

Abstract
To facilitate the study of pneumococcal genes that are essential for viability or normal cell growth, we sought to develop a tightly regulated, titratable gene depletion system that interferes minimally with normal cellular functions. A possible candidate for such a system is the recently discovered signal transduction pathway regulating competence for natural transformation in Streptococcus thermophilus. This pathway, which is unrelated to the ComCDE pathway used for competence regulation in Streptococcus pneumoniae, has not been fully elucidated, but it is known to include a short unmodified signaling peptide, ComS*, an oligopeptide transport system, Ami, and a transcriptional activator, ComR. The transcriptional activator is thought to bind to an inverted repeat sequence termed the ECom box. We introduced the ComR protein and the ECom box into the genome of S. pneumoniae R6 and demonstrated that addition of synthetic ComS* peptide induced the transcription of a luciferase gene inserted downstream of the ECom box. To determine whether the ComRS system could be used for gene depletion studies, the licD1 gene was inserted behind the chromosomally located ECom box promoter by using the Janus cassette. Then, the native versions of licD1 and licD2 were deleted, and the resulting mutant was recovered in the presence of ComS*. Cultivation of the licD1 licD2 double mutant in the absence of ComS* gradually affected its ability to grow and propagate, demonstrating that the ComRS system functions as intended. In the present study, the ComRS system was developed for use in S. pneumoniae. In principle, however, it should work equally well in many other Gram-positive species.
INTRODUCTION
Gene disruption studies have shown that the genome of Streptococcus pneumoniae R6 contains at least 133 essential genes, 32 of which have no known function (23, 25). As these studies were carried out with laboratory-grown pneumococci, it is reasonable to assume that additional genes are essential for survival under natural conditions. For obvious reasons, functional studies of essential genes are experimentally demanding. The best approach is probably to express essential genes ectopically under the control of a tightly regulated, titratable promoter. This allows deletion of the native gene, while the level of transcription of the ectopically expressed gene can be manipulated to gain insight into its function. The same technique ought to be applied to studies of growth-defective genes whose absence affects bacterial growth and proliferation. In this way it should be possible to avoid the selection pressure exerted by deletion of growth-defective genes that gives rise to suppressor mutations which mask or distort the real phenotype of the mutant.
Ideally, gene expression/depletion systems should not interfere with the normal physiology of the host bacterium. The ComRS signal transduction pathway, which regulates competence for natural transformation in Streptococcus thermophilus (9), has no close homologs in S. pneumoniae. We therefore considered it a promising tool for gene depletion studies of the pneumococcus. The licD1 and licD2 genes, which are involved in the synthesis of teichoic acid in S. pneumoniae (26), were selected as target genes in an initial test of the system. The licD genes are located on a transcriptional unit consisting of tacF, licD1, and licD2. The first gene in the operon, tacF, encodes a transporter that is required for the transport of teichoic acid subunits (or polymerized chains) across the cytoplasmic membrane (1, 5). In S. pneumoniae, the chemical structure of the repeating units in cell wall teichoic acid (WTA) and lipoteichoic acid (LTA) are identical. In general, each repeat unit contains two phosphoryl choline residues, one attached to the central N-acetyl-d-galactosamine residue and the other to the ribitol-linked N-acetyl-d-galactosamine residue (8). LicD1 and LicD2 attach phosphoryl choline residues to the teichoic acid subunits on the cytoplasmic side of the membrane, before they are exported by TacF (5). As TacF has a strict specificity for subunits containing phosphoryl choline, extracellular synthesis of WTA and LTA will probably be arrested in the absence of LicD1 and LicD2. It has been reported that the licD2 gene of S. pneumoniae can be readily deleted, while attempts to construct a licD1 deletion mutant were unsuccessful (26). This result suggests that licD1 might be an essential gene. Alternatively, the observed lethal phenotype of the ΔlicD1 mutant could be due to a polar effect on the downstream licD2 gene which is exerted by the pIH1 plasmid used to disrupt the licD1 gene by insertion-duplication mutagenesis. Zhang and coworkers (26) favored the latter alternative and predicted that deletion of both licD genes is lethal for S. pneumoniae. Our results confirmed this prediction but showed that it is possible to construct a pneumococcal licD1 licD2 double deletion mutant if licD1 is ectopically expressed by the ComRS system. Removal of ComS* from the growth medium had no immediate effect, but after 4 to 6 h severe morphological abnormalities were observed. A few hours later the stress imposed by the gradual reduction in LicD1 expression culminated in growth arrest followed by autolysis. We believe the ComRS system will become a valuable addition to the genetic toolbox available for S. pneumoniae.
MATERIALS AND METHODS
Construction of S. pneumoniae mutants.
S. pneumoniae strains and plasmids used in this work are described in Table 1. All transformations and experiments were carried out in C medium (18) at 37°C. However, Todd-Hewitt (Difco) agar plates containing the appropriate antibiotic were used for selection of transformants. The sequences of all primers used are given in Table 2. To construct mutant strains, DNA was introduced into the recipients by natural transformation. Bacterial cultures were grown to an optical density at 550 nm (OD550) of ∼0.05 to 0.1 and induced to competence by adding synthetic competence-stimulating peptide (CSP) (11) to a final concentration of 250 ng ml−1. Then, the transforming DNA was added, and the cultures were further incubated for 120 min at 37°C. Selection of transformed cells was carried out on Todd-Hewitt agar containing the appropriate antibiotic at the following concentrations: streptomycin (Sm), 200 μg ml−1; kanamycin (Kan), 400 μg ml−1. When needed, 2 μM synthetic ComS* (NH2-LPYFAGCL-COOH) (Genosphere Biotechnologies) was included in the C medium during growth and transformation as well as in the Todd-Hewitt agar plates.
Table 1.
Bacterial strains and plasmids
| Strain or plasmid | Genotype/relevant featuresa | Reference/source |
|---|---|---|
| S. pneumoniae strains | ||
| R704 | R6 derivative, comA::ermAM; Eryr | J. P. Claverysc |
| RH1 | R704, but ebg::spc; Eryr Spcr | 13 |
| RH426 | RH425, but ΔIS1167::Janusb; Eryr Kanr | 14 |
| SPH124 | RH426, but with replacement of Janus cassette by PcomR::comR; Eryr Smr | This study |
| SPH125 | SPH124, but with Janus cassette inserted between cpsO and cpsN; Eryr Kanr | This study |
| SPH126 | SPH125, but with replacement of Janus cassette by PcomX::luc; Eryr Smr | This study |
| SPH127 | SPH126, but with replacement of spr0324 by Janus cassette; Eryr Kanr | This study |
| SPH128 | SPH127, but with replacement of Janus cassette by amiA3; Eryr Smr | This study |
| SPH129 | SPH126, but with replacement of comR by Janus cassette; Eryr Kanr | This study |
| SPH130 | SPH129, but with replacement of Janus cassette with P1::PcomR::comR; Eryr Smr | This study |
| SPH131 | SPH130, but with replacement of luc by Janus cassette; Eryr Kanr | This study |
| SPH132 | SPH131, but with replacement of Janus cassette by licD1; Eryr Smr | This study |
| SPH135 | SPH132, but with replacement of native licD1 and licD2 genes by Janus cassette; Eryr Kanr | This study |
| SPH136 | SPH130, but with replacement of luc by the aacA-aphD kanamycin resistance gene; Eryr Smr Kanr | This study |
| Plasmids | ||
| pR424 | ColE1 (pEVP3) derivative; Cmr; carries the luc gene | 4 |
| pFW13 | Carries the aacA-aphD kanamycin resistance gene | 22 |
Cm, chloramphenicol; Ery, erythromycin; Kan, kanamycin; Sm, streptomycin.
Janus indicates the presence of a kan::rpsL+ cassette (24).
Gift from Jean-Pierre Claverys.
Table 2.
Primers
| Primer | Oligonucleotide sequence (5′→3′) | Reference |
|---|---|---|
| 16S F | ACCGCATAAGAGTGGATG | 20 |
| 16S R | CAACGCAGGTCCATCTGGTA | 20 |
| AmiF | CGGTGAAGGAAGTAAGAAGTTT | 14 |
| AmiR | TCAAACTTATCAAGCGCAATGC | This study |
| AmiR1 | AATGGAACCTCCACAAGTATTTTCTAGTATTATAGCACATTTAATCAAACTTATCAAGCGCAATGCCTTTG | 14 |
| cbpDF | ATGAAAATTTTACCGTTTATAG | This study |
| cbpDR | ATTTCCTCTGGAATAGGCATAG | This study |
| Kan484.F | GTTTGATTTTTAATGGATAATGTG | 13 |
| khb29 | GGGTAAATCCCTTATAGATATTATGGAGTTTCTATAAACCAT CTGCCAATT | This study |
| khb31 | ATAACAAATCCAGTAGCTTTGG | This study |
| khb32 | GGTCTAGAGATGATTTTAATTAC | This study |
| khb33 | TTTCTAATATGTAACTCTTCCCAAT | This study |
| khb34 | CATCGGAACCTATACTCTTTTAG | This study |
| khb35 | ATTAAAATCATCTCTAGACCGGTTTTGACTCTATCTCGCTT | This study |
| khb36 | TGAACCTCCAATAATAAATATAAAT | This study |
| khb37 | ATTTATATTTATTATTGGAGGTTCAATGAGATCCGCCAAAAACATA | This study |
| khb38 | ATTGGGAAGAGTTACATATTAGAAATTACAATTTGGGCTTTCCGCC | This study |
| khb41 | ATTAAAATCATCTCTAGACCGTTTGATTTTTAATGGATAATGTG | This study |
| khb42 | ATTGGGAAGAGTTACATATTAGAAACTTTCCTTATGCTTTTGGAC | This study |
| khb43 | GCATTGCGCTTGATAAGTTTGAGGATAATCAAGATTTATCTTATAAA | This study |
| khb50 | CCGATGCAGAAATGGTTGAG | This study |
| khb51 | GATAATCGTACATCTGAAGCTC | This study |
| khb52 | TTGGTAAACTACGAACCGCTAG | This study |
| khb53 | TCATTGTAAGCGCCCAATAAC | This study |
| khb54 | GAGCTTCAGATGTACGATTATCGTTTGATTTTTAATGGATAATGTG | This study |
| khb55 | CTAGCGGTTCGTAGTTTACCAACTTTCCTTATGCTTTTGGAC | This study |
| khb58 | GAGCTTCAGATGTACGATTATCAGGAATTACAGTTGTTATCGTG | This study |
| khb59 | CTAGCGGTTCGTAGTTTACCAACGTGTCTTCATCACACTGAAC | This study |
| khb60 | ATTTATATTTATTATTGGAGGTTCAGTTTGATTTTTAATGGATAATGTG | This study |
| khb61 | ATTTATATTTATTATTGGAGGTTCAATGAAACAACTAACCGTTGAAG | This study |
| khb62 | ATTGGGAAGAGTTACATATTAGAAACCTAATCCTCCAATTTATAAGC | This study |
| khb63 | TTTAGATCGCCTCTTCCTCG | This study |
| khb64 | TTTAAACTCCTATGATTTTTTA | This study |
| khb67 | TAAAAAATCATAGGAGTTTAAAGTTTGATTTTTAATGGATAATGTG | This study |
| khb68 | ATTTCTTTTTTTTCTAAATATTGCATCTTTCCTTATGCTTTTGGAC | This study |
| khb69 | ATTTCTTTTTTTTCTAAATATTGCATTTTAAACTCCTATGATTTTTTA | This study |
| khb70 | TAAAAAATCATAGGAGTTTAAAATGCAATATTTAGAAAAAAAAGAAAT | This study |
| khb72 | TTAAATGTGCTATAATACTAGAAAATACTTGTGGAGGTTCCATTGGATAATCAAGATTTATCTTATAAA | This study |
| khb79 | ATATTTATTATTGGAGGTTCAATGAATATAGTTGAAAATGAAATATG | This study |
| khb80 | AGAGTTACATATTAGAAACTCCGGATGCATATGCATGC | This study |
| khb84 | GTCCAAAAGCATAAGGAAAGCTATAAGTATGTGGCTCTTCG | This study |
| khb85 | CTGCTGACGAAGATGGTGAG | This study |
| khb86 | GTCGACAATGCTCAAGTCTGTTTTAAACTCCTATGATTTTTTA | This study |
| khb87 | TAAAAAATCATAGGAGTTTAAAACAGACTTGAGCATTGTCGAC | This study |
| khb88 | ATTAAAATCATCTCTAGACCAAAGCCGGGAATTTTCCCGGCTTTTTTCTTGGTTTTGACTCTATCTCGCTT | This study |
| khb89 | AAGAAAAAAGCCGGGAAAATTCCCGGCTTTGGTCTAGAGATGATTTTAATTAC | This study |
| lucF | ACCAGAGTCCTTTGATCGTG | This study |
| lucR | GAAAATAGGGTTGGTACTAGC | This study |
| RpsL41.R | CTTTCCTTATGCTTTTGGAC | 13 |
| TreF | CTCCATAATATCTATAAGGGATTTA | 14 |
| TreR | GTGACGGCAGTCACATTCTC | 14 |
Pneumococcus strain SPH124 was derived from strain RH426 by replacing its Janus cassette (24) with the stu0270 gene (designated comR) from S. thermophilus by double-crossover homologous recombination. Janus is a kan rpsL+ DNA cassette that confers resistance to kanamycin and dominant sensitivity to streptomycin in a streptomycin-resistant background. Replacement of the Janus cassette restores streptomycin resistance and kanamycin sensitivity. Fragments corresponding to the ∼1,000-bp upstream and downstream regions of the Janus cassette in RH426 were amplified using the primer pairs AmiF/AmiR and TreF/TreR, respectively. comR, including its upstream promoter region, was amplified using the primer pair khb43/khb29 with genomic DNA from S. thermophilus LMG18311 (2) as template and then fused to the upstream and downstream fragments by overlap extension PCR using the primers AmiF and TreR according to the method of Higuchi et al. (12). All PCR-based DNA-fragment fusions described in the present work were carried out using the method of Higuchi and coworkers.
Strain SPH125 was constructed by inserting the Janus cassette between the capsular genes cpsO (spr0323) and cpsN (spr0322) of the SPH124 strain. The Janus cassette was amplified by PCR using the primer pair khb41/khb42 with genomic DNA from RH426 as template. An ∼800-bp DNA fragment corresponding to the 5′ end of the cpsO gene was amplified using the primers khb31 and khb32, whereas an ∼800-bp DNA fragment corresponding to the 3′ region of the cpsN gene was amplified using the primers khb33 and khb34. These two fragments were subsequently fused to the 5′ and 3′ ends of the Janus cassette, respectively, using the primers khb31 and khb34.
Next, the Janus cassette in SPH125 was replaced by PcomX::luc, giving rise to strain SPH126. First, the promoter PcomX was amplified from S. thermophilus LMG18311 genomic DNA using the primers khb35 and khb36. The luc gene was amplified from pR424 (4) using the primer pair khb37/khb38. PcomX was then fused to the 5′ end of luc using the primers khb35 and khb38. The resulting fragment was subsequently fused to the cpsO upstream and cpsN downstream fragments described above, using the primers khb31 and khb34.
To explore a possible effect of expression of the oligopeptide-binding protein AmiA3, the amiA3 (stu1445) gene from S. thermophilus was inserted into the capsular locus of SPH126, replacing the spr0324 gene. spr0324 was first replaced by a Janus cassette, giving rise to SPH127. The Janus cassette was amplified from RH426 using the primer pair khb54/khb55. Fragments corresponding to the ∼1,000-bp upstream and downstream regions of stu0324 were amplified using the primer pair khb50/khb51 and khb52/khb53, respectively. The two fragments were fused to the Janus cassette using the primers khb50/khb53. Next, amiA3 and its promoter were amplified from S. thermophilus LMG18311 using the primer pair khb58/khb59. The resulting fragment was fused to the upstream and downstream flanking fragments described above, using the primers khb50 and khb53. SPH127 was transformed with the resulting DNA fragment, resulting in strain SPH128.
To construct the strain SPH130, which expresses comR constitutively from the synthetic promoter P1 (14), comR in SPH126 was first replaced with the Janus cassette, giving rise to SPH129. The fragment used was amplified from RH426 using the primer pair AmiF/TreR. The Janus cassette was then replaced by a fragment consisting of comR, its native promoter PcomR, and the synthetic P1 promoter (P1::PcomR::comR), resulting in SPH130. The P1::PcomR::comR fragment was constructed as follows: The S. thermophilus comR gene, including its promoter, was amplified with the primers khb72 and khb29, giving rise to a fragment with the P1 promoter added at its 5′ end. Fragments corresponding to the ∼1,000-bp upstream and downstream regions of the Janus cassette in SPH129 were amplified using the primer pairs AmiF/AmiR1 and TreF/TreR, respectively. The two fragments were then fused to the P1::PcomR::comR fragment by using the primers AmiF and TreR.
To express licD1 (spr1151) ectopically from the PcomX promoter, the luc gene was first replaced by a Janus cassette in SPH130, giving rise to SPH131. The Janus cassette was amplified from RH426 using the primers khb60 and khb42. The primers khb31 and khb36 were then used to amplify an ∼950-bp DNA fragment corresponding to the cpsO::PcomX region flanking the 5′end of luc, whereas the primers khb33 and khb34 were used to amplify an ∼800-bp DNA fragment corresponding to the cpsN region flanking the 3′ end of luc. The two fragments were then fused to the 5′ and 3′ ends of the Janus cassette, respectively, using the primers khb31 and khb34. SPH132 was constructed by transforming SPH131 with a PCR fragment consisting of the licD1 gene flanked by appropriate targeting regions. licD1 was amplified from S. pneumoniae RH1 using the primers khb61and khb62 and fused to the same flanking regions as described for the Janus cassette.
Strain SPH135 was constructed by replacing both native licD genes (licD1 [spr1151] and licD2 [spr1152]) in SPH132 with the Janus cassette. The Janus cassette was amplified by PCR using the primer pair khb67/RpsL41.R with genomic DNA from RH426 as template. The primers khb63 and khb64 were used to amplify an ∼800-bp DNA fragment corresponding to the region flanking the 5′end of the native licD1 gene, whereas the primers khb84 and khb85 were used to amplify an ∼800-bp DNA fragment corresponding to the region flanking the 3′ end of licD2. The two fragments were then fused to the 5′ and 3′ ends of the Janus cassette, respectively, using the primers khb63 and khb85. When SPH132 was transformed with the resulting PCR product, 2 μM ComS was supplied to the growth medium and the agar plates used for selection of kanamycin-resistant transformants to ensure that licD1 was expressed ectopically from the PcomX promoter.
Strain SPH136 was derived from SPH130 by replacing luc with a kanamycin resistance gene (aacA-aphD), so that kanamycin resistance was induced by ComS induction. aacA-aphD was amplified from pFW13 (22) using the primers khb79 and khb80. The resulting DNA fragment was fused to the cpsO upstream and cpsN downstream fragments described above, by using the primers khb31 and khb34.
Luciferase reporter assays.
Mutants harboring the luc gene fused to PcomX were grown in C medium to an OD492 of ∼0.3. Cultures of the different mutants were then diluted to an OD492 of ∼0.05, from which 280 μl of each diluted culture was mixed with 20 μl d-luciferin (10 mM) from Photinus pyralis (Thermo Scientific) in a 96-well Corning NBS clear-bottom plate. The plate was incubated in a Fluostar Optima luminometer (BMG Labtech) at 37°C. The OD492 and luminescence were measured automatically every 10 min throughout the experiment. When performing the ComS* titration experiments, the following concentrations of ComS* were added to cell cultures that had reached an OD492 of 0.1: 10 μM, 1.25 μM, 0.63 μM, 0.31 μM, 0.16 μM, 0.08 μM, or 0 μM.
Depletion assays.
Bacterial cultures were grown to an OD492 of 0.3 in the presence of 2 μM ComS*, pelleted by centrifugation, and washed once with C medium to remove excess ComS*. The washed cells were resuspended in fresh C medium with (2 μM) or without ComS* to an OD492 of ∼0.05 and 2-fold diluted in a 96-well NBS clear-bottom plate (Corning). The plate was incubated in a Fluostar Optima luminometer (BMG Labtech) at 37°C. Luciferin-luciferase luminescence and the OD492 were measured automatically by the luminometer at 10-min intervals.
Western analysis.
Pneumococcal cells (SPH128) were grown to an OD550 of 0.1 before splitting the culture into eight parallels that were subjected to 10 μM, 2.5 μM, 0.63 μM, 0.16 μM, 0.04 μM, 0.01 μM, 0.0025 μM, or no ComS*. After 1 h at 37°C, 10-ml culture aliquots were harvested by centrifugation at 4°C at 4,000 × g for 10 min. The cells were washed once with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4; pH 7.2) before their total protein contents were analyzed by SDS-PAGE as described by Laemmli (19). After gel electrophoresis, the separated proteins were electroblotted onto a polyvinylidene fluoride membrane (Bio-Rad) using a Trans-Blot SD semidry transfer cell from Bio-Rad. Following blocking using 5% (wt/vol) skim milk powder dissolved in Tris-buffered saline–Tween (TBST; 150 mM NaCl, 0.1% (vol/vol) Tween 20, Tris-HCl [pH 7.6]), the membrane was incubated overnight with the primary antibody (antiluciferase antibody produced in rabbit from Sigma) at 4°C. After washing the filter 3 times in TBST, it was incubated with a secondary antibody (polyclonal goat anti-rabbit immunoglobulin–alkaline phosphatase [Dako]) at room temperature for 1 h, followed by 3 washes in TBST. The primary antibody was diluted 1:6,000 in TBST containing 5% (wt/vol) skim milk powder and 15% (vol/vol) soluble protein extract from S. pneumoniae RH1 to reduce unspecific background signals. The extract was made by passing 20 ml of an RH1 culture (OD550, 0.3) through a French press. The secondary antibody was diluted 1:2,000 in TBST containing 5% (wt/vol) skim milk powder. The immunoblots were developed using the 5-bromo-4-chloro-3-indolylphosphate–Nitro Blue Tetrazolium liquid substrate from Sigma as described by the manufacturer.
Microscopy.
To examine the effect of reducing the level of ectopically expressed licD1 in the mutant lacking both licD1 and licD2 (SPH135), cells grown in the presence (2 μM) or absence of ComS* were compared by differential interference contrast (DIC) microscopy. Cells were fixed in a paraformaldehyde-glutaraldehyde solution (2.5:1; 7.5%:0.018% [wt/vol] in PBS [pH 7.2]). After fixation on ice for 30 to 60 min, the cells were examined using a Zeiss LSM 700 microscope.
Transcriptional analyses.
Pneumococcal RNA was isolated by using the RNeasy kit from Qiagen. Cells were grown to an OD550 of 0.1 and then cultures were split in two; one cell culture was induced with ComS* (2 μM) for 30 and 60 min, while the parallel culture was allowed to grow without ComS*. Cells from 10-ml culture aliquots of induced and noninduced cells were collected by centrifugation at 4°C at 4,000 × g for 5 min. The cell pellets were first treated with RNAlater (Qiagen) to stabilize the RNA and then suspended in a mixture of 700 μl RLT buffer (Qiagen) and 500 μl chloroform. This mixture was transferred to a FastPrep tube (MP Biomedicals Europe) containing 0.5 g of ≤106-μm acid-washed glass beads (Sigma). The cells were lysed in a FastPrep-24 apparatus (MP Biomedicals Europe) at 6.5 m s−1 five times for 30 s each. Insoluble cell debris was removed by centrifugation at 16,000 × g for 5 min before the water phase was transferred to a fresh Eppendorf tube and mixed with 500 μl ethanol. RNA from this mixture was isolated using an RNeasy minikit column as described by the manufacturer (Qiagen).
Prior to cDNA synthesis, the RNA was treated with DNase I to remove unwanted genomic DNA as follows: 20 μg RNA (the RNA concentration in samples varied between 1 and 2 μg/μl) was mixed with 1 μl RNaseOut (Invitrogen) and 10 μl DNase I (RNase-free; Qiagen) in 70 μl RDD buffer (Qiagen) and incubated at 37°C for 30 min. After DNA digestion, the DNase I was removed by performing a phenol-chloroform extraction. Phenol (pH 7.6), chloroform, and sample were mixed in a 1:1:1 ratio and vortexed for 1 min. Following centrifugation at 10,000 × g for 5 min, and RNA in the water phase was precipitated in 960 μl ethanol containing 40 μl 3 M sodium acetate and left overnight at −20°C. The precipitated RNA was collected by centrifugation at 16,000 × g for 30 min at 4°C, washed with 70% ice-cold ethanol, air dried, and resolved in RNase-free water. cDNA was synthesized from 1 μg DNase I-treated RNA using the Superscript III reverse transcriptase kit as described by the manufacturer (Invitrogen). Real-time PCR was carried out using the StepOnePlus real-time PCR system and the SYBR Green PCR master mix from Applied Biosystems. Twenty-five nanograms of cDNA was used as template, together with the primer pairs 16sF and 16sR targeting the 16S rRNA gene, lucF and lucR targeting the luc gene, and cbpDF and cbpDR targeting cbpD. The level of 16S rRNA expression was used to normalize the data for the different samples, and the relative levels of luc were determined based on the change in luc expression relative to the level of cbpD.
RESULTS
Establishment of the ComRS system in the pneumococcal genome.
In a recent study, Fontaine et al. (9) proposed a simple model for induction of natural transformation in S. thermophilus. According to their model the Ami system transports the ComS* peptide into the cytoplasm, where it directly interacts with the ComR transcriptional activator. Upon binding of ComS*, ComR is thought to undergo a conformational change that allows it to bind to the ECom box in the comX promoter and activate transcription of the comX gene. Previous studies have shown that the alternative sigma factor ComX controls the competence regulon in S. thermophilus, i.e., the genes involved in binding, uptake, and integration of exogenous DNA (2). How the ComS* precursor is processed and how the mature peptide pheromone is exported from the cell are not known.
To determine if the proposed model is correct, we inserted the comR gene and the comX promoter fused to the firefly luciferase gene (PcomX::luc) into neutral sites of the pneumococcal genome. The comR gene, including its own promoter (Fig. 1), was inserted between the amiF and treR genes, whereas the PcomX::luc fragment was inserted between cpsO and cpsN. Due to a nonfunctional capsule locus, the S. pneumoniae strain R6 is unencapsulated. Insertion of a foreign fragment between cpsO and cpsN should therefore have no biological consequences. We did not transfer the ComS* uptake system to S. pneumoniae, as we considered it likely that the native oligopeptide permease is able to translocate ComS* across the pneumococcal membrane. To evaluate the performance of the ComRS expression system, the resulting pneumococcal mutant strain (SPH126), carrying the comR gene and the PcomX::luc fusion, was grown in 96-well Corning NBS plates at 37°C inside a Fluostar Optima luminometer. When an OD492 of 0.1 was reached, ComS* was added at concentrations ranging from 0 to 16 μM. In cultures treated with ComS*, light emission started to rise above background levels 10 min postinduction and continued to increase for about 1 h. Based on our previous experience with the luc reporter and the Fluostar Optima luminometer, the observed maximum level of luminescence was relatively weak. Hence, before evaluating the ComRS system for its potential use in gene depletion studies, we decided to investigate whether it was possible to improve the level of ComS*-induced expression of target genes.
Fig. 1.

Schematic diagram depicting two genetic regions of S. pneumoniae mutant SPH130 containing the inserted PcomX::luc and P1::PcomR::comR constructs. The promoters PcomX, PcomR, and P1 are indicated. The sequence of PcomX is shown below the diagram, with the predicted ComR-binding site (ECom box) indicated by arrows (9). PcomR represents the native ComR promoter, while P1 represents a synthetic constitutive promoter inserted upstream of PcomR. The Pribnow box and the ribosome-binding site (RBS) are underlined.
Improved expression of the luc reporter.
Uptake of ComS* in S. thermophilus LMD-9 relies on a multisubunit ABC-type transporter consisting of two integral membrane proteins and two cognate ATP-binding proteins located on the cytoplasmic side of the membrane. In addition, the genome of strain LMD-9 encodes two extracellular oligopeptide-binding proteins that are responsible for capturing peptides from the external medium. Gardan et al. (10) showed that one of the oligopeptide-binding proteins, AmiA3, translocates ComS* more efficiently than the other. A possible explanation for the modest ComS*-induced luc expression observed with the SPH126 strain, therefore, was that the native oligopeptide-binding protein of S. pneumoniae has low affinity for ComS*, causing inefficient uptake of ComS* and reduced luc expression. We tested this possibility by incorporating a copy of the amiA3 gene and its promoter into the genome of the SPH126 strain between the cpsO and spr0325 genes. The resulting mutant strain (SPH128) produced the same amount of light as the parental strain, demonstrating that addition of the amiA3 gene did not contribute to increased luc expression (data not shown).
Another factor that might limit ComS*-induced luc expression is the amount of ComR produced in the pneumococcal cell. It is quite conceivable that the native comR promoter functions poorly in S. pneumoniae and that ComR therefore is weakly expressed. The synthetic constitutive promoter P1 (14) was therefore introduced upstream of the native comR promoter (Fig. 1), giving rise to the SPH130 strain. Characterization of this strain revealed that it emitted about 5-fold more light than the parental strain when equal amounts of ComS* were used to induce luc expression (data not shown).
The level of luc expression can be modulated by ComS*.
The properties of the ComRS system were further investigated by subjecting the SPH130 strain to different levels of ComS*. The results presented in Fig. 2A show that no light emission above background level was detected by the Fluostar Optima luminometer in uninduced cultures. Interestingly, addition of different concentrations of ComS* gave rise to different levels of luciferase activity, demonstrating that expression of target genes driven by PcomX can be controlled by varying the peptide pheromone concentration in the growth medium (Fig. 2A). This dose-dependent activation of luc expression was also clearly seen in the Western analysis depicted in Fig. 2B. Maximum luciferase production was obtained with external ComS* concentrations in the range of 1 to 2 μM. In the presence of ComS*, expression of the luc gene persisted throughout the growth phase but dropped as the culture approached stationary phase (Fig. 2A). Importantly, our results showed that transfer of SPH130 cells from ComS*-containing medium (2 μM) to ComS*-free medium brought about a gradual reduction in light emission after a lag period of 45 to 60 min (Fig. 3). This result was very promising with respect to the potential use of the ComRS system as a tool for gene depletion studies.
Fig. 2.

Bioluminescence (A) and Western analyses (B) showing the level of Luc expression after subjecting SPH130 cells to various concentrations of ComS*. (A) SPH130 cell cultures were induced at an OD492 of 0.1 with the following concentrations of ComS*: 0 (◆), 0.08 μM (□), 0.16 μM (■), 0.31 μM (Δ), 0.63 μM (▴), 1.25 μM (○), or 10 μM (●). Luminescence relative to cell density (in relative light units [RLU]/OD492) is indicated by lines with symbols, while lines without symbols indicate bacterial growth. Concentrations of ComS* higher than 1 to 2 μM did not increase the emitted light intensity, suggesting a saturation of the system, whereas induction with lower concentrations of ComS* displayed less production of light in a dose-dependent manner. The data shown are from a representative experiment of several replicates. (B) Detection of the luciferase enzyme by Western analysis after induction for 1 h with different concentrations of ComS*. ComS* was added to SPH130 cell cultures at an OD492 of 0.1. Luciferase was detected by using a polyclonal antiluciferase antibody produced in rabbits. The concentrations of ComS* used are indicated in μM above the respective protein bands. The bands appearing immediately below the full-size luciferase bands represent degradation products. Our results showed that the luciferase enzyme is very unstable in S. pneumoniae.
Fig. 3.

Decay of luciferase activity over time after removal of ComS* from the growth medium. The SPH130 strain was grown either in the presence of 2 μM ComS* (circles) or in the absence of ComS* (triangles). Bacterial growth is indicated by open symbols, whereas luminescence is indicated by filled symbols. After shifting the cells at time zero from a medium containing 2 μM ComS* to a ComS*-free medium, it took about 70 min before the luciferase activity started to decline (▴). In the parallel culture grown in the presence of ComS*, the luciferase activity started to decline as the culture approached stationary phase (●).
Depletion of LicD1.
As mentioned in the introduction, previous investigations had suggested that the pneumococcus can grow normally with a functional licD1 or licD2 gene but will not survive deletion of both. It was shown by Zhang and coworkers (26) that the licD2 gene can be readily knocked out by insertion-duplication mutation, while this strategy failed with the licD1 gene. The reason for this is probably that insertion of a plasmid in the licD1 gene has a polar effect on licD2 expression. To determine whether the ComRS system is suitable for depletion studies of lethal genes, we decided to construct a licD1 licD2 double-knockout mutant. First, a PCR fragment corresponding to the licD1 gene was inserted behind the PcomX promoter essentially as described for the luc gene above. Then, the resulting strain (SPH132) was transformed with a PCR fragment designed to replace licD1 and most of licD2 with the Janus cassette (24), giving rise to SPH135. During transformation and subsequent selection of transformants on agar plates containing kanamycin, 2 μM ComS* was added to the growth medium. After incubation at 37°C, overnight colonies were picked and seeded into medium containing 2 μM ComS*. Analysis of the mutants by PCR and sequencing showed that the Janus cassette had been inserted correctly. As a control experiment, wild-type cells were transformed in parallel with the same fragment. In this case, no transformants were obtained. These results show that S. pneumoniae can survive with only licD1 but is not able to grow in the absence of both licD genes. They also showed that the PcomX promoter is able to drive expression of an essential gene after the native gene has been deleted.
Next we wanted to determine if removal of ComS* from the growth medium would deplete the amount of LicD1 in the pneumococcal cells to a physiologically critical level. A culture of the SPH135 mutant grown in C medium containing 2 μM ComS* was pelleted and washed once in plain C medium. After washing, the bacterial pellet was suspended in ComS*-free C medium to an OD492 of 0.05 and then serially diluted 2-fold in the same medium in a 96-well Corning NBS plate with a clear bottom. Following dilution of the cells, 2 μM ComS* was added to the wells in one row (Fig. 4A), while the wells in the parallel row contained no ComS* (Fig. 4B). The plate was incubated at 37°C in an Optima Fluostar luminometer for 11 h, during which the OD492 was measured automatically by the luminometer every 10 min. The results showed that cells cultivated in the absence of ComS* grew normally for about 5 h, after which they started to lag behind the positive control. After about 7 to 8 h, the cultures stopped growing and started to lyse. Light microscopic examination of LicD1-depleted pneumococcal cultures exhibiting decreasing growth rates revealed morphologically abnormal bacteria (Fig. 4C). The cells were much larger than normal, and many had a grossly deformed elongated shape. In addition, the LicD1-depleted cells grew in short chains.
Fig. 4.

Effects of licD1 depletion on growth and morphology of SPH135 cultures. (A) A culture of SPH135 cells grown to an OD492 of 0.3 in C medium supplemented with 2 μM ComS* was pelleted, washed once in plain C medium, and resuspended to an OD492 of 0.05 in fresh C medium containing 2 μM ComS*. Then, the culture was 2-fold diluted in the same medium in a 96-well plate and incubated in a Fluostar Optima luminometer at 37°C for 11 h. (B) The same culture of SPH135 cells was washed and resuspended in ComS*-free medium but otherwise treated as describe for panel A. In cells growing in the presence of ComS* (A), ectopic expression of licD1 is driven by the ComRS system. In cells growing in ComS*-free medium (B), ectopic expression of licD1 is gradually reduced. About 5 h after the cells were shifted from a ComS*-containing to a ComS*-free medium, growth of LicD1-depleted cells started to slow down. A few hours later the growth stopped completely, and the cells started to lyse (B). The data shown are from a representative experiment of several replicates. (C) Examination of licD1-proficient (+ComS*) and licD1-deficient (−ComS*) SPH135 cells by DIC microscopy. Samples of licD1-proficient and licD1-deficient cells were collected at the transition between logarithmic and stationary phases (at 480 min) from the cultures represented by open triangles (see panels A and B). The pictures shown are representative several independent experiments. The morphology of SPH135 cells grown in the presence of ComS* was indistinguishable from that of wild-type pneumococci, while the morphology of licD1-depleted cells was clearly abnormal.
To further test the performance of the ComRS system, we introduced a kanamycin resistance gene (aacA-aphD) from the plasmid pFW13 (22) behind the PcomX promoter (SPH136) to show that depletion of this Kanr gene by removal of ComS* rendered the cells sensitive to kanamycin (400 μg ml−1). In this context, the Kanr gene can be defined as essential for SPH136. As Fig. 5 shows, the cells became highly sensitive to kanamycin during the Kanr depletion experiment. ComS*-induced SPH136, however, remained resistant to kanamycin and grew normally to stationary phase (data not shown).
Fig. 5.
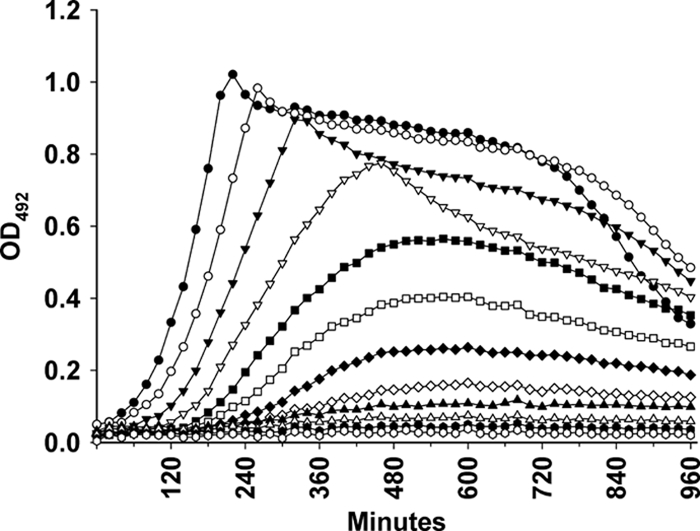
Depletion of the Kanr gene makes the SPH136 mutant sensitive to kanamycin. SPH136 cell cultures were grown in C medium containing 2 μM ComS* until they reached an OD492 of 0.3. Then, they were pelleted, washed once, and resuspended to an OD492 of 0.05 in ComS*-free C medium containing kanamycin (400 μg ml−1). The resuspended cells were 2-fold diluted in the same medium in a 96-well plate and incubated in a Fluostar Optima luminometer at 37°C for 16 h. Growth (measured as the OD492) was determined automatically by the luminometer at 10-min intervals. As Kanr was depleted over time, the growth rate of the SPH136 cells gradually slowed down. After being cultivated for about 8 h in ComS*-free medium, their growth was completely inhibited by kanamycin.
How tightly regulated is the comX promoter?
The time it took before removal of ComS* from the medium had an effect on the growth rate of the SPH135 mutant was longer than expected. We suspected that the long reaction time was partially caused by low-level background transcription of the licD1 gene in the absence of the ComS* inducer. Such background transcription might originate from the PcomX promoter itself, or from a promoter located further upstream of the licD1 gene. To investigate these matters, we performed transcriptional analyses on the SPH130 strain, which contains the luc gene inserted behind the PcomX promoter. The level of luc transcription was examined before and after induction with the ComS* peptide (2 μM) and compared with the expression level of the late competence gene cbpD. Transcription of cbpD, which is controlled by the alternative sigma factor ComX, is shut off in noncompetent cells (15). By real-time reverse transcription-PCR (RT-PCR), it was demonstrated that the basal level of luc transcripts was 10 to 30 times higher than the corresponding level of cbpD transcripts (Fig. 6). In addition, it was shown that luc expression in cultures treated with 2 μM ComS* increased approximately 1,500-fold compared to uninduced cultures. Further experiments are needed to determine whether the observed basal level of luc expression originates from the PcomX promoter itself or from regions upstream of this promoter.
Fig. 6.

Expression levels of luc relative to the expression levels of cbpD measured by real-time RT-PCR. The late competence gene cbpD is only expressed in cells that are competent for natural genetic transformation. (A) Background transcription of luc was examined in SPH130 cells grown without ComS* to an OD550 of 0.1. (B and C) Additional samples from the same culture were collected 30 (B) and 60 min (C) later and analyzed in the same way. The results showed that the background expression of luc in uninduced cells was 10 to 30 times higher than the background expression of cbpD in noncompetent cells. Comparison of luc expression levels in cells induced with 2 μM ComS* for 30 and 60 min (D and E) with those of noninduced cells run in parallel (B and C) showed that addition of ComS* increased luc expression about 1,500-fold.
DISCUSSION
In the present study we exploited the transcriptional activator ComR, its inducer ComS*, and the PcomX promoter from S. thermophilus to construct a gene depletion system for use in S. pneumoniae. The system also represents a useful tool for controlled expression of selected genes at physiological levels. However, since the PcomX promoter appears to be only moderately strong when fully induced by ComS*, the ComRS system is probably not suitable for high-level overexpression of genes. Two other gene depletion systems devised for use in S. pneumoniae have been described. One of them, which was developed by Chan et al. (3), is based on the fucose-regulated promoter PfcsK. In this system, a PCR-generated cassette consisting of the PfcsK promoter, a selectable antibiotic resistance marker, and appropriate flanking regions is introduced upstream of the target gene by homologous double-crossover recombination. A disadvantage of this strategy is that codepletion of downstream genes will take place if they are located on the same transcription unit as the target gene. Since all S. pneumoniae strains tested so far have been unable to grow on fucose as a sole carbon and energy source, the role of this sugar in pneumococcal metabolism remains unclear (3). An alternative method based on the Zn2+-inducible promoter PczcD was described recently (7, 17). To perform a gene depletion experiment with the Zn2+ system, a DNA cassette containing a selectable antibiotic resistance marker and the gene of interest under the control of the PczcD promoter was inserted into the pneumococcal bgaA locus via homologous double-crossover recombination. Then, in the presence of the relatively high Zn2+ concentration needed to drive ectopic expression of the target gene, the native copy of this gene can be deleted from the genome by replacing it with another antibiotic resistance marker. Depletion of the selected gene takes place when the Zn2+ concentration in the medium is reduced. As zinc plays an important role in pneumococcal physiology, the expression and function of a number of gene products are potentially affected by the levels of this metal ion (6, 16, 17, 21). The Zn2+ depletion system should therefore be used with caution. In contrast to the gene depletion techniques described above, which are based on the native PfcsK and PczcD promoters, the ComRS system is of heterologous origin. As no close homologues of the ComRS proteins are encoded in the pneumococcal genome, it is unlikely that the presence of the ComRS system interferes with the normal physiology of the cell.
When the SPH130 mutant was shifted from a medium containing 2 μM ComS* to a ComS*-free medium, it took about 1 h before Luc-generated light emission leveled off and started to decline (Fig. 3). However, following the same procedure, it took about 5 h before depletion of LicD1 affected the growth rate of the SPH135 mutant (Fig. 4B). To determine whether it was possible to reduce the response time, we tried to grow the SPH135 mutant at a lower concentration of ComS* before shifting the culture to ComS*-free medium. It turned out that 0.02 μM ComS* was sufficient to result in a normal growth rate and morphology. In this case the response time was reduced to 3 to 4 h (result not shown). This result indicates that residual ComS* is removed relatively slowly in cultures that have been shifted to ComS*-free medium. A possible explanation is that intracellular ComS* is highly stable and is removed from the cytoplasm very slowly, either by peptidases or by dilution as the cells grow and divide. Whatever the correct explanation, our results demonstrate that gene depletion can easily be achieved using the ComRS system. In fact, the delayed response might be an advantage when performing depletion studies of essential genes. A rapid decrease in gene expression would probably result in growth arrest and cell death before any distinguishable phenotype had time to develop. A slow reduction, on the other hand, leads to a gradual buildup of stress that allows the cells to develop phenotypic changes before they stop growing. It should be noted, though, that the observed background transcription could cause problems if the gene selected for depletion studies were expressed at a very low level. We hope to eliminate this potential problem in future studies by identifying and removing the sequence element(s) responsible for the observed background transcription.
Depletion of LicD1 in the SPH135 mutant, which lacks the native licD1 and licD2 genes, caused striking morphological alterations. Almost all of the LicD1-depleted SPH135 cells grew to a much larger size than corresponding cells grown in the presence of ComS*. This result indicates that LicD1-deficient cells have lost the ability to divide normally. This view is substantiated by other morphological abnormalities characteristic of SPH135 cells grown in the absence of ComS*. The LicD1-deficient cells are morphologically heterogeneous and include some very long, misshapen cells. The elongated cells are evidently able to incorporate new peptidoglycan into their cell walls, but they struggle to divide. Since the TacF flippase is strictly specific for choline-containing subunits (5), depletion of LicD1 will reduce the amount of WTA and LTA in the cell wall of SPH135 cells. Thus, the observed morphological abnormalities are most likely caused by suboptimal levels of WTA and/or LTA. However, it is possible that peptidoglycan synthesis could also be affected by severe LicD1 depletion. The reason for this is that teichoic acid as well as peptidoglycan synthesis depend on the membrane-anchored undecaprenyl carrier lipid. During LicD1 depletion, undecaprenyl-linked choline-free teichoic acid precursors are trapped at the inside of the cytoplasmic membrane. This might exhaust the supply of free carrier that is available for peptidoglycan synthesis. However, the fact that LicD1 depletion results in oversized cells indicates that peptidoglycan synthesis is not critically affected. The observed morphological abnormalities are therefore most likely caused by reduced incorporation of teichoic acid in the pneumococcal cell wall.
ACKNOWLEDGMENT
This work was supported by The Research Council of Norway.
Footnotes
Published ahead of print on 29 July 2011.
REFERENCES
- 1. Baur S., Marles-Wright J., Buckenmaier S., Lewis R. J., Vollmer W. 2009. Synthesis of CDP-activated ribitol for teichoic acid precursors in Streptococcus pneumoniae. J. Bacteriol. 191:1200–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blomqvist T., Steinmoen H., Håvarstein L. S. 2006. Natural genetic transformation: a novel tool for efficient genetic engineering of the dairy bacterium Streptococcus thermophilus. Appl. Environ. Microbiol. 72:6751–6756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chan P. F., et al. 2003. Characterization of a novel fucose-regulated promoter (PfcsK) suitable for gene essentiality and antibacterial mode-of-action studies in Streptococcus pneumoniae. J. Bacteriol. 185:2051–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chastanet A., Prudhomme M., Claverys J. P., Msadek T. 2001. Regulation of Streptococcus pneumoniae clp genes and their role in competence development and stress survival. J. Bacteriol. 183:7295–7307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Damjanovic M., Kharat A. S., Eberhardt A., Tomasz A., Vollmer W. 2007. The essential tacF gene is responsible for the choline-dependent growth phenotype of Streptococcus pneumoniae. J. Bacteriol. 189:7105–7111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dintilhac A., Alloing G., Granadel C., Claverys J. P. 1997. Competence and virulence of Streptococcus pneumoniae: Adc and PsaA mutants exhibit a requirement for Zn and Mn resulting from inactivation of putative ABC metal permeases. Mol. Microbiol. 25:727–739 [DOI] [PubMed] [Google Scholar]
- 7. Eberhardt A., Wu L. J., Errington J., Vollmer W., Veening J. W. 2009. Cellular localization of choline-utilization proteins in Streptococcus pneumoniae using a novel fluorescent reporter system. Mol. Microbiol. 74:395–408 [DOI] [PubMed] [Google Scholar]
- 8. Fischer W., Behr T., Hartmann R., Peter-Katalinic J., Egge H. 1993. Teichoic acid and lipoteichoic acid of Streptococcus pneumoniae possess identical chain structures. A reinvestigation of teichoic acid (C polysaccharide). Eur. J. Biochem. 215:851–857 [DOI] [PubMed] [Google Scholar]
- 9. Fontaine L., et al. 2010. A novel pheromone quorum-sensing system controls the development of natural competence in Streptococcus thermophilus and Streptococcus salivarius. J. Bacteriol. 192:1444–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gardan R., Besset C., Guillot A., Gitton C., Monnet V. 2009. The oligopeptide transport system is essential for the development of natural competence in Streptococcus thermophilus strain LMD-9. J. Bacteriol. 191:4647–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Håvarstein L. S., Coomaraswami G., Morrison D. A. 1995. An unmodified heptadecapeptide pheromone induces competence for genetic transformation in Streptococcus pneumoniae. Proc. Natl. Acad. Sci. U. S. A. 92:11140–11144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Higuchi R., von Beroldingen C. H., Sensabaugh G. F., Erlich H. A. 1988. DNA-typing from single hairs. Nature 332:543–546 [DOI] [PubMed] [Google Scholar]
- 13. Johnsborg O., Eldholm V., Bjørnstad M. L., Håvarstein L. S. 2008. A predatory mechanism dramatically increases the efficiency of lateral gene transfer in Streptococcus pneumoniae. Mol. Microbiol. 69:245–253 [DOI] [PubMed] [Google Scholar]
- 14. Johnsborg O., Håvarstein L. S. 2009. Pneumococcal LytR, a protein from the LytR-CpsA-Psr family, is essential for normal septum formation in Streptococcus pneumoniae. J. Bacteriol. 191:5859–5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kausmally L., Johnsborg O., Lunde M., Knutsen E., Håvarstein L. S. 2005. Choline-binding protein D (CbpD) in Streptococcus pneumoniae is essential for competence-induced cell lysis. J. Bacteriol. 187:4338–4345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kloosterman T. G., van der Kooi-Pol M. M., Bijlsma J. J. E., Kuipers O. P. 2007. The novel transcriptional regulator SczA mediates protection against Zn2+ stress by activation of the Zn2+-resistance gene czcD in Streptococcus pneumoniae. Mol. Microbiol. 65:1049–1063 [DOI] [PubMed] [Google Scholar]
- 17. Kloosterman T. G., Witwicki R. M., van der Kooi-Pol M. M., Bijlsma J. J. E., Kuipers O. P. 2008. Opposite effects of Mn2+ and Zn2+ on PsaR-mediated expression of the virulence genes pcpA, prtA, and psaBCA of Streptococcus pneumoniae. J. Bacteriol. 190:5382–5393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lacks S., Hotchkiss R. D. 1960. A study of the genetic material determining an enzyme in pneumococcus. Biochim. Biophys. Acta 39:508–518 [DOI] [PubMed] [Google Scholar]
- 19. Laemmli U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 20. LeMessurier K. S., Ogunniyi A. D., Paton J. C. 2006. Differential expression of key pneumococcal virulence genes in vivo. Microbiology 152:305–311 [DOI] [PubMed] [Google Scholar]
- 21. Ogunniyi A. D., et al. 2009. Pneumococcal histidine triad proteins are regulated by the Zn2+-dependent repressor AdcR and inhibit complement deposition through the recruitment of complement factor H. FASEB J. 23:731–738 [DOI] [PubMed] [Google Scholar]
- 22. Podbielski A., Spellerberg B., Woischnik M., Pohl B., Lütticken R. 1996. Novel series of plasmid vectors for gene inactivation and expression analysis in group A streptococci (GAS). Gene 177:137–147 [DOI] [PubMed] [Google Scholar]
- 23. Song J. H., et al. 2005. Identification of essential genes in Streptococcus pneumoniae by allelic replacement mutagenesis. Mol. Cell 19:365–374 [PubMed] [Google Scholar]
- 24. Sung C. K., Li H., Claverys J. P., Morrison D. A. 2001. An rpsL cassette, Janus, for gene replacement through negative selection in Streptococcus pneumoniae. Appl. Environ. Microbiol. 67:5190–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thanassi J. A., Hartman-Neumann S. L., Dougherty T. J., Dougherty B. A., Pucci M. J. 2002. Identification of 113 conserved essential genes using a high-throughput gene disruption system in Streptococcus pneumoniae. Nucleic Acids Res. 30:3152–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang J. R., Idanpaan-Heikkila I., Fischer W., Tuomanen E. I. 1999. Pneumococcal licD2 gene is involved in phosphorylcholine metabolism. Mol. Microbiol. 31:1477–1488 [DOI] [PubMed] [Google Scholar]