

Abstract
Evolution of the env gene in transmitted R5-tropic human immunodeficiency virus type 1 (HIV-1) strains is the most widely accepted mechanism driving coreceptor switching. In some infected individuals, however, a shift in coreceptor utilization can occur as a result of the reemergence of a cotransmitted, but rapidly controlled, X4 virus. The latter possibility was studied by dually infecting rhesus macaques with X4 and R5 chimeric simian simian/human immunodeficiency viruses (SHIVs) and monitoring the replication status of each virus using specific primer pairs. In one of the infected monkeys, both SHIVs were potently suppressed by week 12 postinoculation, but a burst of viremia at week 51 was accompanied by an unrelenting loss of total CD4+ T cells and the development of clinical disease. PCR analyses of plasma viral RNA indicated an env gene segment containing the V3 region from the inoculated X4 SHIV had been transferred into the genetic background of the input R5 SHIV by intergenomic recombination, creating an X4 virus with novel replicative, serological, and pathogenic properties. These results indicate that the effects of retrovirus recombination in vivo can be functionally profound and may even occur when one of the recombination participants is undetectable in the circulation as cell-free virus.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) infections of humans and simian immunodeficiency virus (SIV) infections of macaques are both initiated by the binding of virions to CD4 receptors present on the surface of target cells. In addition to CD4, both primate lentiviruses require a second receptor (or coreceptor) for successful cell entry (1). For HIV-1, the chemokine receptors CCR5 and CXCR4 are the major coreceptors. In vivo, the predominant HIV-1 population detected in the blood in recently infected individuals utilizes the CCR5 receptor (and are designated R5 viruses) (51, 55, 60). In a large fraction of individuals infected with HIV-1 subtypes B and D, virus strains capable of using CXCR4 (X4 viruses) or both CCR5 and CXCR4 (R5/X4 viruses) have been recovered from the plasma during the late phase of the chronic infection (6, 22). This shift has been termed “coreceptor switching” and is frequently associated with a more rapid loss of CD4+ T cells and an accelerated clinical course (6, 44).
Several mechanisms have been proposed to explain the change in HIV-1 coreceptor utilization (40). Genetic evolution of the env gene present in the transmitted R5 virus, facilitated by the error-prone nature of the HIV-1 reverse transcriptase, is the most widely accepted explanation for coreceptor switching. This mechanism is consistent with the predominance of R5 HIV-1 strains during the asymptomatic phase of the infection, as well as the resistance of individuals, homozygous for a 32-bp deletion of the CCR5 allele (ccr5-Δ32), to HIV infection (7, 50). Point mutations affecting the gp120 V3 loop that introduce positively charged amino acids at positions 11, 24, and 25 have been associated with the shifted utilization of the CXCR4 chemokine receptor, although changes in other regions of the env gene may also contribute to this process (8, 11, 14, 21, 39, 43). A second mechanism to explain coreceptor switching proposes that X4 and R5 HIV-1 strains are both transmitted to new recipients, but the X4 viruses are more readily controlled and are not detected in the plasma during the asymptomatic phase of the infection, remaining suppressed as long as the immune system is functional (40, 53). As immune competence wanes, the previously constrained X4 and/or dualtropic viruses replicate more freely and begin targeting naive CD4+ T lymphocytes for depletion.
Although pathogenic SIVs have been reported to use a plethora of non-CXCR4 coreceptors in addition to CCR5 (27), they are predominantly R5-tropic in macaque cells, and coreceptor switching has rarely been reported (25, 33, 59). In contrast, there have been several reports of coreceptor switching in rhesus monkeys infected with R5-tropic SHIVs (12, 13, 34, 42). However, in contrast to the delayed emergence of X4- or R5/X4-tropic viruses in HIV-1-infected individuals, all of the R5-SHIV-infected animals have been rapid progressors and the coreceptor switch occurred within a few months of virus inoculation (2).
In this report, we have studied the reemergence mechanism of coreceptor switching by dually infecting rhesus monkeys with X4 and R5 SHIVs. Envelope-specific primer pairs and probes were used to individually monitor each virus strain in vivo. In one of the inoculated animals, the replication of the input X4 and R5 SHIVs, measured by levels of viral RNA in the plasma, was suppressed to the limits of detection by week 12 postinoculation (p.i.). Nonetheless, this monkey experienced a transient burst of virus production at week 25 and a sustained surge of viremia after week 51 p.i., which was accompanied by a marked decline in the number of circulating CD4+ T lymphocytes and death from immunodeficiency at week 100 p.i. In this normally progressing macaque, a recombination event occurred between weeks 25 and 50, which transferred an env gene segment that included the V3 region from the starting X4-tropic SHIV into the genetic background of the input R5 SHIV, and created a novel X4 virus with augmented replicative and pathogenic properties. Single genome amplification (SGA) revealed additional recombination events affecting two regions of gp41 and a 142-nucleotide (nt) segment encompassing tat, rev, and vpu gene sequences. In all cases, the gene sequences were transferred unidirectionally from the X4 SHIV input virus into the R5 SHIV genome. Interestingly, high levels of neutralizing antibodies (NAbs) directed against the starting X4-tropic SHIV contributed to its durable suppression by week 10 p.i. In contrast, no NAbs were detected against the starting R5-tropic SHIV or the novel recombinant X4 SHIV that emerged after week 50 p.i., which accelerated progression to AIDS.
MATERIALS AND METHODS
Virus and animal experiments.
The construction and characterization of the SHIVDH12R-Clone 7 (SHIVDH12R-CL-7), SHIVDH12R-CL-8, and SIVmac239 molecular clones and their use to generate virus stocks have been described previously (18, 46, 52). The origin and preparation of the tissue culture-derived SHIVAD8#2 have been previously reported (34). The 50% tissue culture infective doses (TCID50) of SHIVDH12R-CL-8 and SHIVAD8#2 were determined by infecting rhesus macaque peripheral blood mononuclear cells (PBMC) in quadruplicate with serial 4-fold dilutions of the animal challenge stocks and then assaying for the reverse transcription (RT) activities in the culture supernatants on day 14 for RT activity (58). Rhesus macaques (Macaca mulatta) were maintained in accordance with the guidelines of the Committee on the Care and Use of Laboratory Animals (30) and were housed in a biosafety level 2 facility; biosafety level 3 practices were followed. Phlebotomies, intravenous virus inoculations, euthanasia, and tissue sample collections were performed as previously described (10).
Quantitation of plasma viral RNA and cell associated viral DNA levels.
SIV gag RNA levels in plasma or cell-associated viral DNA frequencies in PBMC were determined by real-time RT-PCR (ABI Prism 7700 sequence detection system; Applied Biosystems, Foster City, CA), respectively, as previously reported (10). SIV-, SHIVDH12-, or SHIVAD8-derived RNAs were separately monitored in dually infected rhesus monkeys by using primer pairs and probes specific for the env gene present in each virus, as shown in Table S1 in the supplemental material.
Lymphocyte immunophenotyping and intracellular cytokine assays.
EDTA-treated blood samples were stained for flow cytometric analysis as described previously using combinations of the following fluorochrome-conjugated monoclonal antibodies (MAbs): CD3 (fluorescein isothiocyanate [FITC] or phycoerythrin [PE]), CD4 (PE, peridinin chlorophyll protein-Cy5.5 [PerCP-Cy5.5] or allophycocyanin [APC]), CD8 (PerCP or APC), CD28 (FITC or PE), and CD95 (APC) (31, 32).
For intracellular cytokine assays, immune stimulation using SIVmac239 Gag peptides 15 amino acids in length was performed on frozen lymphocytes as described previously (34, 38).
SGA of plasma virus RNA.
SGA and direct sequencing were used to characterize and quantitate circulating virus populations in macaque CE43 at various times p.i., as previously described (48), with the following modifications. Plasma viral RNA was extracted by using a QIAamp viral RNA minikit (Qiagen) and then reverse transcribed using SuperScript III First-Strand Synthesis SuperMix (Invitrogen). cDNA was serially diluted to identify a dilution at which PCR-positive wells constituted <30% of the total number of reactions. After direct sequencing, any sequence with mixed bases was excluded. First-round PCR amplifications were performed using the following PCR parameters: 1 cycle of 94°C for 2 min; 40 cycles of a denaturing step of 94°C for 20 s, an annealing step of 55°C for 30 s, and an extension step of 68°C for 1 or 2 min; followed by a final extension of 68°C for 5 min. Second-round PCR amplifications were performed with 2 μl from the first-round PCR products. The second-round PCR was performed under the conditions used for the first-round PCR. The primer sets used for the V3, gp41, and vpu regions are indicated in Table S1 in the supplemental material. Amplified gene segments were directly sequenced by using an Applied Biosystems 3130XL genetic analyzer.
Construction of replication-competent viruses used in coreceptor utilization assays.
Full-length infectious molecular clones, containing the entire env genes of SHIVs circulating in macaques DB3T and DB3N at week 2 p.i., were constructed by RT-PCR amplification of plasma viral RNA, as previously described (34). The 3,040-bp amplified products, which included the entire env gene, were cloned into the EcoRI and SalI (newly created) sites of pSHIVAD8. The two replication-competent SHIVs clones obtained were designated SHIVAD8-ES3T7 and SHIVAD8-ES3N6.
Coreceptor utilization assays in macaque PBMC.
Rhesus monkey PBMC (105 cells in 100 μl of RPMI 1640) were dispensed in triplicate in 96-well round-bottom plates as previously described (17). CCR5 (AD101)- or CXCR4 (AMD3100)-specific inhibitors (8 μM) in 25 μl was added to each well, followed by incubation for 1 h at 37°C. Test virus (multiplicity of infection = 0.01) in 75 μl was then spinoculated (36) onto the cells at 1,200 × g for 1 h at 37°C, and the cultures were maintained for 14 days. One-half (100 μl) of the culture supernatant was replaced daily with fresh medium containing 1 μM coreceptor inhibitor in 0.2% dimethyl sulfoxide (DMSO). Control cultures without inhibitors were maintained with medium containing 0.2% DMSO. The daily supernatant samples collected were monitored for progeny virus production by 32P RT assay as previously described (58).
Virus replication assay in rhesus monkey PBMC.
The preparation and infection of rhesus monkey PBMC have been previously described (34). Briefly, PBMC stimulated with concanavalin A and cultured in the presence of recombinant human interleukin-2 were spinoculated (1,200 × g for 1 h) (36) with virus normalized for RT activity. Virus replication was assessed by an RT assay of the culture supernatant as described above.
Virus neutralization assays.
Virus neutralization assays were performed as previously described (34). Briefly, plasma samples (1:20 dilution) from the monkey CE43 were incubated with virus in quadruplicate in 96-well flat-bottom culture plates in a total volume of 50 μl for 1 h at 37°C. Prechallenge plasma samples from each animal served as controls. Freshly trypsin-treated TZM-bl cells were added, cultures were maintained for an additional 28 h, and intracellular luciferase activity was measured as previously described (57). Any sample resulting in a 50% reduction of luciferase activity compared to that obtained with the uninfected control sample was considered positive for NAbs.
RESULTS
Emergence of a highly pathogenic virus in a macaque dually inoculated with R5 and X4 SHIVs.
Although it is currently accepted that the slow evolution of R5-tropic HIV-1 env genes is the major pathway driving coreceptor switching in chronically infected individuals, the reemergence of cotransmitted, but durably suppressed, X4 viral strains has also been proposed as an alternative mechanism (28, 40). To evaluate this second mechanism, monkeys were dually infected with an attenuated derivative of the X4-tropic SHIVDH12 (SHIVDH12R-CL-8 [46]) and the recently described R5-tropic SHIVAD8#2 (34). In some of these inoculated macaques, this virus combination resulted in the rapid and complete loss of both naive and memory CD4+ T cells during the acute infection, a finding reminiscent of the “CD4 crash” observed with the highly pathogenic X4 SHIV89.6P and SHIVDH12R (19, 41). To circumvent this problem, a low dose (75 TCID50) of SHIVDH12R-CL-8 was coinoculated with the R5-tropic SHIVAD8#2 (7,500 TCID50) to minimize the massive loss of naive and memory CD4+ T cells. Three different primer pair sets were used to measure plasma viral RNA in these infected monkeys: (i) an HIV-1 gp41 membrane-spanning pair, specific for the R5 SHIV; (ii) an HIV-1 gp120 V5 pair, specific for the X4-tropic SHIV; and (iii) an SIV gag gene pair for detecting both SHIVs.
One of the dually infected animals (CE43), which did not experience the complete loss of CD4+ T cells acutely, generated peak levels of R5 SHIV viremia on day 10 p.i. and peak X4 SHIV viral RNA loads on 14 p.i. (Fig. 1). A composite viral RNA profile was observed using the SIV gag gene primers. Both SHIVs were rapidly and effectively controlled by week 10 p.i., as monitored with the env gene primer pairs, and at week 15 the total viral RNA in the plasma was barely (200 RNA copies/ml) above the limits of detection (100 RNA copies/ml) using the gag primer pairs. At the conclusion of this early phase of the infection, the numbers of circulating CD4+ T cells were similar to those measured at the time of virus inoculation (Fig. 1).
Fig. 1.

Plasma viremia and CD4+ T cell levels in a rhesus monkey dually infected with X4- and R5-tropic SHIVs. Macaque CE43 was inoculated intravenously with the X4-tropic SHIVDH12 and the R5-tropic SHIVAD8. Plasma viral RNA levels were measured using env gene primer pairs specific for each SHIV or gag gene primer pairs able to detect both viruses. The numbers of total circulating CD4+ T cells were determined during the 100 weeks of infection by flow cytometry.
After week 15, virus replication gradually increased and peaked (104 RNA copies/ml) at week 27 p.i., using the gag gene primers to monitor plasma viral RNA loads. This increase in plasma viremia was not detectable with env gene primer pairs specific for either the X4-tropic or the R5-tropic SHIVs. A second much larger and sustained burst of virus replication, also only detectable with the gag primers, occurred after week 51 and peaked (2.2 × 105 RNA copies/ml) at week 59 p.i. The resulting elevated levels of plasma viremia were accompanied by a marked decline of naive CD4+ T lymphocytes (from 168 cells/μl at week 59 to 23 cells/μl at week 71 and 8 cells/μl at week 75). This precipitous loss of naive CD4+ T cells was followed by a downhill clinical course characterized by protracted anorexia, severe diarrhea, and marked weight loss, beginning at week 90 p.i., and required euthanasia of monkey CE43 at week 100. Necropsy revealed the existence of Pneumocystis carinii pneumonia; in addition, Entamoeba species were visualized on direct fecal smear samples.
The sudden and sustained increase in plasma viremia after week 51 p.i., coupled with the irreversible loss of naive CD4+ T lymphocytes and the onset of clinical disease, suggested that a highly pathogenic virus population had emerged in macaque CE43. Accordingly, 10 ml of blood collected at the time of euthanasia was inoculated intravenously into two recipient animals (DB3T and DB3N). High levels of peak viremia were measurable in both of these monkeys between weeks 2 and 3 p.i. (Fig. 2). An extremely rapid and complete loss of naive and memory CD4+ T lymphocytes occurred in macaque DB3T, requiring euthanasia at week 22 because of intractable diarrhea and marked weight loss. Animal DB3N also experienced a massive depletion of its naive CD4+ T cells (from 1,200 cells/μl at week 0 to 40 cells/μl at week 7) and yet was able to control its set-point viremia to more modest levels and to preserve its memory CD4+ T cells. The rapid elimination of naive CD4+ T lymphocytes in these two recipients of blood from macaque CE43 is similar to the results described previously for animals dually inoculated with high doses of X4-tropic SHIVs.
Fig. 2.

The transfer of blood from macaque CE43 causes the rapid loss of naive CD4+ T cells in two recipient animals. Portions (10 ml) of blood, collected from macaque CE43 at the time of euthanasia, were transfused into macaques DB3T and DB3N, and the levels of plasma viremia and CD4+ T cell subsets were determined.
A coreceptor switch mediated by intergenomic X4 and R5 SHIV recombination occurred in macaque CE43.
The extraordinarily rapid depletion of naive CD4+ T lymphocytes in the dually infected macaque CE43 and in two of its immediate blood transfusion recipients suggested that a highly virulent X4 SHIV had emerged in animal CE43 even though the X4 SHIVDH12R-CL-8 it was inoculated with was undetectable after week 10 p.i. (see Fig. 1). SGA was therefore used to characterize the virus present in plasma at the time of euthanasia (week 100 p.i.). As shown in Fig. 3, this analysis revealed the existence of two SHIV populations bearing distinctive env genes. One population, the product of X4 SHIV/R5 SHIV intergenomic recombination, carried a 220+-nt segment, which included the entire 105-nt V3 region from the starting X4 SHIVDH12 that had been inserted into SHIVAD8 env gene. The second population carried a V3 region similar to that present in the original R5-tropic SHIVAD8. At week 100 when macaque CE43 was euthanized, 59% (16 of 27 amplicons) carried a V3 region resembling that of the inoculated X4-tropic SHIVDH12 and 41% (11 of 27 amplicons) bore a V3 gene segment similar to that present in the original R5-tropic SHIV. In addition, two gene segments from the gp41 coding region of the starting X4-tropic SHIV (Fig. 4, underlined DH12 [X4]-A and DH12 [X4]-B amino acid sequences) had been inserted into analogous positions of the SHIVAD8 env genes of both virus populations.
Fig. 3.

Two populations of circulating SHIVs bearing distinctive V3 regions are present in the dually infected macaque CE43 at the time of its euthanasia. SGA and direct sequencing of plasma samples collected at week 100 p.i. were used to generate amplicons containing the gp120 V3 region. The bracketed sequences include the V3 region derived from the starting X4-tropic SHIVDH12. The gp120 amino acid sequences present in the inoculated SHIVAD8 and SHIVDH12 are shown in the top and bottom lines, respectively. The ES3T7 and ES3N6 sequences were amplified from the plasma of macaques DB3T and DB3N (see Fig. 2), respectively, at week 2 p.i. The opposing arrows indicate the location of the V3 loop.
Fig. 4.

Two regions of the input X4 SHIV gp41 env gene region have been transferred by intergenomic recombination into the genetic background of SHIVAD8. SGA and direct sequencing of gp41 env sequences present in plasma viral RNA at week 100 p.i. are shown. The gp41 sequences in the input SHIVAD8 and SHIVDH12 are presented in the top and bottom lines, respectively. The heptad repeat 2 (HR2), transmembrane (TM), and lentivirus lytic peptide (LLP) regions are annotated at the top. The origin of the gp41 sequences are indicated at the bottom.
Coreceptor usage properties of the recombinant SHIVs that emerged in macaque CE43.
Not unexpectedly, RT-PCR, cloning, and sequence analyses of plasma collected at week 2 p.i. from the recipients (monkeys DB3N and DB3T) of the CE43 blood transfusion revealed the presence of nearly equal numbers of the two circulating recombinant SHIV populations (data not shown). Full-length replication competent SHIV molecular clones were constructed, which contained an entire env gene, amplified from the week 2 plasma samples collected from macaque DB3T or macaque DB3N (designated ES3T7 and ES3N6, respectively, in the gp120 V3 alignments presented in Fig. 3). The coreceptor utilization properties of the resulting SHIVs (SHIVAD8-ES3T7 and SHIVAD8-ES3N6) were assayed in a 14-day replication assay in the presence of CCR5 or CXCR4 small molecule inhibitors. As shown in Fig. 5, replication of the R5-tropic SIVmac239 and the X4-tropic SHIVDH12R-CL-7 controls was completely blocked by the CCR5- and CXCR4-specific inhibitors, respectively, as previously reported (17). In this assay, the R5 inhibitor suppressed replication of SHIVAD8-ES3T7, which carries a gp120 V3 region similar to that present in the R5-tropic SHIVAD8 inoculum. In contrast, the replication of SHIVAD8-ES3N6, which contains a gp120 V3 region similar to the input X4-tropic SHIVDH12R-CL-8, was blocked by the X4 inhibitor. Taken together, these results show that the recombination-mediated insertion of an X4 gp120 determinant into the genetic background to a CCR5-utilizing env gene was sufficient to shift coreceptor utilization.
Fig. 5.

Coreceptor utilization by SHIVs containing recombinant env genes. The indicated viruses were spinoculated onto rhesus PBMC in the presence of CCR5 (AD101)- or CXCR4 (AMD3100)-specific inhibitors. Particle-associated reverse transcriptase activity released into the medium during a 14-day infection was measured in the presence or absence (dashed lined) of inhibitor.
Timing of the X4/R5 SHIV intergenomic recombinations during in vivo infection.
SGA and direct sequencing were used to characterize and quantitate circulating virus populations at various times following the dual inoculation of macaque CE43 with X4-tropic and R5-tropic SHIVs. The results obtained are summarized in Fig. 6. At week 31 p.i., a single circulating SHIV species was identified in the burst of virus replication that was undetectable by RT-PCR of the plasma samples, using either of the two env-specific primer pairs (see Fig. 1). All of the amplicons (18/18) generated at this time contained the two gp41 segments from the starting X4 SHIV, embedded into the R5-SHIV env gene, as previously seen in the week 100 plasma sample (Fig. 3). The remainder of the original R5-tropic env gene present in the week 31 SHIV remained essentially unchanged from that present in the starting inoculum. Interestingly, the recombination events involving the gp41 gene segment had eliminated the target sequences used to amplify the starting R5-tropic SHIV, explaining our inability to detect this virus with isolate-specific gp41 primer pairs after week 20 p.i. All amplicons of the week 31 plasma virus also contained a previously unrecognized 147-nt segment containing overlapping tat, rev, and vpu gene sequences (Fig. 7) from the input X4-tropic SHIV, which had been inserted into the genetic background of the R5-tropic SHIV (Fig. 6). No amplicons representing the original X4-tropic SHIV env gene were detectable in plasma at week 31 p.i. by SGA.
Fig. 6.

Time line of recombination events occurring in macaque CE43 following the inoculation of X4 and R5 SHIVs. Sequences present in the starting R5-tropic SHIVAD8 are indicated in white, and those from the X4-tropic SHIVDH12 are indicated in red. The black rectangles denote HIV-1 gp120 variable regions, and the yellow bar denotes the SIVmac239 nef gene. The locations of primer pairs and probe sequences used to detect the R5 and X4 SHIVs are indicated by the pair of opposing arrows. The numbers in parentheses represent the percentage of each recombinant at the indicated times, as determined by SGA. Note that the recombination-mediated insertion of gp41 sequences from SHIVDH12 into SHIVAD8 at week 31 eliminated sequences used for detecting the starting R5 SHIVAD8.
Fig. 7.

A short gene segment containing overlapping tat and rev and adjacent vpu sequences was transferred from the input X4 SHIVDH12 into R5 SHIVAD8 by intergenomic recombination. SGA and direct sequencing of plasma samples collected at weeks 31 and 64 p.i. were used to generate amplicons containing the first exons of the tat, rev, and vpu genes shown. The black bars below each sequence indicate the region contributed by the starting X4-tropic SHIVDH12. The numbers in parentheses denote the amplicons with the indicated sequence.
At week 64 p.i., SGA revealed the presence of two circulating viral populations (Fig. 6). One was indistinguishable from the single circulating R5 virus species identified at week 31. The second virus population at week 64 p.i. (56% of the amplicons) carried the V3 region from the starting X4-tropic SHIV embedded into the genetic background of the R5 env gene. A similar ratio of the two SHIV species persisted in the plasma from week 64 to the time of euthanasia at week 100. Taken together, these results suggested that the rapid loss of CD4+ T cells accompanying the increased plasma virus loads in macaque CE43 after week 51 was associated with recombination-mediated coreceptor switching that introduced novel CXCR4 entry determinants into the circulating monophyletic R5-tropic virus.
The emergence of the X4 V3 recombinant SHIV at week 64 p.i. raised the possibility that the earlier tat, rev, or vpu gene and gp41 recombinant virus might have been a necessary intermediate because it conferred augmented replicative properties that facilitated recombination. This was evaluated by infecting rhesus monkey PBMC with the original SHIVAD8 and recombinant SHIVs containing the tat, rev, and vpu gene and gp41 inserts with (designated SHIVAD8-ES3N6) or without (designated SHIVAD8-ES3T7) the V3 sequences. As shown in Fig. 8, the SHIV carrying only the tat, rev, and vpu gene plus gp41 inserts exhibited robust infectivity in macaque PBMC compared to the original SHIVAD8. The addition of V3 sequences to this recombinant greatly accelerated virus production in PBMC. Taken together, these results suggest that the week 31 SHIV recombinant may be more fit in vivo, than the input R5 SHIV. Thus the incorporation of the tat, rev, and vpu gene plus gp41 sequences may have facilitated the subsequent coinfection of CD4+ T cells and resulted in the generation of the week 64 recombinant X4 SHIV.
Fig. 8.

Replication of recombinant SHIVs in rhesus PBMC. Rhesus monkey PBMC were infected with original SHIVAD8 and recombinant SHIVs containing the tat, rev, or vpu gene and gp41 inserts with (SHIVAD8-ES3N6) or without (SHIVAD8-ES3T7) the V3 sequences, normalized for RT activity. Virus production was monitored by RT activity released into the medium.
X4 SHIV proviral DNA was detectable in PBMC but not as cell-free virus in the blood of macaque CE43.
The SGA analyses reported above indicated that after week 20 p.i., the predominant viruses circulating in plasma were derivatives of the starting R5 SHIV into which segments of the original X4 SHIV have been additively inserted. The absence of detectable X4 SHIV RNA in the plasma after week 10 prompted us to examine infected cells for the presence of the input X4 SHIV provirus. Since lymph node specimens were not available, preserved PBMC were analyzed by real-time DNA PCR, using the same primers and probe previously used in real-time RT-PCRs for detection of the starting X4-tropic SHIV RNA in plasma. As shown in Fig. 9, the cell-associated X4 SHIV DNA peaked at 334 copies/105 PBMC at week 3 p.i. and then gradually declined over the next 12 months. At week 27 p.i., just prior to the emergence of the tat, rev, and vpu gene and gp41 recombinants, the frequency of cell-associated X4 SHIV DNA was 53 copies/105 PBMC. The detection of cell-associated X4 SHIV proviral DNA in circulating PBMC suggested that low levels of virus-infected CD4+ T cells were present in some body compartment of macaque CE43, most likely secondary lymphoid tissues.
Fig. 9.
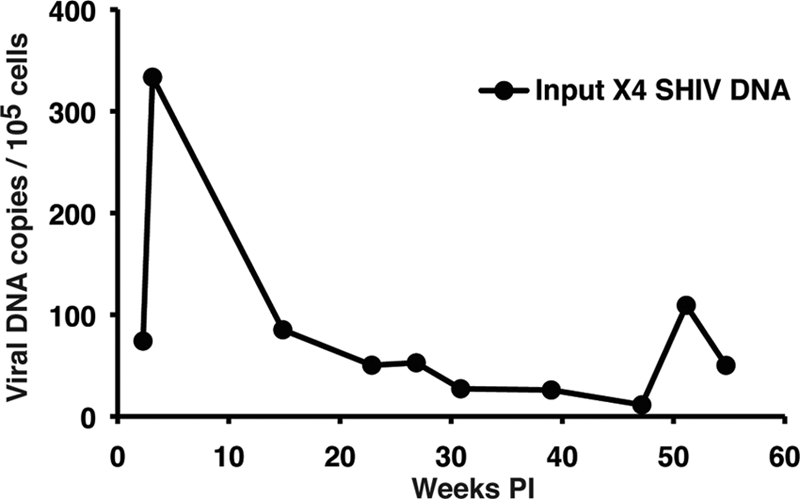
Cell-associated X4 SHIVDH12 DNA loads in PBMC. The frequencies of PBMC-associated viral DNA in macaque CE43 were determined over a 55-week period by quantitative real-time PCR using the primers and probe specifically detecting the starting X4 SHIVDH12 env gene.
Immune responses directed against the starting and recombinant SHIVs in macaque CE43.
Antiviral SIV Gag-specific (present in both X4 and R5 SHIVs) cytotoxic T lymphocyte responses were measured by flow cytometry by intracellular staining for tumor necrosis factor alpha and or gamma interferon at weeks 19, 35, and 43 p.i. The levels of anti-Gag specific CD8+ T cells at these times were 0, 3.1, and 3.6%, respectively, indicating that a strong antiviral cell-mediated response had been elicited during the first year of the dual SHIV infection.
Antiviral NAbs directed against the starting X4 and R5 SHIVs were also assessed. A 1:20 dilution of plasma samples, collected at different times throughout the 100-week infection of macaque CE43, was used in the TZM-bl cell assay system to determine the presence of neutralization activity against each virus. As shown in Fig. 10, high levels of NAbs directed against the starting X4-tropic SHIVDH12 were detected by week 8 and peaked at week 14 p.i. This result is consistent with previous reports from our laboratory, using a different virus neutralization assay system, showing that X4 SHIVDH12 derivatives generate NAbs between weeks 4 and 10 p.i. (10, 20). A limiting dilution assay indicated a 50% neutralization titer of 1:121 against the input X4 SHIVDH12 at week 14 p.i. After week 45 p.i., the levels of NAb activity against the starting SHIVDH12, measured at a 1:20 dilution of plasma, had declined to near background levels (<30% neutralization).
Fig. 10.

Neutralizing antibodies are directed against the input X4 SHIVDH12 but not against the recombinant X4 SHIV that emerged in macaque CE43. Plasma samples (1:20 dilution) were collected from dually infected animal CE43 at the indicated times, incubated with the input X4 SHIVDH12, the input SHIVAD8, or the recombinant X4 SHIVES3N6 for 1 h at 37°C, and assayed for infectivity in TZM-bl cells. Plasma viral RNA levels were determined by using gag gene primer pairs and probes.
We recently reported that NAbs directed against SHIVAD8 became detectable after week 25 p.i. in some infected rhesus monkeys, and the titers measured correlated with the levels of set point viremia (34). In general, anti-SHIVAD8 NAbs were not detected when plasma viral loads were lower than 103 RNA copies/ml. As shown in Fig. 8, NAbs directed against the starting R5 SHIVAD8 were never detected during the 100-week observation period, even when plasma viremia exceeded 105 RNA copies/ml at week 59 p.i. The TZM-bl cell assay system was also used to monitor any NAb activity directed against the recombinant X4 SHIV, which emerged at week 51 p.i. As represented by the reconstructed SHIVAD8-ES3N6, which exclusively uses CXCR4 to enter rhesus PBMC, no NAbs were detected during the entire course of infection in macaque CE43. These results suggest that both cell-mediated and humoral immune responses potently and durably suppressed the input X4 SHIV, which became undetectable by RT-PCR in plasma after week 10 p.i. On the other hand, the late-emerging recombinant X4 SHIV elicited no NAbs, due to either continued masking of envelope glycoprotein epitopes (derived primarily from the original R5 SHIV) or to irreversible damage to the immune system accompanying the rapid depletion of CD4+ T cells after week 70 p.i.
DISCUSSION
Coreceptor switching has rarely been reported during pathogenic SIV infections of rhesus macaques, although a molecular clone (SIVmac155/T3), carrying the env gene recovered from the tissues of an infected monkey, has been constructed, which uses CXCR4 in cell entry assays and causes the depletion of naive CD4+ T lymphocytes in inoculated rhesus macaques (37). In contrast, coreceptor switching has been reported in rhesus monkeys infected with R5 SHIVs. Cheng-Mayer and coworkers have described the emergence of dualtropic or X4-tropic SHIVs in four rapid progressors following inoculations of SHIVSF162P3 derivatives (12, 13, 42). We previously reported that one of three R5 SHIVAD8 rapid progressors had experienced coreceptor switching (34). In all of these animals, coreceptor switching occurred within months of virus inoculation rather than during the late symptomatic phase of infection as reported for HIV-1.
The overwhelming evidence accumulated over more than 2 decades of HIV-1 natural history studies points to the evolution of a transmitted R5-tropic env gene, particularly its V3 loop, as the driving force for coreceptor switching in chronically infected individuals. Numerous reports have identified R5 HIV-1 strains, but not X4 viruses, in plasma following seroconversion or during the asymptomatic phase of the in vivo infection. The X4 virus that emerges during the late stages of the HIV-1 infection usually bears an env gene, which is genetically related to the homologue present in the coexisting R5 strain except for changes in the V3 region. These results are consistent with SGA analyses that have identified only one or two transmitted virus species in plasma of recently HIV-1-infected individuals, the vast majority of which were R5-tropic (23, 47, 49).
It is still possible that the reemergence of cotransmitted X4-tropic HIV-1 strains may contribute to coreceptor switching in some infected individuals. SGA analyses performed to date have only evaluated virus strains circulating in the blood during the acute infection and not in tissues such as the gut-associated lymphoid tissues and draining lymph nodes, the primary site of virus replication during the acute infection. A recent clonal analysis of HIV-1 present in 150 recent seroconverters revealed that CXCR4 using viruses, validated in cell entry assays, were present in seven individuals (15). Even if the establishment of dual X4 and R5 HIV-1 infections occurs at low frequencies in recently exposed individuals, the generation of dualtropic/X4 strains by substitutional evolution of the input R5 virus can occur at anytime following virus acquisition. The SHIV/macaque system described here could therefore also model the intrapatient recombination between R5 HIV-1 env gene NAb escape variants and suppressed X4 HIV strains, which emerged postacquisition. An X4 HIV-1 recombinant arising from such an event might be able to change its neutralization phenotype and target both naive and memory CD4+ T lymphocytes in vivo, thereby further compromising the immune system. In this regard, several studies, published since 2005, have reported a high prevalence of circulating dualtropic or X4-tropic viral strains (50% or greater) in heavily treatment experienced HIV-1-infected patients (9, 16, 56) compared to antiretroviral naive individuals (18 to 19%) (3, 29). In one of these studies, 11 of 23 patients on highly active antiretroviral therapy (HAART) for 5 years with undetectable levels of viral RNA experienced a switch from R5 (at baseline) to X4-tropic strains (9). It is currently unclear why HAART suppression of HIV-1 replication unmasks preexisting CXCR4 utilizing virus strains. Taken together, these studies suggest that previously unrecognized X4 HIV-1 variants may be present throughout the infection.
Our results demonstrate that the potential consequences of recombination in vivo may be functionally profound. Retrovirus recombination is predicated on the dual infection of a single cell by two viruses, the copackaging of nonhomologous genomic RNAs into progeny particles, and template switching during subsequent rounds of reverse transcription. For HIV-1, recombination events have given rise to circulating recombinant forms (45), augmented the spread of resistance to antiretroviral drugs (35), and accelerated disease progression (26). Although we have described recombination between X4 and R5 env gene segments in a single dually infected animal, several studies have reported X4 and R5 intrapatient recombination events in HIV-1-infected individuals (4, 24, 54). Moreover, a recent study examining the genotype and coreceptor usage of clones derived from sequential isolates from four patients experiencing coreceptor switching determined that (i) 9% of clones using the CXCR4 receptor arose by recombination and (ii) a majority of breakpoints identified were located in the gp120 C2 region and involved the transfer of the V3 region of gp120 (28).
In the present study, the recombination events observed in macaque CE43 were multistep and invariably unidirectional (transfer of gene segments from the input X4 SHIV into the genetic background of the starting R5-tropic SHIV). Based on coreceptor usage, a plausible scenario to explain our results would involve a SHIV coinfection of memory CD4+ T lymphocytes, which, unlike the naive CD4+ subset, are susceptible to both X4 and R5 SHIVs. The transfers of tat, rev, or vpu and gp41 gene segments from the X4 SHIV into the genetic background of the starting R5 SHIV between weeks 15 and 31 p.i. were the first recombination events identified (Fig. 6). Infectivity analyses carried out in rhesus monkey PBMC demonstrated that these early recombinations greatly improved the replication capacity of the input R5 SHIV and was reflected by a transient increase of set-point viremia during this phase of the infection.
The subsequent transfer of the gp120 V3 region from the X4-tropic SHIV parent to the monophyletic recombinant R5 SHIV circulating at week 50 p.i. greatly increased plasma viremia and irreversibly altered the clinical course. This second phase of recombination created a virus with two novel properties: (i) the capacity of infect both memory and naive CD4+ T lymphocytes and (ii) a neutralization-resistant X4-tropic gp120. In contrast to the starting X4 SHIV, which possessed neutralization epitopes mapping to the V1/V2 and V4 regions of gp120 (5), the novel recombinant X4 SHIV that emerged had acquired the neutralization resistant properties of the input R5 SHIV (Fig. 10). The unidirectionality of the recombinations described (X4 SHIV to R5 SHIV) were all stochastic events, very likely driven by the prior in vivo passaging of the X4-tropic SHIVDH12 that optimized replication in macaques (19), as well as the NAb sensitivities of the starting X4 and R5 SHIVs. The products of these recombinations (a nearly 50/50% mixture of recombinant X4 and R5 SHIVs, each possessing robust replication properties) were highly virulent, accelerating progression to AIDS in macaque CE43 and causing an extraordinarily rapid depletion of CD4+ T cells following intravenous transfer to two recipient animals.
Supplementary Material
ACKNOWLEDGMENTS
We thank Robin Kruthers and Ronald Plishka for nucleotide sequence analyses determining viral RNA levels and Vanessa Hirsch for critical comments during preparation of the manuscript. We thank Boris Skopits, Amy Fuse, and Rahel Petros for diligently assisting in the care and maintenance of our animals. We are indebted to the NIH AIDS Research and Reference Reagent Program for providing AMD3100 and to Julie Strizki, Shering Plough, for providing AD101.
This study was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 3 August 2011.
REFERENCES
- 1. Berger E. A., Murphy P. M., Farber J. M. 1999. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 17: 657–700 [DOI] [PubMed] [Google Scholar]
- 2. Brown C. R., et al. 2007. Unique pathology in simian immunodeficiency virus-infected rapid progressor macaques is consistent with a pathogenesis distinct from that of classical AIDS. J. Virol. 81: 5594–5606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brumme Z. L., et al. 2005. Molecular and clinical epidemiology of CXCR4-using HIV-1 in a large population of antiretroviral-naive individuals. J. Infect. Dis. 192: 466–474 [DOI] [PubMed] [Google Scholar]
- 4. Charpentier C., Nora T., Tenaillon O., Clavel F., Hance A. J. 2006. Extensive recombination among human immunodeficiency virus type 1 quasispecies makes an important contribution to viral diversity in individual patients. J. Virol. 80: 2472–2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cho M. W., Lee M. K., Chen C. H., Matthews T., Martin M. A. 2000. Identification of gp120 regions targeted by a highly potent neutralizing antiserum elicited in a chimpanzee inoculated with a primary human immunodeficiency virus type 1 isolate. J. Virol. 74: 9749–9754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Connor R. I., Sheridan K. E., Ceradini D., Choe S., Landau N. R. 1997. Change in coreceptor use correlates with disease progression in HIV-1-infected individuals. J. Exp. Med. 185: 621–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dean M., et al. 1996. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 273: 1856–1862 [DOI] [PubMed] [Google Scholar]
- 8. De Jong J. J., De Ronde A., Keulen W., Tersmette M., Goudsmit J. 1992. Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J. Virol. 66: 6777–6780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Delobel P., et al. 2005. R5 to X4 switch of the predominant HIV-1 population in cellular reservoirs during effective highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 38: 382–392 [DOI] [PubMed] [Google Scholar]
- 10. Endo Y., et al. 2000. Short- and long-term clinical outcomes in rhesus monkeys inoculated with a highly pathogenic chimeric simian/human immunodeficiency virus. J. Virol. 74: 6935–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fouchier R. A., et al. 1992. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J. Virol. 66: 3183–3187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ho S. H., et al. 2007. Coreceptor switch in R5-tropic simian/human immunodeficiency virus-infected macaques. J. Virol. 81: 8621–8633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ho S. H., Trunova N., Gettie A., Blanchard J., Cheng-Mayer C. 2008. Different mutational pathways to CXCR4 coreceptor switch of CCR5-using simian-human immunodeficiency virus. J. Virol. 82: 5653–5656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang W., et al. 2008. Coreceptor tropism can be influenced by amino acid substitutions in the gp41 transmembrane subunit of human immunodeficiency virus type 1 envelope protein. J. Virol. 82: 5584–5593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang W., et al. 2009. Characterization of human immunodeficiency virus type 1 populations containing CXCR4-using variants from recently infected individuals. AIDS Res. Hum. Retrovir. 25: 795–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hunt P. W., et al. 2006. Prevalence of CXCR4 tropism among antiretroviral-treated HIV-1-infected patients with detectable viremia. J. Infect. Dis. 194: 926–930 [DOI] [PubMed] [Google Scholar]
- 17. Igarashi T., et al. 2003. Macrophage-tropic simian/human immunodeficiency virus chimeras use CXCR4, not CCR5, for infections of rhesus macaque peripheral blood mononuclear cells and alveolar macrophages. J. Virol. 77: 13042–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Igarashi T., et al. 2007. Although macrophage-tropic simian/human immunodeficiency viruses can exhibit a range of pathogenic phenotypes, a majority of isolates induce no clinical disease in immunocompetent macaques. J. Virol. 81: 10669–10679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Igarashi T., et al. 1999. Emergence of a highly pathogenic simian/human immunodeficiency virus in a rhesus macaque treated with anti-CD8 MAb during a primary infection with a nonpathogenic virus. Proc. Natl. Acad. Sci. U. S. A. 96: 14049–14054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Igarashi T., et al. 2003. Early control of highly pathogenic simian immunodeficiency virus/human immunodeficiency virus chimeric virus infections in rhesus monkeys usually results in long-lasting asymptomatic clinical outcomes. J. Virol. 77: 10829–10840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jensen M. A., et al. 2003. Improved coreceptor usage prediction and genotypic monitoring of R5-to-X4 transition by motif analysis of human immunodeficiency virus type 1 env V3 loop sequences. J. Virol. 77: 13376–13388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karlsson A., Parsmyr K., Sandstrom E., Fenyo E. M., Albert J. 1994. MT-2 cell tropism as prognostic marker for disease progression in human immunodeficiency virus type 1 infection. J. Clin. Microbiol. 32: 364–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keele B. F., et al. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 105: 7552–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kemal K. S., et al. 2003. HIV-1 in genital tract and plasma of women: compartmentalization of viral sequences, coreceptor usage, and glycosylation. Proc. Natl. Acad. Sci. U. S. A. 100: 12972–12977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kirchhoff F., et al. 1997. Simian immunodeficiency virus variants with differential T-cell and macrophage tropism use CCR5 and an unidentified cofactor expressed in CEMx174 cells for efficient entry. J. Virol. 71: 6509–6516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu S. L., et al. 2002. Selection for human immunodeficiency virus type 1 recombinants in a patient with rapid progression to AIDS. J. Virol. 76: 10674–10684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Margulies B. J., Hauer D. A., Clements J. E. 2001. Identification and comparison of eleven rhesus macaque chemokine receptors. AIDS Res. Hum. Retrovir. 17: 981–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mild M., Esbjornsson J., Fenyo E. M., Medstrand P. 2007. Frequent intrapatient recombination between human immunodeficiency virus type 1 R5 and X4 envelopes: implications for coreceptor switch. J. Virol. 81: 3369–3376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moyle G. J., et al. 2005. Epidemiology and predictive factors for chemokine receptor use in HIV-1 infection. J. Infect. Dis. 191: 866–872 [DOI] [PubMed] [Google Scholar]
- 30. National Institutes of Health 1985. Guide for the care and use of laboratory animals. Department of Health and Human Services publication no. NIH 85-23 National Institutes of Health, Bethesda, MD [Google Scholar]
- 31. Nishimura Y., et al. 2005. Resting naive CD4+ T cells are massively infected and eliminated by X4-tropic simian-human immunodeficiency viruses in macaques. Proc. Natl. Acad. Sci. U. S. A. 102: 8000–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nishimura Y., et al. 2007. Loss of naive cells accompanies memory CD4+ T-cell depletion during long-term progression to AIDS in simian immunodeficiency virus-infected macaques. J. Virol. 81: 893–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nishimura Y., et al. 2004. Highly pathogenic SHIVs and SIVs target different CD4+ T cell subsets in rhesus monkeys, explaining their divergent clinical courses. Proc. Natl. Acad. Sci. U. S. A. 101: 12324–12329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nishimura Y., et al. 2010. Generation of the pathogenic R5-tropic simian/human immunodeficiency virus SHIVAD8 by serial passaging in rhesus macaques. J. Virol. 84: 4769–4781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nora T., et al. 2007. Contribution of recombination to the evolution of human immunodeficiency viruses expressing resistance to antiretroviral treatment. J. Virol. 81: 7620–7628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. O'Doherty U., Swiggard W. J., Malim M. H. 2000. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J. Virol. 74: 10074–10080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Picker L. J., et al. 2004. Insufficient production and tissue delivery of CD4+ memory T cells in rapidly progressive simian immunodeficiency virus infection. J. Exp. Med. 200: 1299–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pitcher C. J., et al. 2002. Development and homeostasis of T cell memory in rhesus macaque. J. Immunol. 168: 29–43 [DOI] [PubMed] [Google Scholar]
- 39. Pollakis G., et al. 2001. N-linked glycosylation of the HIV type-1 gp120 envelope glycoprotein as a major determinant of CCR5 and CXCR4 coreceptor utilization. J. Biol. Chem. 276: 13433–13441 [DOI] [PubMed] [Google Scholar]
- 40. Regoes R. R., Bonhoeffer S. 2005. The HIV coreceptor switch: a population dynamical perspective. Trends Microbiol. 13: 269–277 [DOI] [PubMed] [Google Scholar]
- 41. Reimann K. A., et al. 1996. A chimeric simian/human immunodeficiency virus expressing a primary patient human immunodeficiency virus type 1 isolate env causes an AIDS-like disease after in vivo passage in rhesus monkeys. J. Virol. 70: 6922–6928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ren W., et al. 2010. Different tempo and anatomic location of dualtropic and X4 virus emergence in a model of R5 simian-human immunodeficiency virus infection. J. Virol. 84: 340–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Resch W., Hoffman N., Swanstrom R. 2001. Improved success of phenotype prediction of the human immunodeficiency virus type 1 from envelope variable loop 3 sequence using neural networks. Virology 288: 51–62 [DOI] [PubMed] [Google Scholar]
- 44. Richman D. D., Bozzette S. A. 1994. The impact of the syncytium-inducing phenotype of human immunodeficiency virus on disease progression. J. Infect. Dis. 169: 968–974 [DOI] [PubMed] [Google Scholar]
- 45. Robertson D. L., et al. 2000. HIV-1 nomenclature proposal. Science 288: 55–56 [DOI] [PubMed] [Google Scholar]
- 46. Sadjadpour R., et al. 2004. Induction of disease by a molecularly cloned highly pathogenic simian immunodeficiency virus/human immunodeficiency virus chimera is multigenic. J. Virol. 78: 5513–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sagar M., et al. 2009. Selection of HIV variants with signature genotypic characteristics during heterosexual transmission. J. Infect. Dis. 199: 580–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Salazar-Gonzalez J. F., et al. 2008. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol. 82: 3952–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Salazar-Gonzalez J. F., et al. 2009. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 206: 1273–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Samson M., et al. 1996. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382: 722–725 [DOI] [PubMed] [Google Scholar]
- 51. Schuitemaker H., et al. 1992. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 66: 1354–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shibata R., et al. 1997. Infection and pathogenicity of chimeric simian-human immunodeficiency viruses in macaques: determinants of high virus loads and CD4 cell killing. J. Infect. Dis. 176: 362–373 [DOI] [PubMed] [Google Scholar]
- 53. Tersmette M., Miedema F. 1990. Interactions between HIV and the host immune system in the pathogenesis of AIDS. AIDS 4(Suppl. 1): S57–S66 [PubMed] [Google Scholar]
- 54. van Rij R. P., Worobey M., Visser J. A., Schuitemaker H. 2003. Evolution of R5 and X4 human immunodeficiency virus type 1 gag sequences in vivo: evidence for recombination. Virology 314: 451–459 [DOI] [PubMed] [Google Scholar]
- 55. van't Wout A. B., et al. 1994. Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission. J. Clin. Invest. 94: 2060–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wilkin T. J., et al. 2007. HIV type 1 chemokine coreceptor use among antiretroviral-experienced patients screened for a clinical trial of a CCR5 inhibitor: AIDS Clinical Trial Group A5211. Clin. Infect. Dis. 44: 591–595 [DOI] [PubMed] [Google Scholar]
- 57. Willey R., Nason M. C., Nishimura Y., Follmann D. A., Martin M. A. Neutralizing antibody titers conferring protection to macaques from a simian/human immunodeficiency virus challenge using the TZM-bl assay. AIDS Res. Hum. Retrovir. 26: 89–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Willey R. L., et al. 1988. In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J. Virol. 62: 139–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang Y., et al. 2000. Use of inhibitors to evaluate coreceptor usage by simian and simian/human immunodeficiency viruses and human immunodeficiency virus type 2 in primary cells. J. Virol. 74: 6893–6910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhu T., et al. 1993. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 261: 1179–1181 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.