DOI] [PubMed] [Google Scholar]
- 17. Han X., et al. 2008. Twitching motility is essential for virulence in Dichelobacter nodosus. J. Bacteriol. 190:3323–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johansson A., et al. 2004. Worldwide genetic relationships among Francisella tularensis isolates determined by multiple-locus variable-number tandem repeat analysis. J. Bacteriol. 186:5808–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khan A. S., Morse S., Lillibridge S. 2000. Public-health preparedness for biological terrorism in the USA. Lancet 356:1179–1182 [DOI] [PubMed] [Google Scholar]
- 20. Kirn T. J., Bose N., Taylor R. K. 2003. Secretion of a soluble colonization factor by the TCP type 4 pilus biogenesis pathway in Vibrio cholerae. Mol. Microbiol. 49:81–92 [DOI] [PubMed] [Google Scholar]
- 21. Laemmli U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 22. Milton D., O'Toole R., Horstedt P., Wolf-Watz H. 1996. Flagellin A is essential for the virulence of Vibrio anguillarum. J. Bacteriol. 178:1310–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nudleman E., Kaiser D. 2004. Pulling together with type IV pili. J. Mol. Microbiol. Biotechnol. 7:52–62 [DOI] [PubMed] [Google Scholar]
- 24. Pavlov V., Mokrievich A., Volkovoy K. 1996. Cryptic plasmid pFNL10 from Francisella novicida-like F6168: the base of plasmid vectors for Francisella tularensis. FEMS Immunol. Med. Microbiol. 13:253–256 [DOI] [PubMed] [Google Scholar]
- 25. Petersen J., Schriefer M. 2005. Tularemia: emergence/re-emergence. Vet. Res. 36:455–467 [DOI] [PubMed] [Google Scholar]
- 26. Power P. M., Seib K. L., Jennings M. P. 2006. Pilin glycosylation in Neisseria meningitidis occurs by a similar pathway to wzy-dependent O-antigen biosynthesis in Escherichia coli. Biochem. Biophys. Res. Commun. 347:904–908 [DOI] [PubMed] [Google Scholar]
- 27. Qutyan M., Paliotti M., Castric P. 2007. PilO of Pseudomonas aeruginosa 1244: subcellular location and domain assignment. Mol. Microbiol. 66:1444–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rohmer L., et al. 2007. Comparison of Francisella tularensis genomes reveals evolutionary events associated with the emergence of human pathogenic strains. Genome Biol. 8:R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Salomonsson E., et al. 2009. Functional analyses of pilin-like proteins from Francisella tularensis: complementation of type IV pilus phenotypes in Neisseria gonorrhoeae. Microbiology 155:2546–2559 [DOI] [PubMed] [Google Scholar]
- 30. Salomonsson E., et al. 2009. Reintroduction of two deleted virulence loci restores full virulence to the live vaccine strain of Francisella tularensis. Infect. Immun. 77:3424–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sambrook J., Fritsch E. F., Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 32. Simon R., Priefer U. B., Pühler A. 1983. A broad host range mobilization system for in vitro genetic engineering transposon mutagenesis in gram negative bacteria. Biotechnology 1:784–791 [Google Scholar]
- 33. Skerker J. M., Berg H. C. 2001. Direct observation of extension and retraction of type IV pili. Proc. Natl. Acad. Sci. U. S. A. 98:6901–6904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Strom M., Lory S. 1993. Structure-function and biogenesis of the type IV pili. Annu. Rev. Microbiol. 47:565–596 [DOI] [PubMed] [Google Scholar]
- 35. Strom M., Nunn D., Lory S. 1993. A single bifunctional enzyme, PilD, catalyzes cleavage and N-methylation of proteins belonging to the type IV pilin family. Proc. Natl. Acad. Sci. U. S. A. 90:2404–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tärnvik A. 1989. Nature of protective immunity to Francisella tularensis. Rev. Infect. Dis. 11:440–451 [PubMed] [Google Scholar]
- 37. Twine S. M., et al. 2009. Motility and flagellar glycosylation in Clostridium difficile. J. Bacteriol. 191:7050–7062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37a. Verma A., et al. 2006. Glycosylation of b-type flagellin of Pseudomonas aeruginosa: structural and genetic basis. J. Bacteriol. 188:4395–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weiss D., et al. 2007. In vivo negative selection screen identifies genes required for Francisella virulence. Proc. Natl. Acad. Sci. U. S. A. 104:6037–6042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Whipp M., et al. 2003. Characterization of a novicida-like subspecies of Francisella tularensis isolated in Australia. J. Med. Microbiol. 52:839–842 [DOI] [PubMed] [Google Scholar]
- 40. Winther-Larsen H. C., et al. 2001. Neisseria gonorrhoeae PilV, a type IV pilus-associated protein essential to human epithelial cell adherence. Proc. Natl. Acad. Sci. U. S. A. 98:15276–15281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Winther-Larsen H. C., et al. 2005. A conserved set of pilin-like molecules controls type IV pilus dynamics and organelle-associated functions in Neisseria gonorrhoeae. Mol. Microbiol. 56:903–917 [DOI] [PubMed] [Google Scholar]
- 42. Wolfgang M., van Putten J. P., Hayes S. F., Dorward D., Koomey M. 2000. Components and dynamics of fiber formation define a ubiquitous biogenesis pathway for bacterial pili. EMBO J. 19:6408–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wolfgang M., van Putten J. P., Hayes S. F., Koomey M. 1999. The comP locus of Neisseria gonorrhoeae encodes a type IV prepilin that is dispensable for pilus biogenesis but essential for natural transformation. Mol. Microbiol. 31:1345–1357 [DOI] [PubMed] [Google Scholar]
- 44. Zogaj X., Chakraborty S., Liu J., Thanassi D., Klose K. 2008. Characterization of the Francisella tularensis subsp. novicida type IV pilus. Microbiology 154:2139–2150 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.