

Abstract
Improved methods for structural analyses of glycosaminoglycans (GAGs) are required to understand their functional roles in various biological processes. Major challenges in structural characterization of complex GAG oligosaccharides using liquid chromatography-mass spectrometry (LC-MS) include the accurate determination of the patterns of sulfation due to gas-phase losses of the sulfate groups upon collisional activation and inefficient on-line separation of positional sulfation isomers prior to MS/MS analyses. Here, a sequential chemical derivatization procedure including permethylation, desulfation, and acetylation was demonstrated to enable both on-line LC separation of isomeric mixtures of chondroitin sulfate (CS) oligosaccharides and accurate determination of sites of sulfation by MSn. The derivatized oligosaccharides have sulfate groups replaced with acetyl groups, which are sufficiently stable to survive MSn fragmentation and reflect the original sulfation patterns. A standard reversed-phase LC-MS system with a capillary C18 column was used for separation, and MSn experiments using collision-induced dissociation (CID) were performed. Our results indicate that the combination of this derivatization strategy and MSn methodology enables accurate identification of the sulfation isomers of CS hexasaccharides with either saturated or unsaturated nonreducing ends. Moreover, derivatized CS hexasaccharide isomer mixtures become separable by LC-MS method due to different positions of acetyl modifications.
Keywords: Chondroitin, Glycosaminoglycans, GAGs, MSn, Carbohydrates
Introduction
Glycosaminoglycans (GAGs) are linear polysaccharides often linked to the protein component of proteoglycans, which can be divided into classes consisting of different repeating disaccharide units: hyaluronan, chondroitin sulfate (CS), dermatan sulfate, heparan sulfate, heparin, and keratan sulfate [1]. The repeating disaccharide unit of CS is comprised of glucuronic acid (GlcA) linked to N-acetylgalactosamine (GalNAc), where sulfations may occur at 4- and/or 6-positions of GalNAc, and/or 2-position of GlcA, yielding five common CS units with different sulfation patterns: [GlcA-GalNAc(4S)] (A-unit), [GlcA-GalNAc(6S)] (C-unit), [GlcA(2S)-GalNAc(6S)] (D-unit), [GlcA-GalNAc(4S6S)] (E-unit), and the nonsulfated unit [GlcA-GalNAc] (O-unit), along with other rare types [2]. The monosulfated units, A- and C-units, are commonly found in great proportions in typical CS sources such as chondroitin sulfate-A (CS-A) and chondroitin sulfate-C (CS-C), respectively, with small proportions of other units also present.
Interactions between GAGs and functional proteins are involved in many biological processes, including cell signaling, migration, and tissue development [3–7]. It is believed that particular saccharide domains with specific sulfation patterns play crucial rules in these biological functions. For example, studies have shown that significant structural changes in CS chains, caused by alterations in their expressions, lead to functional changes of CS-proteoglycans involved in diseases like atherosclerosis and cancer [8–10]. Therefore, structural investigation of longer biologically active oligosaccharides rather than just disaccharide compositional analyses is quite necessary for further understanding functional domains of GAGs [11, 12]. Refined structural characterization of GAG oligosaccharides, especially the identification of their sulfation patterns, is imperative to understand the structure–function relations of GAGs in complexes with target proteins.
The high degree of heterogeneity in GAG polymers caused by the variety of chain lengths and diverse sulfation patterns makes their structural determination a very challenging task. Various techniques have been applied for this purpose, including nuclear magnetic resonance (NMR) and mass spectrometry (MS). While NMR techniques have been demonstrated as a powerful tool for detailed structural information [13], their application is typically limited to highly enriched, relatively abundant samples that are difficult to obtain from many GAG preparations and often exhibits difficulties with spectral complexity. Thus, MS methods are generally more desirable for applications involving low sample amounts and/or heterogeneous samples due to their higher sensitivity and easier compatibility with on-line liquid chromatography separations. The possibility to isolate and structurally interrogate analytes from complex mixtures using tandem MS techniques is an additional important advantage. The major challenges involved in the fine structural characterization of GAG oligosaccharides using MS include the difficulty to obtain accurate identification of the sulfation patterns due to gas-phase losses of the sulfate groups upon collisional activation, and the inefficiency of on-line separation prior to MS/MS analyses of sulfation isomers having identical elemental compositions and saccharide sequences but differing solely in the position of sulfation.
Multiple ionization techniques have been used for structural analyses of GAGs including fast-atom bombardment (FAB) [14, 15], matrix-assisted laser desorption-ionization (MALDI) [16, 17], as well as electrospray ionization (ESI) coupled with different tandem MS techniques [18–20]. ESI is preferable for its gentle ionization and has been used for structural analysis of GAGs by several groups. Zaia and co-workers have shown that the loss of labile sulfate groups during collision-induced dissociation (CID) can be minimized by deprotonation of these groups, using a combination of charge state manipulation and metal ion adduction. Their tandem mass spectra are often useful in differentiation of sulfation isomers of CS in enriched samples, and even in differentiating DS-like oligosaccharides from CS oligosaccharides with additional MS3 stage [21, 22]. However, unique optimized conditions are required for each different GAG oligosaccharide. Sulfate losses are still quite common, making mixture analysis highly problematic due to the presence of both sulfated and nonsulfated versions of many product ions. Sufficient fragmentation information for definitive assignment of sulfation sites is not always available, even under optimized conditions, which makes these methods often insufficient for complete sulfation position analysis of GAG oligosaccharide mixtures with high degrees of heterogeneity.
Another way to overcome the limitations of sulfation losses during CID fragmentations in structural analysis of longer oligosaccharides is disaccharide sequencing, in which enzymatic digestions followed by sequential stages of tandem mass spectrometry analyses (MSn) are employed for disaccharide compositional analysis as well as disaccharide sequencing [20, 23]. Sequencing of heparan sulfate hexasaccharides has been successfully achieved by using this methodology coupled with a heparin oligosaccharide sequencing tool for automatic MSn data interpretation [24]. However, pre-purified oligosaccharides or samples with very few contaminants are still required in order to generate promising structural information. Digestion with multiple enzymes and quantification of the resulting disaccharides mixtures are required prior to this MS-coupled sequencing, increasing the required amount of sample handling.
In addition to CID, electron-detachment dissociation (EDD) has also been applied to the analysis of GAG oligomers. EDD has been demonstrated to produce much higher abundances of cross-ring cleavages and able to distinguish glucuronic acid from iduronic acid based on diagnostic product-ions [25]. However, this dissociation technique also requires relatively high levels of purity of the analytes for accurate structural assignment, and the time-scale for EDD makes coupling with on-line LC separations difficult, limiting the extension of this technique to complex mixtures of oligomers.
On-line LC method coupled with MS has been used to simplify mixtures of GAG oligosaccharides in MSn analysis while limiting sample losses [26]. Those LC techniques include hydrophilic interaction (HILIC), size-exclusion (SEC), porous graphitized carbon (PGC), and ion-pairing reversed-phase (IPRP) chromatography. HILIC and SEC have been shown to effectively separate heparin oligosaccharides based on size, and HILIC is also capable of separation based on sulfation and acetylation content. However, both are incapable of separation of sulfation isomers, limiting their effectiveness in analysis of GAG oligosaccharide mixtures [27–29]. IPRP and PGC give superior retention of sulfated GAGs than using standard C18 reversed-phase chromatography, and have been shown to be capable in separation of isomeric oligosaccharides with small size and less sulfation such as heparan disaccharides, CS disaccharides, or monosulfated hexasaccharides, including the conserved linkage tetrasaccharide where the GAG chain is linked in proteoglycans [30–33]. However, little success has been demonstrated in analyses of larger oligosaccharides with sulfation patterns more complex than one sulfate per disaccharide unit.
While all these techniques have their own advantages in GAG oligosaccharide analyses, there is still no effective methodology that can achieve accurate detailed identification of oligosaccharides in a complex mixture of GAG isomers with different sulfation patterns. Here, we present an approach involving sequential chemical derivatizations to modify native CS oligosaccharides prior to structural MS analysis enabling accurate identification of GAG components in a mixture. This methodology consists essentially of replacing the labile sulfate groups with stable acetyl groups, while maintaining the positional information of the original sulfation pattern. This method enables the combination of on-line high-resolution reversed phase LC (RPLC) separation with MSn fragmentation schemes that allow us to separate sulfation isomers and interrogate them using a step-wise, modular dissociation scheme. In our method, CS hexasaccharides were first permethylated to protect free hydroxyl and carboxyl groups. The sulfate groups were then removed, and the resulting hydroxyl groups were acetylated. The chemically derivatized isomers were successfully separated by on-line capillary LC using a C18 column, and the original positions of sulfates were determined based on identification of sites of acetyl groups in the MSn spectra. Purified oligosaccharides thoroughly characterized by 2D NMR were used to verify the accuracy of the derivatization/MSn technique, and the ability of our protocol to separate and characterize complex mixtures of CS hexamers.
Experimental
Materials
Chondroitin sulfate-A sodium salt from bovine trachea, chondroitin sulfate-C sodium salt from shark cartilage, hyaluronidase from sheep testes type V, chondroitin C lyase from Flavobacterium heparinum, Sephadex G-15 resin (fractionation range of dextrans<1.5 kDa), and Dowex 50WX8-100 ion-exchange resin were purchased from Sigma-Aldrich Inc. (St. Louis, MO, USA). Bio-Gel P-10 gel resin (Fractionation range of dextrans from ~1.5 to ~20 kDa) and the polypropylene chromatographic columns (1.5×120 cm for size-exclusion chromatography, and 50×1.0 cm for desalting) were obtained from Bio-Rad Laboratories (Hercules, CA, USA). A prepacked Spherisorb S10 SAX column (10×250 mm, 5 μm) was from Waters Corporation (Milford, MA, USA). Chondroitin disaccharide ΔDi-diSE, (sodium salt, with structure of ΔGlcA-GalNAc4S6S) was purchased from Dextra Laboratories Ltd. (Reading, UK). All other regular chemical reagents used in the chemical derivatization, section below, were purchased from Sigma-Aldrich Inc. (St. Louis, MO, USA).
Preparation of CS Hexasaccharides
CS hexasaccharides were prepared as described previously [13]. Briefly, three enzymatically digested samples were applied separately for further chromatography purification including C lyase-digested CS-C, C lyase-digested CS-A, and hyaluronidase digested CS-A. The depolymerized samples were subjected to size exclusion chromatography on a Bio-Gel P-10 column using 1 M NaCl containing 10% ethanol as a mobile phase to obtain oligosaccharides fractions with uniform length. The fraction containing hexasaccharides was concentrated, desalted through Sephadex G-15 column using distilled water mobile phase, and then lyophilized. The desalted, dried hexasaccharide fraction was then reduced using sodium borohydride as previously described [13], followed by desalting and lyophilization. The reduced hexasaccharide faction was further separated by strong anion exchange (SAX) chromatography using a linear gradient of NaCl (10 mM/min) in H2O (pH 5.0). Major fractions were collected, desalted, and dried.
Structural Determination by NMR
The purified CS hexasaccharide fractions from SAX chromatography were fully characterized by two-dimensional NMR experiments, 1H/1H double quantum filtered correlation spectroscopy (DQF-COSY), 1H/1H total correlation spectroscopy (TOCSY), and 1H/13C gradient heteronuclear single quantum coherence (gHSQC). The different patterns of sulfation were assigned primarily via 1H/13C-chemical shifts of cross-peaks in the 13C-gHSQC spectra that were indicative of specific sulfation positions. The 13C-assignment in 13C-HSQC cross-peaks were assigned by the 1H-chemical shifts obtained previously from DQF-COSY or TOCSY experiments. More details about parameters in these NMR experiments have been addressed in previous work [13].
Chemical Derivatization of CS Hexasaccharides
The permethylation of sulfated CS oligosaccharides was a modified from those standard protocols used for unsulfated glycans because the high degree of sulfation groups and carboxyl groups prevented the dissolution of CS oligosaccharides in DMSO [34]. Reduced CS hexasaccharides were converted to triethylamine (TEA) salts by a self-packed cation-exchange column with Dowex 50 W resin, followed by lyophilization. The dried TEA salts (10–50 μg) were resuspended in 200 μL DMSO and 200 μL anhydrous suspension of NaOH in DMSO (150 μg/μL) followed by addition of 100 μL iodomethane. After 5 min vortexing and 10 min sonicating, the reaction was stopped by adding 2 mL water and spared with nitrogen to remove iodomethane, followed by desalting using a C18 Sep-Pak cartridge (Waters Co.). The permethylated products were dried and then made as pyridinium salts using the same type of cation-exchange column and lyophilization. The pyridinium salts (10–50 μg) of hexasaccharides were dissolved in 10 μL DMSO containing 10% methanol and incubated for 3 h at 80 °C to remove the sulfate groups [35]. The desulfated product was lyophilized and then resuspended in 175 μL pyridine, 25 μL acetic anhydride, and incubated at 50 °C overnight to acetylate the hydroxyl groups [36]. The solvents were then removed by using a Speed-Vac concentrator, and the samples were resuspended in water at a concentration of 0.2 μg/μL for later analysis.
LC-MSn Analysis
RPLC was performed on a regular capillary C18 column (Michrom Bioresources, Auburn, CA, USA, 0.2×50 mm, 3 μm, 200Å), using a linear gradient of acetonitrile from 20% to 60% over 60 min in 1 mM sodium acetate, with a flow rate of 3 μL/min and a 10 μL injection at a sample concentration of 0.2 μg/μL water. Mass spectrometry was performed on a Thermo LTQ-FT instrument using Captive-Spray ionization with an Advance Ion Source for Thermo MS (Michrom Bioresources Inc.). The spray voltage was set at 1.9 kV; capillary temperature was set to 250 °C. A full mass scan was acquired under FT mode followed by several directed MSn scans of precursors with ion trap mode to direct MSn analysis. The range of the collision energy for all stages of ion activation was set as 35–40 V.
Results and Discussion
Homogeneous fractions of CS hexasaccharides were obtained by a combination of SEC and SAX chromatography (Figure S1, online supporting information). The structures of these isomers were then determined by 2D-NMR (Figure S2). Previous studies had already shown that commercial CS sources like CS-A and CS-C are not composed of only A-unit or C-unit but always with substantial amounts of other disaccharide units. The proper validation of any new analytical method requires the use of standards of known structure, therefore the purification and rigorous structural analysis of CS oligomers by LC and NMR spectroscopy, while difficult and laborious, is a key part of the effort to develop analytical methods, especially for heterogeneous polysaccharides such as GAGs.
Two types of enzymes were used to obtain CS oligosaccharides with either unsaturated nonreducing end (by C lyase) or with saturated nonreducing end (by hyaluronidase). Initial work was performed using hexasaccharides generated by lyase digestion, which introduces a double bond at the nonreducing terminus. The asymmetry introduced by the double bond makes interpretation of product ion spectra easier. Four homogeneous CS hexasaccharides were isolated: two trisulfated hexasaccharides [ΔGlcA-GalNAc(4S)-GlcA-GalNAc(4S)-GlcA-GalNAc(4S)-ol] (ΔC4;4;4S-ol) and [ΔGlcA-GalNAc(6S)-GlcA-GalNAc(6S)-GlcA-GalNAc(6S)-ol] (ΔC6;6;6S-ol) from C lyase digested CS-A, one disulfated hexasaccharide [GlcA-GalNAc(6S)-GlcA-Gal-NAc(4S)-GlcA-GalNAc-ol] (C6;4;0S-ol) from hyaluronidase digested CS-A, and one tetra-sulfated hexasaccharide [ΔGlcA-GalNAc(4S)-GlcA(2S)-GalNAc(6S)-GlcA-GalNAc(6S)-ol] (ΔC4;2,6;6S-ol) from C lyase digested CS-C. The symbol Δ indicates 4,5-unsaturation on GlcA at the non-reducing end, the “S” refers to sulfation group, and the “-ol” indicates GalNAc at the reducing end is reduced. A hexasaccharide fraction isolated from hyaluronidase-digested CS-A was collected after SEC separation and without further SAX separation in order to test the method’s ability to separate and characterize structures from mixtures.
Chemical Derivatizations of CS hexasaccharides
The initial part of our methodology is the chemical derivatization of the oligosaccharides, which include sequential permethylation, desulfation, and acetylation. The purpose of the chemical derivation strategy is to substitute the labile sulfate groups with a more stable and distinguishable group that can be differentiated from permethylated groups, both by mass and by LC retention time. This allows the LC separation and structural identification of sulfated GAG oligomers differing only by one or more positions of sulfation. Thus, we substitute the sulfate groups with acetyl groups, which are more stable and distinguishable from methyl groups. The position of acetyl groups determined by CID tandem mass spectrometry will reflect the original position of sulfate groups.
The first obstacle encountered for the derivatization scheme was the dissolution of highly sulfated CS hexasaccharides in DMSO, which is the common solvent used in the standard permethylation procedure. Unlike unsulfated or less densely-sulfated glycans, the high density of the sulfate and carboxyl groups make GAG oligosaccharides highly negative charged and usually exist as sodium salts, which prevents their complete dissolution in DMSO. Therefore, the counter-ions in our CS hexasaccharides were exchanged to form a TEA salt prior to the permethylation reaction. The TEA-CS salts were easily dissolved in DMSO, even for highly sulfated GAG oligomers. The mass of the products showed that the CS hexasaccharides were fully permethylated, including the hydroxyl groups of the carboxyls on the GlcA and the hydrogen of the acetamine group on the GalNAc, leaving the sulfate groups unmodified as shown in the supporting information (Figure S3a and b).
For desulfation reaction, two methods were experimentally compared in order to increase sulfation removal efficiency: methanolic desulfation and the solvolytic desulfation. The former was performed by incubating the permethylated GAG in methanol/HCl, where the sulfate groups are removed by acid methanolysis [37]. However, the acidic conditions seemed to cause the cleavage of some glycosidic bonds concurrently with desulfation, as previously observed with sulfated N-linked glycans [38]. Even though some glycosidic bond cleavages in our products were decreased by optimizing conditions, the amounts of glycolysis were still leading to undesirable levels of sample losses. Adoption of a pyridinium salt-mediated solvolytic desulfation protocol described in the Experimental section reduced the sample loss considerably as determined by direct infusion analysis in both positive and negative ESI-MS (data not shown).
After desulfation of the hexameric mixture, three different derivatization schemes were comparatively tested for determination of the original sites of sulfation. One aliquot was derivatized by acetylation, labeling the original sulfation sites with a group that could be differentiated from permethylated sites by mass and, potentially, by retention on a C18 column. A second aliquot was derivatized by trideuteropermethylation due to the higher efficiency of permethylation compared with peracetylation methods. In this aliquot, the original site of sulfation was labeled with a group that could be easily differentiated from a standard permethylation site by mass, but probably not by retention time. Finally, a third aliquot was kept underivatized at the hydroxyl groups left after desulfation.
All three derivatized versions were retained on a standard C18 column and analyzed in positive ion mode. The subsequent MSn analyses of the aliquot with underivatized hydroxyl groups at the original sites of sulfation exhibited extensive water loss during the fragmentation procedure. This resulted in fewer informative product ions than the other two derivatized types. The chromatograms of the unmodified hexasaccharides also gave broader peaks and shorter retention times, making the separation less efficient than for the acetylated product, as shown in Figure S4a. Trideuteromethylation of former sites of sulfation solved the issues pertaining to water loss during ion activation. The permethylated-desulfated-trideuteromethylated products gave valuable information from the MSn spectra, and enabled differentiation of isomeric hexasaccharide species based on diagnostic product ions. However, the efficient separation of isomers based on differential position of trideuteromethyl groups in the fully permethylated GAG was not possible, which makes confident identification of specific CS hexamers from an isomeric mixture difficult at best (Figure S4b). For products where the original sites of sulfation were replaced by acetyl groups, CS sulfation isomers were readily distinguishable through MSn analyses, as detailed below. Additionally, relatively efficient separations were achieved using standard RPLC based on the different positions of the acetyl groups (Figure S4c).
After reduction, permethylation, desulfation, and acetylation, the total sample loss is estimated at 50%. The masses of the permethylated-desulfated products and the permethylated-desulfated-acetylated products showed that CS hexasaccharides were properly derivatized (Figure S3c and d). The majority of byproducts were tetrasaccharides and disaccharides caused primarily by the β-elimination reaction between the carbon-4 and –5 of the GlcA during the permethylation (Figure S3b), with a smaller amount of byproducts apparently formed during the desulfation procedure. These byproducts are notable for not introducing species that would lead to the false identification of sites of sulfation; their major effect is reducing the apparent sensitivity of the technique.
Another problem caused by permethylation is the undermethylation, giving a product (m/z of 544.16 in Figure S3b) with one free hydroxyl group left. This undermethylation product can also be acetylated in subsequent acetylation step, giving a final derivatized product with m/z of 804.84 as shown in Figure S3d. While such product might give a false identification of one additional sulfation site, the amount is very low compared with the fully derivatized product (m/z of 790.84), and the LC chromatogram also gives a separation of 3 min between the two products (data not shown).
Structural Differentiation of CS Isomers ΔC4;4;4S-ol and ΔC6;6;6S-ol
As CS molecules vary mainly at 4- and 6-sulfation of the GalNAc, homogeneous trisulfated hexasaccharides ΔC4;4;4S-ol and ΔC6;6;6S-ol were selected as initial standards. They allow differentiation during the MSn analyses between the A-unit and C-unit at each region of the hexamer, including nonreducing end, internal region, and reducing end. As illustrated in Figure 1, all free hydroxyl groups of the oligosaccharides are now methylated after derivatization, and the previous sites of sulfation are labeled by acetyl groups. Fragmentation nomenclature is followed here as described by Domon and Costello [39]. After all derivatizations, ΔC4;4;4S-ol and ΔC6;6;6S-ol were first analyzed by manual MSn experiments by direct infusion of solutions of the sample at a concentration of 10 μM in 50/50 acetonitrile/water with a flow rate of 3μL/min. Four ions, [M+H]+, [M+2H]2+, [M+Na]+, and [M+2Na]2+, were detected and analyzed by thorough MSn up to MS4 for the most abundant product ions for each round of fragmentation (data not shown). While it is relatively simple to drive permethylated sugar ions toward sodium adducts by the introduction of sodium salts into the running buffers, it is very difficult to wholly eliminate sodium adduct ions and drive permethylated sugars to protonated ions. Additionally, the [M+2Na]2+ ion yielded more thorough and reliable structural information than [M+Na]+ under corresponding optimized condition. Therefore, selection of the doubly charged sodium adduct as the preferred precursor ion allows us to increase our sensitivity by driving our ion population toward sodium adducts.
Figure 1.

Derivatized product of [ΔGlcA-GalNAc(6S)-GlcA-GalNAc(6S)-GlcA-GalNAc(6S)-ol] (ΔC6;6;6S-ol), where the symbol Δ indicates 4,5-unsaturation on GlcA at the nonreducing end, the “S” refers to sulfation group, and the “-ol” indicates GalNAc at the reducing end is reduced. The structure shows that there is one acetyl group on the 6 position for each GalNAc indicating the original sulfation sites, while other positions are all methylated
As illustrated in Figure 2a, the product ions [B2+Na]+, [B4Y4+Na]+, and [Y2+Na]+ with different m/z values represent the three disaccharides units of each hexasaccharide, which enable us to compare the differences between 4- and 6-sulfation on different positions along the oligomeric chain in a systematic, modular scheme by doing MSn individually of the respective product ions. The MS2 of [M+2Na]2+ gave three major product ions [0,2X5+2Na]2+, [B2+Na]+, and [Y4+Na]+ (Figure 2b) regardless of the original pattern of sulfation. MS3 of the [Y4+Na]+ product ion gave two product ions [B4Y4+Na]+ and [Y2+Na]+ (Figure 2c). The three unique ions representing each disaccharide unit along the hexamer allows for individual determination of the site(s) of sulfation of each disaccharide unit of the oligosaccharide in a systematic manner.
Figure 2.

Fragmentation path for derivatized CS trisulfated hexasacharides (a) and MSn spectra for derivatized oligosaccharide ΔC6;6;6S-ol: (b) MS2 of [M+2Na]2+ (m/z 774.8) and (c) MS3 of [Y4+Na]+ (m/z 1053.4). Symbols:  ΔGlcA,
ΔGlcA,  GlcA,
GlcA,  GalNAc. All product ions labeled on the spectra are singly charged and monosodiated unless otherwise annotated
GalNAc. All product ions labeled on the spectra are singly charged and monosodiated unless otherwise annotated
As part of the development of the initial MSn parameters for 6- and 4-sulfation at each disaccharide unit of the hexasaccharide by direct infusion of these two enriched NMR-characterized standards, identification of diagnostic MSn ions was performed by on-line RPLC-MS. The additional LC-based separation step was performed prior to diagnostic MSn product ion analysis since the purity of these standards was only verified by NMR. Low levels of contaminant isomers missed by NMR analysis might lead to incorrect identification of diagnostic product ions in the MSn spectra. Online LC separation also isolated the target products (hexasaccharides) from those by-products (tetrasaccharides and disaccharides) resulting from the chemical derivatization steps. This also helped to increase the sensitivity of the analysis by reducing ion suppression in the electrospray process from by-products. In order to get [M+2Na]2+ as the predominant parent ion, 1 mM sodium acetate was added to the LC mobile phases, which did not seem to significantly compromise electrospray stability.
The MS method was set up as six events including one full mass scan followed by five MSn scans for five ions shown in Figure 2a:MS2 of [M+2Na]2+, MS3 of [B2+Na]+, MS3 of [Y4+Na]+, MS4 of [B4Y4+Na]+, and MS4 of [Y2+Na]+. Three MSn spectra of ΔC4;4;4S-ol (Figure 3a–c) and ΔC6;6;6S-ol (Figure 3d–f) were compared. From the MS3 of [B2+Na]+, we noticed that the B2Z5 ion with m/z of 278.2 was abundant only for the 6-sulfated disaccharide on the nonreducing end (Figure 3d), while 4-sulfation disaccharide at the nonreducing end resulted in a MS3 product ion of m/z 196.2 (Figure 3a). Similarly for the internal disaccharide, the B4Z3 ion of m/z 278.2 was also observed for the 6-sulfated GalNAc (Figure 3e) on the MS4 spectra of [B4Y4+Na]+ ion and not for the 4-sulfated GalNAc (Figure 3b), where the product ion of m/z 196.2 was observed. The trend did not continue with the disaccharide on the reducing end where the GalNAc was reduced. As shown in Figure 3c, the MS4 of [Y2+Na]+ ion gave two diagnostic product ions with m/z of 480.2 and 502.2 for 4-sulfated GalNAc, which were practically undetectable for the 6-sulfated GalNAc at the reducing end (Figure 3f). For the reduced GalNAc at the reducing end, the acetyl group on the 4-position appeared more likely to be lost during the fragmentation than it did on the 6-position, resulting in the more intense product ions as observed. Overall, by fragmenting the three target product ions corresponding to disaccharides units in a sequential manner, we were able to differentiate between the 4- and 6-sulfated GalNAcs at any disaccharide position along the CS hexasaccharides using a systematic predetermined MSn scheme.
Figure 3.

MSn spectra for two derivatized hexasaccharide standards ΔC4;4;4S-ol (a), (b), (c) and ΔC6;6;6S-ol (d), (e), (f). From top row to bottom row, they are (a), (d) MS3 of [B2+Na]+ (m/z 496.2), (b), (e) MS4 of [B4Y4+Na]+ (m/z 514.2) and (c), (f) MS4 of [Y2+Na]+ (m/z 562.2) spectra, representing the three disaccharide units respectively at the nonreducing end, in the middle and at the reducing end. The peak marked with an asterisk represents the diagnostic product ions used for differentiation between 4-sulfated and 6-sulfated GalNAc
LC-MSn Analysis of CS Trisulfated Hexasaccharides Mixture
In order to explore the separation capabilities of RPLC for derivatized CS hexasaccharides, as well as to demonstrate the ability to differentiate isomers by MSn on an LC time-scale, a mixture of trisulfated CS hexasaccharides was prepared as described in the Experimental section, and analyzed by the similar LC-MSn method used for standards. Unlike the standards ΔC4;4;4S-ol and ΔC6;6;6S-ol, having unsaturated nonreducing end residues (due to lyase digestion), the hexasaccharides mixture used here was prepared by hyaluronidase digestion in order to produce oligosaccharides with saturated nonreducing end residue. This would extend the application of our methodology to CS oligosaccharides produced from different workflows. As listed in Table 1, among the five ions analyzed in the MSn method, the [M+2Na]2+ and [B2+Na]+ ion had different m/z values for hexasaccharides having unsaturated and saturated non-reducing ends, while the [Y4+Na]+, [B4Y4+Na]+, and [Y2+Na]+ ion had same m/z values for both species. In order to check if the differentiation of 4- and 6-sulfated for the GalNAc of the saturated disaccharide units would be similar to unsaturated disaccharide units, the MS3 of [B2+Na]+ ion (m/z 496.2) were extracted and two different types of spectra were observed, as shown in Figure 4. The diagnostic ions with m/z of 278.2 and 196.2 were still observed and able to be used for distinguishing the 6- and 4-sulfated GalNAc respectively, even though the MS/MS fragmentation looks different from the standards (Figure 3a versus d) by missing the [B20,2X5+Na]+ ion (m/z 352.2) that was believed to be typically produced by retro-Diels-Alder decomposition of cyclohexene-like structure.
Table 1.
m/z of Relevant Precursor and Product Ions for Derivatized CS Hexamer Identification
| Parent and product ionsa |
Saturated nonreducing end |
Unsaturated nonreducing end |
Mass shift by±one sulfation group |
|---|---|---|---|
| M2+ | 790.8 | 774.8 | ±14 |
| 0,2X52+ | NAb | 703.2 | ±14 |
| B2+ | 528.2 | 496.2 | ±28 |
| Y4+ | 1053.4 | 1053.4 | ±28 |
| B4Y4+ | 514.2 | 514.2 | ±28 |
| Y2+ | 562.2 | 562.2 | ±28 |
| C1+ | 273.2 | NAb | ±28 |
| B2Z5+ | 278.2 | 278.2 | ±28 |
| C3Y4+ | 259.2 | 259.2 | ±28 |
| B4Z3+ | 278.2 | 278.2 | ±28 |
| C5Y2+ | 259.2 | 259.2 | ±28 |
| Z1+ | 326.2 | 326.2 | ±28 |
| B20,2X5+ | NAb | 352.2 | ±28 |
All ions were mono-sodiated except for M2+ and 0,2X52+ which were di-sodiated
The corresponding product ion was not observed
Figure 4.
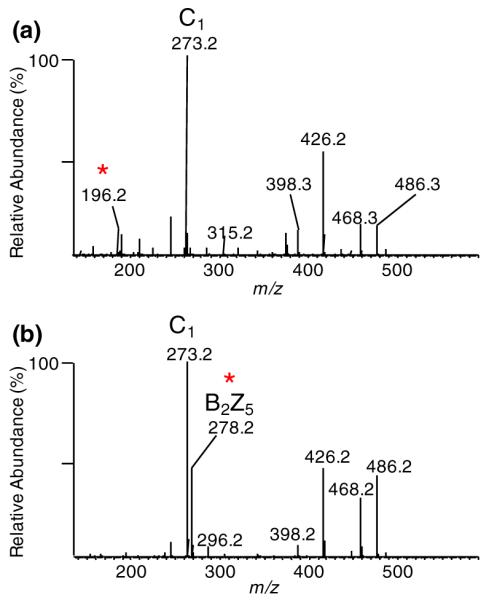
Two different MS3 of [B2+Na]+ (m/z 528.2) spectra extracted from the LC-MSn analysis of mixture of derivatized trisulfated hexasaccharides with a saturated nonreducing end. While the product ion spectra are notably different from the hexasaccharide standards with an unsaturated non-reducing end (Figure 3a/d), the same diagnostic ions were observed at m/z 196.2 and 278.2 representing the 4-sulfated GalNAc (a) and the 6-sulfated (b) GalNAc. The peak marked with an asterisk represents the diagnostic product ions used for differentiation between 4-sulfated and 6-sulfated GalNAc
The base peak chromatograms (BPC) of the hexasaccharide mixture for MS3 of [B2+Na]+, MS4 of [B4Y4+Na]+, and MS4 of [Y2+Na]+ were extracted and shown as a black line in Figure 5 from the top panel to the bottom one. For MS3 of [B2+Na]+, the diagnostic product ions with m/z of 278.2 (6-sulfated) and 196.2 (4-sulfated) indicate 6-sulfation (Figure 5a, red line) or 4-sulfation (Figure 5a, purple line) in GalNAc disaccharide located at the nonreducing end. The same selected ion chromatogram (SIC) was extracted for MS4 of [B4Y4+Na]+ (Figure 5b), where the same diagnostic product ions with m/z of 278.2 and 196.2 were extracted. For MS4 of [Y2+Na]+, the ion with m/z value of 480.2 was extracted to indicate isomers with 4-sulfated GalNAc on the reducing end (Figure 5c, red line). However, there was no diagnostic product ion for 6-sulfated isomers; rather, 6-sulfated GalNAc on the reducing end is signified by a mass consistent with a sulfated reducing end unit combined with a lack of MS4 product ion of m/z 480.2, as shown in Figure 3f. In order to plot the SIC for the 6-sulfated GalNAc on the reducing end, the intensity of the ion of m/z of 326.2 (which is common to both 6-sulfated and 4-sulfated reducing end disaccharide product ions) was subtracted by the sum of the intensities of the ion of m/z of 480.2 and 502.2 (which are abundant for the 4-sulfated reducing end disaccharide product ion but not to the 6-sulfated one) to show the 6-sulfated species (Figure 5c, purple line). Each SIC in Figure 5 represents all isomers with the annotated modification; therefore, in order to identify a particular isomer, data from all three panels must be superimposed, with the data in panel a indicating the sulfation site at the nonreducing end GalNAc, panel b indicating the sulfation site at the internal GalNAc, and panel b indicating the sulfation site at the reducing end GalNAc. In this manner, even though the chromatography alone is insufficient to resolve the isomers to baseline, combination of the chromatography with the MSn data are capable of resolving all of the different isomers in the trisulfated hexamer.
Figure 5.

LC-MSn analysis for a derivatized mixture of CS trisulfated hexasaccharides with saturated nonreducing ends. Base peak chromatograms were shown for MS/MS spectra of three disaccharide units: (a) MS3 of [B2+Na]+, (b) MS4 of [B4Y4+Na]+ and (c) MS4 of [Y2+Na]+ (black). Extracted single ion chromatograms (SIC) with m/z of diagnostic ions indicate the appropriate GalNAc is 6-sulfated (in red) or 4-sulfated (in purple). The three sulfation sites for each BPC peak can be assigned separately by the color of the three SIC that cover the corresponding peak area
Three major peaks were observed in the BPC shown in Figure 5. For the first peak, the three SICs that covered the peak area at ~71 min for GalNAc at the nonreducing end (panel a), the internal (panel b), and the reducing end (panel c) disaccharides showed that all contain 4-sulfation (purple lines), indicating for the first peak at ~71 min that the structure is C4;4;4S-ol.
The second major peak in the BPC at ~74 min seemed to have more than one species based on the various SICs extracted for the nonreducing end (Figure 5a) and the internal disaccharides (Figure 5b). The SICs of the non-reducing end indicate the presence of a large amount of 6-sulfation, with a small amount of 4-sulfation that elutes toward the front of the peak. Analysis of the SICs from the internal disaccharide indicate the presence of both the 6- and 4-sulfated internal disaccharide, where the 6-sulfated internal disaccharide co-elutes with the 4-sulfated nonreducing disaccharide, and the 4-sulfated internal disaccharide co-elutes with the 6-sulfated nonreducing disaccharide. No significant overlap was found between the 6-sulfated non-reducing disaccharide and the 6-sulfated internal disaccharide, neither between the 4-sulfated nonreducing disaccharide and the 4-sulfated internal disaccharide. All of the hexasaccharides in the second major peak in the BPC at ~74 min exhibited a 4-sulfated reducing end, with no 6-sulfation observed at the reducing end. These results indicate that the second major peak in the BPC at ~74 min consists of two analytes: the major analyte is C6,4,4S-ol, which eluted in the latter portion of the peak, and a minor component identified as C4,6,4S-ol that eluted at the front of the peak.
For the third major peak in the BPC at ~76 min, both the nonreducing end and the internal disaccharide were identified as being solely sulfated at the 6-position. However, the third major peak was determined to consist of two analytes that differed in sulfation at the reducing end: the front of the peak showed the major component to have 4-sulfation at the reducing end, while the tail of the peak showed a relatively minor component with 6-sulfation at the reducing end. These chromatograms allowed the interpretation of the third major peak in the BPC at ~76 min as consisting of both C6,6,4S-ol and C6;6;6S-ol. Even though the chromatography did not separate the CS hexasaccharide isomers to baseline in the BPC, the MSn indicated sufficient separation capability to clearly identify five slightly different CS hexamer isomers.
In total, five isomers were identified even with two minor spices (C4,6,4S-ol and C6;6;6S-ol) that cannot be chromatographically separated and detected by 2D-NMR without derivatization. Generally, the isomers could be separated by the extension of 6-sulfation, eluting in the order of no 6-sulfate (C4;4;4S-ol), one 6-sulfate (C4,6,4S-ol and C6,4,4S-ol), two 6-sulfates (C6,6,4S-ol), and three 6-sulfates (C6;6;6S-ol).
Structural Analysis of CS Disulfated and Tetra-Sulfated Hexasaccharides
While the monosulfated A- and C-unit disaccharides are the most commonly detected disaccharide units in most chondroitin sulfate sources, the nonsulfated [GlcA-GalNAc] O-unit and the disulfated [GlcA(2S)-GalNAc(6S)] D-unit and [GlcA-GalNAc(4S6S)] E-unit are also commonly found. Analyses of hexasaccharides containing the O-unit or the D-unit were performed, and are detailed in online supporting information (Figure S5). While hexasaccharides containing the E-unit could not be identified in our library of CS hexasaccharides, we did derivatize an E-unit disaccharide and compared its fragmentation properties with those of a derivatized D-unit disaccharide, with detailed results presented in online supporting information (Figure S6). To briefly summarize our results, all three units are efficiently derivatized using the protocol detailed above. The derivatized O-unit and D-unit can easily be differentiated from each other, as well as from the A-unit and C-unit, based on the masses of the disaccharide product ions in the MSn fragmentation scheme. While the masses of the D-unit and E-unit disaccharide are identical, our analyses of derivatized disaccharides indicate that these two units can be easily differentiated based on the masses of the abundant monosaccharide product ions present in the final stage of our MSn protocol, allowing for the accurate identification of all five common disaccharide units in CS hexamers.
Conclusions
In this work, a sequential chemical derivatization strategy allowed the combination of online capillary RPLC separation of isomeric trisulfated CS hexasaccharides with subsequent MSn fragmentation for differentiation of isomers by their sulfation patterns. Online separation of sulfation position isomers was easily achieved by using a standard capillary C18 column using a typical reversed phase gradient coupled to an electrospray interface operated in positive ion mode, which greatly simplifies the analytical procedures and readily supports complex samples of CS hexasaccharides. By using MSn fragmentations in a linear ion trap, we could fragment hexasaccharides into three distinguishable individual disaccharide units and analyze each separately in a systematic, modular scheme instead of assigning all sulfation sites in just one single complex MS/MS spectra. Therefore, even without producing an abundance of cross-ring cleavage products, assignments based on product ions primarily generated by glycosidic bond cleavages were still easily capable of differentiation between the 4- and 6-sulfated GalNAcs. When the modular MSn fragmentation scheme is combined with the capillary RPLC separation of sulfation isomers, the resulting method is capable of handling even complex isomeric mixtures of CS oligomers.
In addition, the methodology presented here has been demonstrated to identify CS sulfation patterns for both saturated and unsaturated hexasaccharides by simple mass shift corrections to the MSn spectra of the nonreducing terminus. The method described here can be extended to any CS hexasaccharides, regardless of the sulfation pattern, sulfate composition, or nonreducing end saturation/unsaturation, without using specific synthetic standard. Extension to larger oligosaccharides also is a current issue being investigated. In octasaccharides, for example, a nonreducing end disaccharide product ion and a reducing end hexasaccharide product ion would be generated after performing MS-MS, in which the disaccharide product can be identified by its MS3. The hexasaccharide product can be further fragmented stepwise into positional disaccharides by MSn, with each disaccharide identified as described in present work. Although, the characterization of oligosaccharides larger than hexasaccharides using such a strategy must require a unique multistage fragmentation path as well as higher stages of tandem mass spectrometry, such an extension should not be prohibitive assuming sufficient signal exists to support the additional layers of MSn required. Similarly, we are currently investigating the analysis of other sulfated GAGs, including more diverse GAGs such as heparan sulfate, where we anticipate utilizing trideuterated acetyl groups to differentiate between N-acetylated and N-sulfated amino sugars. We anticipate that with further refinements, this methodology will be expanded to the analysis of sulfation patterns of oligomers from all classes of sulfated GAGs. This is a very efficient analytical tool for characterization of these polysaccharides that are structurally heterogeneous, but widely relevant biomedically.
Supplementary Material
Acknowledgments
The author acknowledge support for this work by a grant from National Center for Research Resources as part of the Research Resources for Integrated Glycotechnology (P41RR005351). V.H.P. was partially supported by a postdoctoral fellowship (PDE 201019/2008-6) from Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq. The authors are grateful to Christian Heiss and Parastoo Azadi for advice on the permethylation of CS oligosaccharides.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s13361-011-0174-0) contains supplementary material, which is available to authorized users.
References
- 1.Perrimon N, Bernfield M. Cellular functions of proteoglycans-an overview. Semin. Cell Dev. Biol. 2001;12:65–67. doi: 10.1006/scdb.2000.0237. [DOI] [PubMed] [Google Scholar]
- 2.Deepa SS, Yamada S, Fukui S, Sugahara K. Structural determination of novel sulfated octasaccharides isolated from chondroitin sulfate of shark cartilage and their application for characterizing monoclonal antibody epitopes. Glycobiology. 2007;17:631–645. doi: 10.1093/glycob/cwm021. [DOI] [PubMed] [Google Scholar]
- 3.Whitelock JM, Iozzo RV. Heparan sulfate: a complex polymer charged with biological activity. Chem. Rev. 2005;105:2745–2764. doi: 10.1021/cr010213m. [DOI] [PubMed] [Google Scholar]
- 4.Bandtlow CE, Zimmermann DR. Proteoglycans in the developing brain: new conceptual insights for old proteins. Physiol. Rev. 2000;80:1267–1290. doi: 10.1152/physrev.2000.80.4.1267. [DOI] [PubMed] [Google Scholar]
- 5.Kirn-Safran CB, D’Souza SS, Carson DD. Heparan sulfate proteoglycans and their binding proteins in embryo implantation and placentation. Semin. Cell Dev. Biol. 2008;19:187–193. doi: 10.1016/j.semcdb.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linhardt RJ, Toida T. Role of glycosaminoglycans in cellular communication. Acc. Chem. Res. 2004;37:431–438. doi: 10.1021/ar030138x. [DOI] [PubMed] [Google Scholar]
- 7.Martel-Pelletier J, Tat SK, Pelletier J-P. Effects of chondroitin sulfate in the pathophysiology of the osteoarthritic joint: a narrative review. Osteoarthr. Cartil. 2010;18:S7–S11. doi: 10.1016/j.joca.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 8.Nasser NJ. Heparanase involvement in physiology and disease. Cell Mol. Lifer Sci. 2008;65:1706–1715. doi: 10.1007/s00018-008-7584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seeberger PH, Werz DB. Synthesis and medical applications of oligosaccharides. Nature. 2007;446:1046–1051. doi: 10.1038/nature05819. [DOI] [PubMed] [Google Scholar]
- 10.Plaas AH, West LA, Wong-Palms S, Nelson FR. Glycosaminoglycan sulfation in human osteoarthritis. Disease-related alterations at the nonreducing termini of chondroitin and dermatan sulfate. J. Biol. Chem. 1998;273:12642–12649. doi: 10.1074/jbc.273.20.12642. [DOI] [PubMed] [Google Scholar]
- 11.Kinoshita A, Sugahara K. Microanalyses of glycosaminoglycan derived oligosacharides labeled with a fluorophore 2-aminobenzamide by HPLC: application to disaccharide compositional analyses and exosequencing of oligosaccharides. Anal. Biochem. 1999;269:367–378. doi: 10.1006/abio.1999.4027. [DOI] [PubMed] [Google Scholar]
- 12.Yamada S, Sugahara K. Preparation of oligosaccharides from sulfated glycosaminoglycans using bacterial enzymes. Methods. Mol. Biol. 2003;213:71–78. doi: 10.1385/1-59259-294-5:71. [DOI] [PubMed] [Google Scholar]
- 13.Pomin VH, Sharp JS, Li X, Wang L, Prestegard JH. Characterization of glycosaminoglycans by 15N NMR spectroscopy and in vivo isotopic labeling. Anal. Biochem. 2010;82:4078–4088. doi: 10.1021/ac1001383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linhardt RJ, Wang HM, Loganathan D, Lamb DJ, Mallis LM. Isolation and characterization of a human heparin. Carbohydr. Res. 1992;225:137–145. doi: 10.1016/0008-6215(92)80045-3. [DOI] [PubMed] [Google Scholar]
- 15.Dell A, Rogers ME, Thomas-Oates JE. Fast atom bombardment mass spectrometric strategies for sequencing sulphated oligosaccharides. Carbohydr. Res. 1988;179:7–19. [Google Scholar]
- 16.Juhasz P, Biemann K. Utility of noncovalent complexes in the matrix-assisted laser desorption ionization mass spectrometry of heparin-derived oligosaccharides. Carbohydr. Res. 1995;270:131–147. doi: 10.1016/0008-6215(94)00012-5. [DOI] [PubMed] [Google Scholar]
- 17.Laremore TN, Murugesan S, Park TJ, Avci FU, Zagorevski DV, Linhardt RJ. Matrix-assisted laser desorption/ionization mass spectrometric analysis of uncomplexed highly sulfated oligosaccharides using ionic liquid matrices. Anal. Chem. 2006;78:1774–1779. doi: 10.1021/ac051121q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chai W, Luo J, Lim CK, Lawson AM. Characterization of heparin oligosaccharide mixtures as ammonium salts using electrospray mass spectrometry. Anal. Chem. 1998;70:2060–2066. doi: 10.1021/ac9712761. [DOI] [PubMed] [Google Scholar]
- 19.Yang HO, Gunay NS, Toida T, Kuberan B, Yu G, Kim YS, Linhardt RJ. Preparation and structural determination of dermatan sulfate-derived oligosaccharides. Glycobiology. 2000;10:1033–1039. doi: 10.1093/glycob/10.10.1033. [DOI] [PubMed] [Google Scholar]
- 20.Saad OM, Leary JA. Compositional analysis and quantification of heparin and heparan sulfate by electrospray ionization ion trap mass spectrometry. Anal. Chem. 2003;75:2985–2995. doi: 10.1021/ac0340455. [DOI] [PubMed] [Google Scholar]
- 21.Zaia J, Costello CE. Tandem mass spectrometry of sulfated heparin-like glycosaminoglycan oligosaccharides. Anal. Chem. 2003;75:2445–2455. doi: 10.1021/ac0263418. [DOI] [PubMed] [Google Scholar]
- 22.Bielik AM, Zaia J. Multistage tendem mass spectrometry of chondroitin sulfate and dermatan sulfate. Int. J. Mass Spectrom. 2010 doi: 10.1016/j.ijms.2010.10.017. doi:10.1016/j.ijms.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saad OM, Ebel H, Uchimura K, Rosen SD, Bertozzi CR, Leary JA. Compositional profiling of heparin/heparan sulfate using mass spectrometry: assay for specificity of a novel extracellular human endosulfatase. J. A. Glycobiology. 2005;15:818–826. doi: 10.1093/glycob/cwi064. [DOI] [PubMed] [Google Scholar]
- 24.Saad OM, Leary JA. Heparin sequencing using enzymatic digestion and ESI-MSn with host: a heparin/hs oligosaccharide sequencing tool. Anal. Chem. 2005;77:5902–5911. doi: 10.1021/ac050793d. [DOI] [PubMed] [Google Scholar]
- 25.Wolff JJ, Chi L, Linhardt RJ, Amster IJ. Distinguishing glucuronic from iduronic acid in glycosaminoglycan tetrasaccharides by using electron detachment dissaciation. Anal. Chem. 2007;79:2015–2022. doi: 10.1021/ac061636x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zaia J. On-line separations combined with MS for analysis of glycosaminoglycans. Mass Spectrom. Rev. 2009;28:254–272. doi: 10.1002/mas.20200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hitchcock AM, Costello CE, Zaia J. Glycoform quantification of chondroitin/dermatan sulfate using an LC/MS/MS platform. Biochemistry. 2006;45:2350–2361. doi: 10.1021/bi052100t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hitchcock AM, Yates KE, Costello CE, Zaia J. Comparative glycomics of connective tissue glycosaminoglycans proteomics. Proteomics. 2008;8:1384–1397. doi: 10.1002/pmic.200700787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naimy H, Leymarie N, Bowman MJ, Zaia J. Characterization of heparin oligosaccharides binding specifically to antithrombin III using mass spectrometry. Biochemistry. 2008;47:3155–3161. doi: 10.1021/bi702043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei W, Ninonuevo MR, Sharma A, Danan-Leon LM, Leary JA. A comprehensive compositional analysis of heparin/heparan sulfate-derived disaccharides from human serum. Anal. Chem. 2011 doi: 10.1021/ac2001077. doi:10.1021/ac2001077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solakyildirim K, Zhang Z, Lindhart RJ. Ultraperformance liquid chromatography with electrospray ionization ion trap mass spectrometry for chondroitin disaccharide analysis. Anal. Biochem. 2010;397:24–28. doi: 10.1016/j.ab.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Estrella RP, Whitelock JM, Pacher NH, Karlsson NG. Graphitized carbon LC-MS characterization of the chondroitin sulfate oligosaccharides of aggrecan. Anal. Biochem. 2007;79:3597–3606. doi: 10.1021/ac0622227. [DOI] [PubMed] [Google Scholar]
- 33.Karlsson NG, Schulz BL, Pacher NH, Whitelock JM. Use of graphitised carbon negative ion LC-MS to analyse enzymatically digested glycosaminoglycans. J. Chromatogr. B. 2005;824:139–147. doi: 10.1016/j.jchromb.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 34.Heiss C, Wang Z, Azadi P. Sodium hydroxide permethylation of heparin disaccharides. Rapid. Commun. Mass. Spectrom. 2011:774–778. doi: 10.1002/rcm.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagasawa K, Inoue Y, Tokuyasu T. An improved method for the preparation of chondroitin by solvolytic desulfation of chondroitin sulfates. J. Biochem. 1979;86:1323–1329. doi: 10.1093/oxfordjournals.jbchem.a132648. [DOI] [PubMed] [Google Scholar]
- 36.Bendiak B, Fang TT, Jones DNM. An effective strategy for structural elucidation of oligosaccharides through NMR spectroscopy combined with peracetylation using doubly 13C-labeled acetyl groups. Can. J. Chem. 2002;80:1032–1050. [Google Scholar]
- 37.Taguchi T, Iwasaki M, Muto Y, Kitajima K, Inoue S, Khoo KH, Morris HR, Dell A, Inoue Y. Occurrence and structural analysis of highly sulfated multiantennary N-linked glycan chains derived from a fertilization-associated carbohydrate-rich glycoprotein in unfertilized eggs of tribolodon hakonensis. Eur. J. Biochem. 1996;238:357–367. doi: 10.1111/j.1432-1033.1996.0357z.x. [DOI] [PubMed] [Google Scholar]
- 38.Lei M, Mechref Y, Novotny MV. Structural analysis of sulfated glycans by sequential double-permethylation using methyl iodide and deuteromethyl iodide. J Am. Soc. Mass Spectrom. 2009;20:1660–1671. doi: 10.1016/j.jasms.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 39.Domon B, Costello CE. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J. 1988;5:397–409. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.