

Abstract
Autophagy is a stress-induced cell survival program whereby cells under metabolic, proteotoxic, or other stress remove dysfunctional organelles and/or misfolded/polyubiquitylated proteins by shuttling them via specialized structures called autophagosomes to the lysosome for degradation. The end result is the release of free amino acids and metabolites for use in cell survival. For tumor cells, autophagy is a double-edged sword: autophagy genes are frequently mono-allelically deleted, silenced, or mutated in human tumors, resulting in an environment of increased oxidative stress that is conducive to DNA damage, genomic instability, and tumor progression. As such, autophagy is tumor suppressive. In contrast, it is important to note that although tumor cells have reduced levels of autophagy, they do not eliminate this pathway completely. Furthermore, the exposure of tumor cells to an environment of increased metabolic and other stresses renders them reliant on basal autophagy for survival. Therefore, autophagy inhibition is an active avenue for the identification of novel anti-cancer therapies. Not surprisingly, the field of autophagy and cancer has experienced an explosion of research in the past 10 years. This review covers the basic mechanisms of autophagy, discusses its role in tumor suppression and cancer therapy, and posits emerging questions for the future.
Keywords: autophagy, cancer, mTOR, Beclin-1, p53, oncogene, tumor suppressor, chloroquine
I. INTRODUCTION
Autophagy (from the Greek term for “self-eating”) is a catabolic process whereby cells degrade via the lysosome proteins and organelles in order to survive periods of stress, especially nutrient deprivation. There are three modes of autophagy that differ in the manner in which the “cargo” (that which will be degraded) is delivered to the lysosome. Microautophagy refers to the nonselective process whereby cytosolic proteins are sequestered by invagination of the lysosomal membrane. Chaperone-mediated autophagy is a selective process whereby proteins with defined consensus sequences are recognized by molecular chaperones, including Hsc70, and delivered to the lysosome. Finally, macroautophagy (the topic of this review, and hereafter referred to as autophagy) is the process whereby bulk proteins and organelles in the cytosol are delivered by autophagosomes to the lysosome for degradation. While it is generally regarded as nonselective, there can be some selectivity in autophagy; for example, mitochondria or the endoplasmic reticulum can be selectively degraded (“mitophagy” and “reticulophagy,” respectively), or misfolded and/or polyubiquitylated proteins can be selectively degraded, such as following proteotoxic stress. Autophagy is a critical mechanism for the adaptation of cells to stress, and it is induced by numerous stimuli, including nutrient deprivation, hypoxia, hormone stimulation, and DNA damage.1,2
The process of autophagy, and the identification of genes critical for this pathway, was originally identified in the yeast Saccharomyces cerevisiae in the early 1990s.3 Over 30 autophagy (Atg) genes have been described in yeast, and over 20 of these have been identified in mammalian cells.4 Because some of the yeast genes influential in autophagy were originally cloned in the pathway involved in vesicular protein sorting, some of these genes are denoted by the acronym Vps instead of Atg. Importantly, the molecular mechanism of autophagy is conserved from yeast to mammals, and the orthologs of the majority of the yeast Atg genes can be found in mammalian cells, with identical or similar function.
II. THE PROCESS OF AUTOPHAGY
Autophagy is a multi-step process that starts with the nucleation of a membrane called the phagophore (Fig. 1). The exact source of the phagophore membrane is not known, but there is evidence that it may be derived from the endoplasmic reticulum and trans-Golgi network5 as well as the mitochondria6,7; one possibility is that the derivation of the phagophore membrane depends upon the nature of the stress that induces autophagy. The phagophore expands and grows, selectively or nonselectively engulfing organelles and proteins during its expansion. At the end of this elongation step, a portion of the cytosol is sequestered into a double-membrane vesicle termed the autophagosome. The autophagosome then fuses with an endosome and/or the lysosome, thereby forming the autolysosome. In the autolysosome, lysosomal hydrolases degrade the protein constituents, resulting in free amino acids and macromolecules that are transported back into the cytosol for reuse.
FIGURE 1.
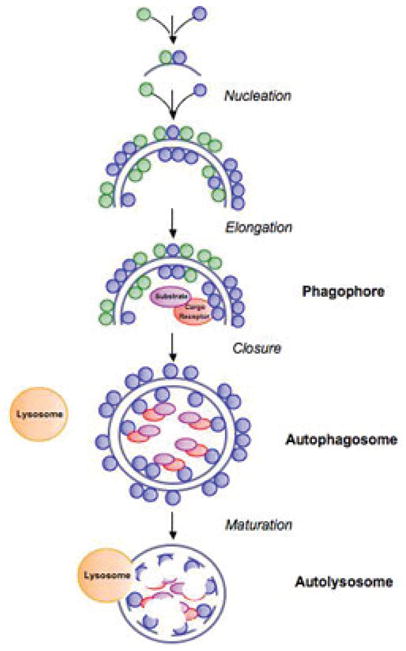
Schematic depiction of the steps of autophagy in mammalian cells. Mammalian autophagy is initiated by the formation of the phagophore (at the nucleation step). ULK1 and class III PtdIns3K complexes are required for this step. During elongation and expansion of the phagophore membrane, Atg12-Atg5 conjugates interact noncovalently with Atg16L, and this complex acts as an E3-like enzyme, determining the sites of Atg8/LC3 lipidation. Substrates are recruited to the inner surface of the growing phagophore by binding to the cargo receptor. At the end of the elongation step, a portion of the cytosol is sequestered into a double-membrane vesicle, termed the autophagosome. The autophagosome fuses with the lysosome, thereby forming the autolysosome, where the substrates are degraded by acid hydrolases.
There are at least four core subgroups of Atg complexes that play key roles in autophagy. The first is the Atg1/Unc-51-like kinase (ULK) complex, which is directly controlled by mTOR phosphorylation. The second is the class III phosphatidylinositol 3-kinase (Pt-dIns3k)/Vps34 complex. The third and fourth are two ubiquitin-like protein conjugation systems, involving the ubiquitin-like proteins Atg12 and Atg8/LC3, respectively. In addition to these four core subgroups, there are also two transmembrane proteins that deserve mention, Atg9 and VMP1.
The yeast serine/threonine kinase Atg1 is a master regulator of autophagy; Atg1 interacts with Atg13 and Atg17, and these are essential for Atg1 activity. The yeast TOR protein is a serine/threonine kinase that is evolutionarily conserved among species and plays a central role in various cellular events like cell cycle, cell growth, and autophagy.8 The yeast TOR protein phosphorylates Atg13, reducing its ability to bind Atg1 and thereby decreasing Atg1 activity.9,10 Inhibition of TOR causes dephosphorylation of Atg13, accumulation of Atg1-Atg13-Atg17 complexes, and activation of autophagy (Fig. 2A). In mammals, ULK1 and ULK2 (Unc-51-like kinases 1 and 2) are the homologues of yeast Atg1; these proteins also exist in complex with Atg13 (mAtg13), along with the scaffold protein FIP200 (an ortholog of yeast Atg17), and mTOR. In mammalian cells, there are two known mTOR complexes, mTORC1 and mTORC2.11 mTORC1 is the predominant form associated with autophagy. mTORC1 is quickly dissociated from the Atg13/FIP200/ULK1/2 complex during starvation.12–14 This dissociation is accomplished by several phosphorylation events, including phosphorylation of mAtg13 by ULK1, ULK2, and mTORC1; phosphorylation of FIP200 by ULK1 and ULK2; and phosphorylation of ULK1 and ULK2 by mTORC1. Overall, however, the regulation of autophagy by mTORC1 follows similar principles: a decrease in mTORC1 activity leads to dephosphorylation of ULK1, ULK2, and mAtg13; this leads to activation of ULK1 and ULK2, which then phosphorylate mAtg13 and FIP200.12,14 Therefore, while the events are seemingly more complicated in mammals, the end result in both yeast and mammals is the direct control of Atg1/ULK, the master regulator of autophagy, by the master sensor of nutritional stress, mTOR.
FIGURE 2.

Master molecular regulators of autophagy. (A) The mammalian complex ULK1/2-mAtg13-FIP200 is required for autophagy. mTORC1 acts as a negative regulator of autophagy by phosphorylating Atg13 and ULK1/2. During starvation, mTORC1 is released from this complex resulting in dephosphorylation of the components and activation of ULK1 and ULK2. (B) The Beclin-1/Vps34/Atg14L complex. This complex has three forms. The Atg14L (Atg14L–Beclin1–hVps34–p150) and UVRAG-Bif-1 (UVRAG–Beclin1–hVps34–p150) complexes induce autophagy. In contrast, the Rubicon complex (Rubicon–UVRAG–Beclin1–hVps34–p150) negatively regulates autophagy. Bcl-xl/Bcl-2 binds to Beclin-1, disrupting its association with hVps34, thereby inhibiting autophagy.
Nucleation and elongation of the pre-autophagosomal membrane is controlled by the class III PI3 kinase (PI3KIII) Vps34. Vps34 exists in a complex with Beclin-1 (Atg6) and p150 (a homolog of Vps15),15, 17 along with the protein UVRAG (ultraviolet irradiation resistance-associated gene, a homolog of Vps38) and Bif-1; the latter two proteins bind and enhance the activity of PI3KIII through their interaction with Beclin-1.16,18 Vps34 also interacts with Atg14, which directs the Vps34 complex to the pre-autophagosomal membrane.19 In vivo, the Vps34 complex phosphorylates phosphatidylinositol to form phosphatidylinositol-3-phosphate (PI3P). PI3P serves as a docking point for proteins necessary for the formation of the autophagic vacuole (Fig. 2B).
Elongation and completion of the autophagic vacuole is mediated by two ubiquitin-like conjugation pathways (Fig. 3). The first pathway consists of the Atg12-Atg5 conjugation system, which is formed by the action of the E1- and E2-like proteins Atg7 and Atg10, respectively.20–23 The Atg12-Atg5 conjugate then interacts noncovalently with a small coiled-coil protein, Atg16L (an ortholog of Atg16 in yeast); these oligomerize to form a large multimeric Atg12-Atg5-Atg16L complex. Atg16L directs this complex to the outer autophagosomal membrane,24 and in turn this oligomerized complex is in part responsible for creating the concave nature of the forming autophagosome membrane.
FIGURE 3.

Ubiquitin-like conjugation systems that play roles in the early steps of autophagy. Early steps in autophagosome formation are regulated by two ubiquitin-like conjugation systems. In both cases, a ubiquitin-like protein (Atg8 and Atg12) is conjugated to an E1-like enzyme (Atg7), and then to an E2-like enzyme (Atg3 and Atg10). These are then used to form phosphatidylethanolamine (PE) conjugates of Atg8 (LC3 II) as well as protein conjugates of Atg5/12/16.
In the second ubiquitin-like pathway, LC3 (the mammalian ortholog of Atg8) is lipidated upon conjugation with phosphatidyl-ethanolamine (PE).25 The carboxy-terminal residue of Atg8/LC3 is cleaved off by a cysteine protease, Atg4, generating the cytosolic LC3-I, which now will contain a glycine residue at the C-terminus.26 LC3-I then conjugates to PE by the formation of an amide bond between the amino group of PE and the C-terminal glycine of LC3-I. This reaction requires the E1 protein Atg7 and the E2 protein Atg3.25 Lipidation of Atg8/LC3 converts the soluble Atg8/LC3-I into the autophagosome associated form LC3-II. LC3-II recruits lipid molecules to expand the autophagosome membrane. When membrane elongation is completed, Atg8/LC3 is detached from PE via Atg4 and then released back to the cytosol.26
Recent studies suggest that these two ubiquitin-like systems regulate each other. For example, recruitment of Atg8/LC3 to the pre-autophagosomal membrane requires the Atg12-Atg5-Atg16 complex; in addition, the Atg12-Atg5 conjugate acts as an E3-like enzyme, determining the sites of Atg8/LC3 lipidation.27,28 Conversely, the Atg8/LC3 conjugation system seems to be required for Atg16 complex formation.29 The importance of this joint regulation is presently unclear, though it likely ensures that this self-eating survival program is tightly regulated.
There are two transmembrane proteins that are required for mammalian autophagy that aid in the function of the four complexes outlined above: mAtg9 (the mammalian homolog of Atg9 in yeast) and vacuole membrane protein 1 (VMP1). mAtg9 protein localizes to the trans-Golgi network and endosomes; during autophagy this protein localizes with GFP-LC3-positive autophagosomes.30 In yeast, the available data suggest that Atg9 may be required for the delivery of membranes to the autophagosome.31 In contrast, VMP1 is localized to the plasma membrane; during autophagy this protein co-localizes with LC3 and Beclin-1, and helps recruit components of class III PtdIns3k to the phagophore.32
A. Sequestration of “Cargo”
Autophagy possesses both selective and non-selective degradation processes. For example, autophagy plays a role in the selective degradation of protein aggregates and polyubiquitylated proteins.33,34 Autophagy also plays a role in the selective degradation of dysfunctional mitochondria, endoplasmic reticulum, and peroxisomes. During selective degradation, it is believed that LC3, which acts as a “receptor” at the phagophore, interacts with “adaptor” molecules on the target (e.g., mitochondria), promoting their selective delivery and degradation. The best-characterized autophagy receptor is p62SQSTM1, a multi-functional adaptor protein that contains an LC3-interacting region (LIR) as well as a ubiquitin-association (UBA) domain, which binds to polyubiquitin molecules.35 Mounting evidence suggests that p62SQSTM1, along with another autophagy receptor NBR1, serve as cargo receptors for selective clearance of misfolded/polyubiquitylated proteins and damaged organelles.35–40 The identification of such cargo receptors is ongoing, and it is highly likely that other similar receptors are yet to be identified.
B. Movement and Fusion of Autophagosomes
In yeast, successful fusion of the autophagosome with the lysosome relies on micro-tubules along with the proteins Ypt7p (the yeast homologue of Rab7),41 Vam3p (a syntaxin homologue),42 Sec18p (the yeast homologue of N-ethylmaleimide sensitive factor, NSF), and Vti1p (a SNARE protein).43 Similar roles for Rab7 and Vti1p in autophagy have been demonstrated in mammalian cells.44–46 The movement of autophagosomes in the cytosol to peri-nuclear regions, where fusion takes place, is mediated by the motor protein dynein.47
C. Autophagy Inducers
It is highly likely that the majority of cellular stresses induce autophagy. The best characterized inducer of autophagy is nutrient deprivation. In cultured mammalian cells, only minutes of nutrient depletion can induce autophagy, and the highest levels can be achieved when cells are cultured in the complete absence of nutrients and growth factors.48 As mentioned briefly, the key molecular regulator of starvation-induced autophagy is the serine-threonine kinase mTOR (mammalian target of rapamycin). Depletion of insulin, insulin-like growth factor, or other growth factors is sufficient to lead to inhibition of mTOR (specifically, mTORC1, or mTOR complex 1) and induce autophagy. A key enzyme in the signaling of nutrient deprivation is AMPK, which monitors the energy status of the cell via its ability to sense the AMP:ATP ratio. Upstream kinases—such as liver kinase B1 (LKB1), calcium/calmodulin kinase kinase-β, and TGF-activated kinase-1—can activate AMPK by phosphorylation.49 AMPK activation induces autophagy through mTORC1 inhibition, via phosphorylation of the tuberous sclerosis complex 2 (TSC2), and the regulatory associated protein of mTOR, Raptor.50
Although much attention has been given to autophagy regulation by mTOR, several signaling pathways seem to influence autophagy independently of mTOR. For example, Akt phosphorylates and activates another autophagy stimulator, the Forkhead box O (FoxO) transcription factor FoxO3, by an mTORC2-dependent but mTORC1-independent mechanism.51,52 Similarly, AMPK has been shown to induce autophagy in an mTORC1-independent manner, by phosphorylating and stabilizing p27 under conditions of metabolic stress.53 AMPK can directly facilitate autophagy by directly phosphorylating and activating ULK1.54,55 Sirtuins are a family of NAD-dependent deacetylases that can deacetylate the key autophagy proteins Atg5, Atg7, and LC3, as well as the transcription factor Forkhead box O3a (FOXO3a).56 Following its deacetylation, FOXO3a translocates into the nucleus and upregulates key autophagy genes, including ULK2, Beclin 1, VPS34, BNIP3, ATG12, ATG4B, and LC3, thereby leading to autophagy induction.52
A second stress that induces autophagy is hypoxia. If oxygen concentrations fall below 5%, the HIF1 (hypoxia-inducible factor) transcriptional regulator is activated,57,58 and this protein transactivates two key autophagy inducers, BNIP3 and BNIP3L (NIX).59 These proteins then function to activate the key autophagy complex containing the class III PI3K Vps34. Hypoxia also increases the transcription of the essential autophagy genes LC3 and Atg5 through the transcription factors ATF4 and CHOP, respectively.60 Notably, there is strong evidence for co-localization of regions of hypoxia with autophagy in vivo.61
III. AUTOPHAGY IN NORMAL DEVELOPMENT
Recent studies indicate that transient activation of autophagy occurs directly after birth in several tissues of the mouse.62 A critical role for autophagy in neonatal survival was shown by inactivation of the autophagy-related genes Atg5 and Atg7 in mice. Atg5 and Atg7 knockout mice were normal at birth (although they had lower body weight than controls), but they could not survive the neonatal starvation period and died within 1 day after birth. Notably, under nonsuckling conditions these mice died much earlier than wild-type mice (after 10–13 h compared to 20–22 h after birth). The concentration of amino acids in the plasma of knockout mice 10 h after birth was approximately 20% lower, compared to controls. In addition, there was no autophagosome formation in these mice.62 The combined data suggest that autophagy is critical for survival during the neonatal starvation period when the transplacental nutrient supply is suddenly ended.
Mice deficient for either Atg5 or Atg7 also accumulate polyubiquitylated protein aggregates and abnormal mitochondria, indicating that this process is required for protein and organelle quality control. Interestingly, these mice also undergo neuronal degeneration with age,63,64 suggesting that autophagy plays a role in normal aging and degeneration. Along these lines, an absence of autophagy has been hypothesized to underlie Huntington and Parkinson’s diseases, due to the accumulation of misfolded proteins.65 The liver is an organ that extensively undergoes autophagy, and the livers of conditional Atg7-deficient mice display several abnormalities, including accumulation of peroxisomes and deformed mitochondria, along with hepatomegaly.66 In tissues of the central nervous system of Atg7-deficient mice, a loss of cerebral and cerebellar cortical neurons takes place and ubiquitin aggregates accumulate in axons, which leads to neurodegeneration and eventual death.63,66 The combined data support the relevance of autophagy in nutrient deprivation, protein quality control, and aging.
IV. MONITORING AUTOPHAGY IN THE CELL
Autophagy was first detected by transmission electron microscopy. The focal degradation of cytoplasmic areas of the cell, sequestered by the phagophore (a specialized type of smooth, ribosome-free double membrane), is one of the hallmarks of autophagy. In general, the use of electron microscopy is considered an important method for the qualitative and quantitative analysis of autophagic structures, such as the phagophore, autophagosome, and the autolysosome.67,68
There are several methods that can be used to monitor autophagy in cells and organisms; some of these are depicted in Fig. 4. For example, the lipidated form of LC3 (LC3 II) is predominantly associated with autophagic organelles,25,69 and can be monitored by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting with LC3 antisera, where LC3 II is the faster mobility form.70–72 One caveat to this approach is that LC3 II is both induced and degraded during autophagy. Therefore, Western blot detection of LC3 II must be accompanied by efforts to halt autophagic flux in the cell, such as by the use of Bafilomycin A1, hydroxychloroquine, or combinations of pepstatin A and E64d. These latter treatments inhibit the Na+/H+ pump at the lysosome, increase lysosomal pH, and inhibit the acidic lysosomal protease, respectively; all prevent the degradation of LC3 II and hence halt autophagic flux.73
FIGURE 4.
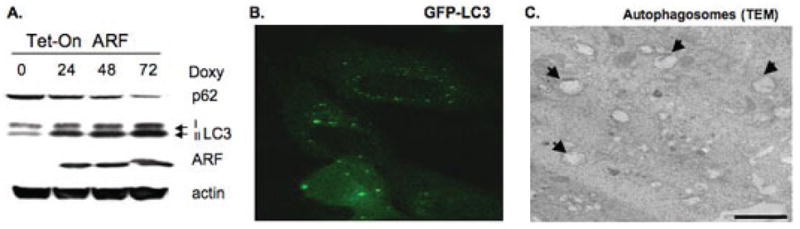
Assays for autophagy. (A) Western blot analysis of tetracycline-inducible p19ARF cells treated with doxycycline for 24 h. The upper band represents p62, which is degraded following ARF-induced autophagy. The bottom LC3 band (LC3 II) represents the form that accumulates during autophagy. (B) Tetracycline-inducible p19ARF cells were transiently transfected with GFP-LC3 for 24 h and treated with doxycycline for 24 h, followed by confocal microscopy to detect GFP-labeled LC3, which is localized to vacuoles (autophagosomes). (C) Transmission electron microscopy of serum-starved H1299 cells. Arrows depict double membrane autophagosomes. The scale bar is 500 nm.
The expression of a GFP-LC3 fusion protein, and its existence in autophagosomes, has also been used extensively to monitor autophagy in cultured cells.69 It has recently been determined that GFP-LC3 is sensitive to acidic pH, which impedes fluorescence after autolysosome formation. For this reason, a new tandem fluorescent-tagged LC3 (tfLC3) has been developed. In this vector, LC3 is expressed tandemly fused with both GFP and RFP (red fluorescent protein); the latter is resistant to lysosomal proteolytic degradation. Co-localization of both GFP and RFP fluorescence in a cell indicates an autophagosome that has not yet fused with a lysosome, whereas vesicle-associated RFP signals denote fused autolysosomes.74,75
p62SQSTM1 (sequestosome 1, and herein referred to as p62) is an adaptor molecule involved in activating autophagy that interacts with polyubiquitinated protein aggregates and targets them to autophagosomes. Because p62 is degraded along with cargo in the autolysosome, Western blotting for steady-state levels of this protein is often used as a reliable indicator of autophagy, and there is no need for cessation of autophagic flux when monitoring p62. Recent studies show that inhibition of autophagy correlates with increased levels of p62.35,76
Measurement of the degradation of long-lived proteins is a reliable measure of autophagic flux. In this approach, intracellular proteins are pulse-labeled for over 24 h with a radiolabeled amino acid, such as [14 C]-leucine or [14 C]-valine. Following a long (>24 h) chase period in unlabeled excess amino acid, the time-dependent release of acid-soluble radioactivity is measured by liquid scintillation counting.77 While this assay is considered a “gold-standard” for the measurement of autophagy, the extensive time courses involved and the use of radioactivity have limited its use. Fortunately, assays that avoid the use of radioactivity have been developed, based upon the starvation-dependent accumulation of betaine homocysteine methyltransferase (BHMT).78–81
V. AUTOPHAGY AND CANCER
That autophagy plays a significant role in cancer initiation and progression is best evidenced by the number of proteins with roles in autophagy that also play key roles in cancer (see Fig. 5). Some of these key players in both cancer and autophagy are denoted here.
FIGURE 5.

Signaling pathways that regulate mammalian autophagy. Autophagy is regulated by a complex signaling network of various stimulatory (arrowheads) and inhibitory (bars) inputs. Stimulation of the class I PtdIns3K complex and small GTPase Ras (via activation of growth factor/insulin receptors) leads to activation of the PtdIns3K-PKB-TOR pathway. PKB phosphorylates and inhibits the tuberous sclerosis complex TSC1/TSC2, which activates mTORC1 through Rheb-GTPase stabilization, causing inhibition of autophagy. Amino acids inhibit the Raf-1-MEK1/2-ERK1/2 signaling pathway, acting as a negative regulator of autophagy. Metabolic stress, such as high AMP/ATP ratios, leads to phosphorylation and activation of AMP-activated protein kinase (AMPK) by LKB1. AMPK then activates TSC1/TSC2, leading to mTORC1 inactivation and autophagy induction. Following genotoxic stress, nuclear p53 causes autophagy induction via transactivation of AMPK subunits or upregulation of the lysosomal autophagy protein DRAM. Cytosolic p53 has an inhibitory effect on autophagy. JNK1, DAPK, BAD, and p19ARF all function to disrupt the association between BCL-2/Bcl-xl and Beclin-1, facilitating Beclin-1-class III PtdIns3K complex formation and thereby promoting autophagy.
A. Beclin-1
The connection between cancer development and autophagy was not firmly established until the discovery and characterization of Beclin-1.82–84 Beclin-1 was first identified as a Bcl-2–binding protein that is structurally similar to Atg6 in yeast and evolutionarily conserved among various species.84–88 Beclin-1 induces autophagy by binding and activating Vps34 through an evolutionarily conserved domain between amino acids 244 and 337.88
Several lines of evidence indicate that Beclin-1 is a bona fide tumor suppressor. The Beclin-1 locus frequently undergoes monoallelic deletion in various human malignancies, including brain tumors and ovarian, prostate, and breast cancers.89,90 Overexpression of Beclin-1 in human breast cancer cell line MCF7 inhibits its proliferation in vitro, as well as tumorigenesis in a mouse xenograft model.82 Moreover, in a genetically engineered mouse model with heterozygous disruption of Beclin-1, spontaneous development of lymphoma, breast, and lung cancers, as well as hepatocellular carcinoma, occur.87,91 The combined data firmly support the identification of Beclin-1 as a haplo-insufficient tumor suppressor (that is, that deletion of only one copy of Beclin-1 is sufficient to drive tumorigenesis). That the role of Beclin-1 in tumor suppression was related to its role in autophagy was first suggested when it was discovered that the same domain required for Beclin-1 to bind Vps34 and induce autophagy was also necessary for its tumor-suppressive activity.88
Several regulators of Beclin-1 have been identified to date. The two major positive regulators of Beclin-1 are UVRAG and Bax-interacting factor-1 (Bif-1). UVRAG is required for Beclin-1–mediated autophagy and enhances Beclin-1 activity by promoting its binding with Vps34.92,93 Monoallelic deletion or mutation of UVRAG has been reported in various human cancers, including colon, gastric, and breast cancers.94–96 Bif-1 also acts as a positive regulator of Beclin-1.18 Similar to UVRAG, Bif-1 enhances the binding between Beclin-1 and Vps34, resulting in increased autophagy. Expression of Bif-1 is markedly reduced in human malignancies, including colon cancer, prostate cancer, urinary bladder cancer, and gastric carcinoma,97–100 and homozygous deletion of Bif-1 was reported in some cases of mantle cell lymphoma.101 Bif-1 knockout mice develop spontaneous tumors at a significantly higher rate compared to wild-type mice, indicating that Bif-1 is also likely to be a tumor suppressor gene. Despite the compelling nature of the evidence implicating UVRAG and Bif-1 as tumor suppressor genes, that they function in tumor suppression through autophagy has yet to be stringently proven. For example, some groups have suggested that Bif-1 suppresses tumorigenesis through its interaction with pro-apoptotic protein Bax.102
Two new Beclin-1 regulators have recently been identified. Rubicon and ATG14L function antagonistically upon interaction with Beclin-1.103,104 Specifically, in a mutually exclusive manner, ATG14L induces Vps34 kinase activity and autophagy independent of UVRAG, whereas Rubicon inhibits autophagy through mediating impaired maturation of the autophagosome. Whether these regulators are altered in expression in cancer has yet to be determined.
B. Bcl2 Family
Perhaps the most important negative regulator of Beclin-1 is Bcl-2 (B-cell CLL/lymphoma-2).105,106 Originally cloned as an anti-apoptotic gene, the role of Bcl-2 in autophagy was not realized until the identification of Beclin-1 as a Bcl-2 interacting protein. Bcl-2 inhibits Beclin-1–mediated autophagy by constitutively binding with Beclin-1 and blocking the interaction between Beclin-1 and Vps34.107–109 For autophagy to be initiated, two possible mechanisms have been described for the release of Beclin-1 from Bcl-2. First, phosphorylation of either Beclin-1 or Bcl-2 has been shown to weaken their association and result in induced autophagy. This can occur either in the N-terminal loop of Bcl-2 or the BH3 domain of Beclin-1, mediated by c-Jun N-terminal protein kinase 1 (JNK1) or death-associated protein kinase (DAPK), respectively.110–112 Alternatively, BH3-only proteins are hypothesized to compete with Beclin-1 for the interaction with Bcl-2, and to induce the release Beclin-1 from the inhibitory effect of Bcl-2. This latter phenomenon has been reported to occur with Bid, Bad, a BH3-mimetic compound, and the Caenorhabditis elegans BH3-only protein EGL-1.109,113,114
With the accumulation of information implicating Bcl-2 in autophagy, it was hypothesized and subsequently found that other Bcl-2 family members also play roles in this process. The Bcl-2 family includes proteins with Bcl-2 homology (BH) domains; anti-apoptotic Bcl-2 family members have four BH domains, and these include Bcl-2 and Bcl-xL. Pro-apoptotic Bcl-2 members either contain three BH domains, such as Bax and Bak, or they possess only the BH3 domain; these include Bad and Noxa. Recently, it has been reported that other anti-apoptotic Bcl-2 family members—like Bcl-xL, Mcl-1, Bcl-w, and viral Bcl-2 homologs encoded by KSHV and γHV68—also suppress autophagy.108,109,113–115 In addition, pro-apoptotic BH3-only Bcl-2 family members—such as Bad, Bik, Noxa, Puma, Bi-mEL, and BNIP3—all exhibit autophagy-inducing abilities.113,116–119 Therefore, the Bcl-2 family serves as a central regulator of both the autophagy and apoptosis pathways.
C. mTOR
The research on molecular pathways regulating autophagy was brought to the forefront with the identification of the role of the target of rapamycin (TOR; or mTOR in mammalian cells).120–122 The mTOR complex mTORC1 is best known for its role in regulating protein synthesis through two of its substrates, 4E-BP1 and p70S6K. Phosphorylation of 4E-BP1 prevents its inhibitory activity on RNA cap-binding protein eIF4F, leading to the induction of cap-dependent mRNA translation.123 Conversely, p70S6K is activated after being phosphorylated. Activation of p70S6K results in increased protein synthesis through induced expression of proteins involved in the translational apparatus, including elongation factors and ribosomal proteins.124,125 Moreover, activated p70S6K phosphorylates and inhibits eEF-2 kinase, which blocks elongation by phosphorylating eEF-2. Therefore, inhibition of eEF-2 kinase removes the block on elongation and promotes protein synthesis.126
Three cancer-relevant upstream signaling pathways have been shown to connect external stimuli to mTOR regulation: these are the PI3K-Akt pathway, the ERK-RSK-DAPK pathway, and the AMPK pathway. All of these pathways regulate mTOR activity through the tuberous sclerosis complex (TSC) 1/TSC2 complex, albeit in different ways. For example, the TSC1/TSC2 complex inhibits mTOR functions by inactivating Rheb; Rheb binds and activates mTOR only when the former is in its GTP-bound form.127 The phosphoinositide-3-kinase (PI3K)-Akt pathway activates mTOR by Akt-mediated phosphorylation of TSC2, followed by disassembly and inhibition of the TSC1/TSC2 complex.128,129 The extracellular signal-related kinase (ERK) stimulates mTOR activation by either directly phosphorylating TSC2 or by inducing TSC2 phosphorylation via its two downstream effectors, ribosomal S6 kinase (RSK) and DAPK.130–132 Finally, AMP-activated protein kinase (AMPK) phosphorylates TSC2 and stabilizes the TSC1/TSC2 complex, thereby inhibiting Rheb-mediated activation of mTOR.133 AMPK can also regulate mTOR function in a TSC-independent manner by directly phosphorylating Raptor, an essential component of mTORC1, to inhibit mTOR activity.134 Notably, TSC1 and 2, PI3K, and Akt are all subject to mutation in human tumors.
Our understanding of the role of mTOR in autophagy is still emerging. For example, it has been unclear why p70S6K, one of the key effectors of mTORC1 signaling, is actually required for autophagy.135,136 One potential explanation for this is that p70S6K is believed to function in a negative feedback loop, by phosphorylating and inhibiting the insulin receptor and PI3K.137–140 It has been suggested that this negative feedback pathway might enable cells to mount a rapid autophagic response upon metabolic stress, followed by a gradual loss of p70S6K activity; this gradual loss would be predicted to prevent cell death induced by excessive autophagy.141,142
D. PI3K
PI3K is heavily involved in autophagy regulation, both positively and negatively. In initial studies, rat hepatocytes were treated with the PI3K inhibitors wortmannin or LY294002, with the expectation that inhibition of PI3K-mTOR–mediated p70S6K phosphorylation would induce autophagy. Somewhat surprisingly, both p70S6K phosphorylation and autophagy were blocked by these inhibitors.143 This unexpected finding prompted the subsequent identification of distinct classes of PI3K with roles in autophagy139; of these, class I PI3K (PI3KI) and class III PI3K (PI3KIII) have negative and positive roles, respectively.144
The effects of PI3KI and PI3KIII on autophagy depend on the products of each kinase. The product of PI3KI, phosphatidylinositol (3,4,5)-trisphosphate (PIP3), has inhibitory effects on autophagy mainly through the regulation of the Akt-mTOR pathway, as described above. In contrast, the product of PI3KIII is phosphatidylinositol 3-phosphate (PI3P), and this molecular is essential for autophagy inhibition. In autophagy, PI3P is believed to serve as the platform for autophagosome biogenesis. Consistent with the distinctive functions between PI3KI and PI3KIII in autophagy regulation, PTEN, an exclusive PI3KI inhibitor, only functions positively in autophagy.145
E. p53
Since its discovery, the tumor suppressor gene p53 has become the most well-studied cancer-related gene due to the fact that it is mutated or deleted in over 50% of human malignancies. In response to various stresses like DNA damage, oxidative stress, and oncogene activation, p53 acts as the guardian of tumorigenesis by inducing cell cycle arrest or apoptosis. Several recent studies have added autophagy to the impressive résumé of molecular events regulated by p53. Interestingly, p53 has been described as both a positive and a negative regulator of autophagy, depending upon the cell type and the stresses that cells are experiencing.
Upon treatment of mouse embryo fibroblasts with the DNA damaging agent etoposide, p53 mediates the activation of AMPK and thus the inhibition of mTOR and induction of autophagy.146 In addition, activation of p53 by genotoxic stress causes the trans-activation of two p53-target genes, Sestrin 1 and 2, both of which are negative regulators of mTOR activity.147,148 Another p53 target gene, DRAM (damage-regulated autophagy modulator), is a lysosomal membrane protein that is also induced by p53 to facilitate autophagy upon genotoxic stress.149
Seemingly contradictory to the aforementioned autophagy-inducing activity of p53, this protein has also been reported to inhibit autophagy. Specifically, inhibition of p53 by chemical inhibitors, siRNA, or genetic deletion was found to increase the basal level of autophagy. In this case, evidence was found that cytosolic, and not nuclear, p53 played this negative regulatory role.150,151 This phenomenon was also observed with certain oncogenic p53 mutants that are preferentially localized in the cytoplasm, suggesting a connection between p53-mediated inhibition of autophagy and tumorigenesis.152 Alternatively, because the p53 tumor suppressor negatively regulates the ARF tumor suppressor, it has also been suggested that silencing or inhibiting p53 induces autophagy because this leads to increased expression of ARF.153
F. ARF
The ARF tumor suppressor (p14ARF in humans, p19ARF in mice) plays a positive role in the induction of autophagy. This was first found with studies of a small-molecular-weight version of ARF (termed “smARF”), which was reported to traffic to mitochondria and induce autophagy.154 Subsequently, other groups reported that full-length ARF was likewise capable of trafficking to mitochondria and inducing autophagy.155–157 The mechanism for ARF-induced autophagy is by virtue of its ability to directly interact with Bcl-xl, and to limit the ability of Bcl-xl to bind and inhibit Beclin-1.156 Notably, and consistent with a role for autophagy in survival, tumors with ARF expressed at high levels were found to survive nutrient depletion in a superior manner, and ARF levels were found, in p53-null tumors, to confer increased transformed properties to tumor cells in vivo.153
G. Other Regulators of Autophagy With Roles in Cancer
Overexpression of DAPK, a serine/threonine kinase, has been shown to induce the formation of autophagic vesicles.158 DAPK has also been identified as a positive autophagy mediator in C. elegans.159 Mechanistically, DAPK is believed to mediate the induction of autophagy partly through releasing Beclin-1 from the inhibitory effects of Bcl-2.112 Interestingly, DAPK has also been shown to induce mTOR signaling by phosphorylating TSC2, which would be predicted to lead to inhibition of autophagy.132 Further studies will be required to understand how DAPK balances these two opposing effects in autophagy regulation.
The first core component of the autophagy machinery that has been identified as a tumor suppressor gene is Atg4C.160 Atg4C is the cysteine protease that mediates the formation of the autophagosome; mice with homozygous deletion of the Atg4C gene are more vulnerable to chemical-induced fibrosarcomas, along with reduced starvation-induced autophagy, providing a connection between Atg4C’s abilities to induce autophagy and to suppress tumorigenesis.161
VI. AUTOPHAGY IS TUMOR SUPPRESSIVE
That autophagy plays a key role in cancer is not disputed. There are, however, some controversies regarding whether autophagy is tumor suppressive or tumor promoting. The difference seems to be in the stage of cancer: autophagy clearly suppresses the initiation and development of tumors. However, it is also a key survival pathway in response to stress, and many established tumors require autophagy in order to survive. Evidence that autophagy is tumor suppressive, and that it can promote the survival of established tumors, is presented here.
Even before the association between cancer and autophagy was made, it was noted that proteolysis, one of autophagy’s hallmarks, is reduced in transformed cells.162 Autophagy was later found to be reduced in cancer tissues compared with their normal counterparts, suggesting that autophagy is tumor suppressive.163 Later, with the identification of Beclin-1 as both a autophagy mediator and tumor suppressor,82 the premise that autophagy is suppressive to tumor development became solidified. Since then, a number of oncogenes/oncogenic pathways and tumor suppressors have been found to play critical roles in the regulation of autophagy (Table 1). Consistent with the hypothesis that autophagy is tumor suppressive, most of the oncogenes on this list are potent inhibitors of autophagy, whereas most of the tumor suppressors positively regulate this process. In support of this premise, many core autophagy genes, including Beclin-1, UVRAG, and Bif-1, can be found mutated in human tumors.16,18,89,91 Moreover, a recent study identified a high incidence of frameshift mutations in Atg2B, Atg5, Atg9B, and Atg12 in human gastric and colon cancer samples with microsatellite instability.164 The combined data firmly support the role of autophagy in tumor suppression. To date, at least three mechanisms have been described to explain the role of autophagy in tumor suppression: 1) inhibition of necrosis-mediated inflammation, 2) maintenance of genome integrity, and 3) autophagy-mediated cell death and senescence.
TABLE 1.
Oncogenes and Tumor Suppressor Genes That Impact Autophagy
| Genes | Oncogenic or tumor suppressive | Autophagy inducer or inhibitor | Reference |
|---|---|---|---|
| Beclin 1 | Tumor suppressive | Inducer | 82–84, 87–91 |
| UVRAG | Tumor suppressive | Inducer | 92–96 |
| Bif-1 | Tumor suppressive | Inducer | 18, 97–102 |
| BH3-only Bcl-2 family | Tumor suppressive | Inducer | 109,113,114,116–119 |
| DRAM | Tumor suppressive | Inducer | 149 |
| LKB1 | Tumor suppressive | Inducer | 49,133 |
| PTEN | Tumor suppressive | Inducer | 145 |
| TSC1/TSC2 | Tumor suppressive | Inducer | 127–129 |
| ATG4C | Tumor suppressive | Inducer | 160,161 |
| ARF | Tumor suppressive | Inducer | 153–157 |
| P53 | Tumor suppressive | Inducer/inhibitor | 146–152 |
| DAPK | Tumor suppressive | Inducer/inhibitor | 112,132,158,159 |
| Antiapoptotic Bcl-2 family | Oncogenic | Inhibitor | 108,109,113–115 |
| mTORC1 | Oncogenic | Inhibitor | 8–14,120–126,135 |
| PI3KI | Oncogenic | Inhibitor | 128,129,139,143,144 |
| Akt | Oncogenic | Inhibitor | 128,129 |
| ERK | Oncogenic | Inhibitor | 130–132 |
| Bcl-2 | Oncogenic | Inhibitor | 105–109 |
A. Inhibition of Necrosis-Mediated Inflammation
Upon oncogene activation, a highly inflammatory microenvironment is often created that favors tumor development because of elevated oxidative stress.165 Similarly, inhibition or inactivation of autophagy in apoptosis-deficient cancer cells was shown to induce an inflammatory response due to enhanced necrotic cell death along with increased secretion of pro-inflammatory factors, such as high mobility group 1 (HMGB1) protein; this increased inflammatory response led to increased tumor growth.166 This hypothesis is consistent with the findings that autophagy deficiency is associated with inflammation-related disease. For example, Atg26L1 deficiency is a risk factor for Crohn’s disease, as is Atg5 deficiency.167 Moreover, autophagy is important for the disposal of aggregated mutant alpha-1-antitrypsin, which causes chronic inflammation and diseases in the lungs and the liver due to its aggregation in the endoplasmic reticulum of hepatocytes.168,169 This is also consistent with the finding that the loss of Beclin-1 results in failure of autophagy-mediated protein quality control and cancer promotion mediated by accumulation of p62.61
B. Maintenance of Genome Integrity
The heterozygous loss of Beclin-1 or loss of Atg7 in the mouse liver causes aberrant accumulation of p62 and other polyubiquitylated protein aggregates.35 Similarly, in mice deficient for Atg5 or Atg7 in the brain, misfolded and aggregated proteins, along with dysfunctional mitochondria, accumulate.63,64,170 White et al. have found that, prior to the onset of tumorigenesis, a major event accompanying aberrant accumulation of p62 and impaired organelle disposal in autophagy-deficient cells is DNA damage and chromosomal instability, indicated by DNA double-strand breaks, centrosome abnormalities, and/or increased DNA content.166,171,172 The link between the failed elimination of aggregated/misfolded proteins and dysfunctional mitochondria with the loss of genomic stability has been suggested to be increased oxidative stress due to increased reactive oxygen species (ROS). In support of this premise, in Beclin-1–deficient tumor-prone cells, p62 accumulation leads to the production of high levels of ROS and DNA damage, which can be reversed by knocking down p62 or introducing ROS scavengers.61
C. Autophagy and Oncogene-Induced Senescence
Senescence is a potent tumor-suppressive mechanism that forces oncogene-expressing cells to permanently exit the cell cycle. Autophagy is activated during oncogene-induced senescence,173 and a subset of autophagy-related genes are upregulated as well. Moreover, inhibition of autophagy delays the senescence phenotype.173 The detailed mechanism of autophagy-mediated senescence is still unclear, and its significance to tumor suppression remains to be determined. Notably, autophagy-mediated cellular senescence has also been described in biliary epithelial cells and in a plant model organism, Arabidopsis thaliana.174,175
D. Autophagy-Mediated Cell Death
There are some researchers who contend that autophagy might suppress tumorigenesis by inducing cell death; in some instances this is mediated by the accumulation of ROS. This cell death can be blocked by knocking down key autophagy proteins like Atg7 or Atg8, and has been shown to be caspase independent.176 In vivo evidence connecting autophagy and cell death can be traced back to reports of the formation of autophagic vacuoles during the destruction of larval tissues in early Drosophila salivary gland development.177,178 This connection was confirmed recently, and an engulfment receptor, Draper, was identified as an important regulator to steer autophagy toward cell destruction instead of survival.179,180 Interestingly, autophagy-mediated cell death has also been suggested to be instrumental in the progression of neurodegenerative diseases like Alzheimer’s and Parkinson’s disease.181,182 It should be noted, however, that there is considerable controversy with the premise that autophagy induces cell death, and to date there is no concrete evidence to suggest that autophagy induces, as opposed to accompanies, cell death. Therefore, whether autophagy inhibits tumorigenesis by inducing programmed cell death remains a highly debated subject.64,183
E. Other Mechanisms for Tumor Suppression by Autophagy
Other mechanisms for autophagy-mediated tumor suppression, including autophagy-regulated immunosurveillance and autophagy-inhibited angiogenesis, have also been suggested. It was known early on that autophagy can be induced by immune factors like interferon gamma (IFN-γ), and autophagy-mediated innate immunity is important for protection against infection of viral and bacterial pathogens.83,184–187 It was only recently that autophagy was found to also play an important role in modulating adaptive immunity, which has more implications in preventing tumorigenesis by increasing tumor antigen presentation. Autophagy has been shown to facilitate both MHC class I and class II antigen presentation.188,189 More importantly, a recent study showed that autophagy is essential for cross-presentation of melanoma-cell antigens by dendritic cells in vitro and in vivo, with autophagosome as the antigen carrier.190 This is the first direct evidence supporting the potential of autophagy to induce immunosurveillance against tumor cells. Interestingly, autophagy was also found to enhance MHC class II presentation of Epstein-Barr virus antigen-1 (EBNA1), an essential protein for the latent infection of Epstein-Barr virus (EBV), a prominent oncogenic virus.191 This finding presents an unconventional way for autophagy to suppress tumorigenesis by controlling the infection of tumor-inducing viruses.
It has also been proposed that autophagy can inhibit cancer development by blocking angiogenesis. Neuropilin 1, a positive regulator of vascular endothelial growth factor (VEGF) signaling, is degraded by autophagy under metabolic stress.192 Inhibition of angiogenesis and tumor growth was also observed in vitro and in vivo with the treatment of mTOR inhibitors, which are known to induce autophagy.193,194
VII. AUTOPHAGY CAN PROMOTE TUMORIGENESIS
Whereas accumulated data suggest that autophagy can inhibit tumorigenesis, there are also data indicating that it can promote cellular transformation and that low levels of autophagy may be necessary for tumor cell survival. Recently, it was found that autophagy promotes Ras-induced transformation. Specifically, autophagy-deficient mouse embryo fibroblasts were found to display decreased soft agar colony formation in response to transformation with Ha-Ras (V12); similar findings were made in immortalized mouse kidney cells.195,196 Intriguingly, autophagy deficiency did not affect the proliferation of nontransformed cells, suggesting that autophagy may possess a unique role in facilitating the proliferation of Ras-transformed cells. These researchers found that autophagy enhanced the glycolytic capacity of Ras-transformed cells195; similarly, other groups showed that autophagy was necessary for efficient oxygen consumption and metabolism in cancer cells.196 In summary, basal levels of autophagy were absolutely required in Ras-transformed cells for efficient metabolism and survival. Therefore, while reducing the levels of autophagy in a nontransformed cell may make the cell conducive for transformation, basal levels of autophagy seem to be required for the proliferation and survival of cancer cells.
VIII. AUTOPHAGY INHIBITION LEADS TO CELL DEATH
Autophagy is frequently down-regulated in many tumors, primarily due to the fact that the PI3K-Akt-mTOR axis is constitutively activated in most tumors. This renders tumor cells with critically low levels of this pathway, and hence increased sensitivity to metabolic stress and/or chemical inhibitor of autophagy. Along these lines, the loss of autophagy promotes sensitivity to metabolic stress in apoptosis-impaired tumor cells.166,197 In addition, inhibition of autophagy is known to promote cell death. In the liver, the induction of cell death in response to autophagy inhibition was shown to rely on the accumulation of the cargo sequestration protein p62, as the silencing of p62 rescued the toxic effects of autophagy inhibition.35 When analyzed further, it was found that autophagy-deficient cells accumulate aggregates of p62; these aggregates bind and inactivate the protein Keap1, which normally targets the transcription factor Nrf2 for degradation. Inactivation of Keap1 frees the Nrf2 transcription factor to transactivate genes involved in the oxidative stress response. Notably, silencing of Nrf2 protected these autophagy-deficient cells from death.198 A similar study showed that aggregated p62 can inactivate the pro-inflammatory and pro-survival transcription factor nuclear factor-kappa B (NF-κB).61 Therefore, while autophagy is tumor suppressive, this pathway remains at low levels in human tumors, rendering these cells very sensitive to inhibitors of the autophagy pathway.
There are considerable data indicating that tumor cells use autophagy for survival in response to cytotoxic agents. Increases in autophagy have been observed after many anti-neoplastic treatments, including the DNA alkylating agent temozolomide,199 the selective estrogen receptor modulator tamoxifen,200 and radiation.201 Autophagy in these instances is considered a tumor protective mechanism, as it allows recycling of protein and damaged cellular components during the cancer treatment, ultimately yielding tumor survival. Indeed, inhibition of autophagy seems to be a viable treatment strategy for tumors that have escaped the cytotoxic effects of anti-apoptotic therapies.197,201,202 In addition, autophagy inhibitors could target the specific subpopulations of tumor cells that are most difficult to treat: the populations within the hypoxic and nutrient-scarce regions of the tumor that are particularly resistant to radiation and chemotherapeutic treatments and most likely to be associated with recurrence or metastasis. The best data implicating autophagy as a survival pathway for tumor cells, particularly in response to treatment with cytotoxic agents, are that inhibitors of autophagy frequently synergize with these agents.
A. Autophagy Inhibition for Cancer Therapy: Chloroquine
Chloroquine (CQ) and its derivative hydroxychloroquine impair lysosomal function by altering lysosomal pH; these drugs thereby inhibit the lysosome-dependent degradation of autophagosomes.203,204 Thompson et al. were the first to demonstrate that CQ inhibited tumor growth and prolonged the tumor refractory period in a B-cell lymphoma model in the mouse.205 The mechanism by which CQ inhibited the tumor growth was inhibition of autophagy, which was confirmed by observation of accumulating autophagosomes by electron microscopy (EM) and by genetic knockout of the essential autophagy gene, Atg5. Notably, combining CQ with the DNA alkylating agent cyclophosphamide significantly increased the rate of tumor regression and, more importantly, significantly delayed tumor recurrence.205 Coincidentally, CQ prophylaxis against malaria in Africa was found to result in a significant reduction in the number of Burkitt’s lymphoma cases.206
Inhibition of autophagy with CQ enhances the apoptotic activity of the histone deacetylase inhibitor, SAHA.207 The combination of autophagy inhibition with SAHA treatments was effective in murine B-cell lymphoma models and in primary cells of chronic myeloid leukemia (CML) patients that were refractory to imatinib. Importantly, the combination treatment of CQ and SAHA was selective for malignant cells even when p53 function was compromised. The ability of CQ to enhance the anti-tumor activity of SAHA was linked to generation of ROS, modulation of cathepsin D, and down-regulation of thio-redoxin, ultimately leading to ROS-mediated cell death.207
More recently, a phase III clinical trial of glioblastoma multiforme utilized hydroxychloroquine (HCQ) treatments in adjuvant settings, following surgery, chemotherapy, and radiation treatments. In a small cohort, patients receiving HCQ had a markedly better median survival compared to placebo-treated patients.208 Currently, there is a number of clinical trials that utilize CQ, either as a single or combination treatment for various malignancies (see Table 2).
TABLE 2.
Clinical Trials Utilizing Chloroquine (CQ) and Hydroxychloroquine (HCQ)
| Clinical trial.gov identifier | Condition | Drug | Phase | Sponsor |
|---|---|---|---|---|
| NCT00933803 | Non-small cell lung cancer | HCQ, paclitaxel, carboplatin, bevacizumab | I/II | University of Medicine and Dentistry, New Jersey (UMDNJ); Cancer Institute of New Jersey (CINJ) |
| NCT01266057 | Advanced cancer | HCQ, sirolimus, vorinostat | I | MD Anderson Cancer Center |
| NCT01273805 | Pancreatic cancer | HCQ | II | Dana Farber Cancer Institute, Brigham and Women’s Hospital, Massachusetts General Hospital |
| NCT01006369 | Colorectal cancer | HCQ, bevacizumab XELOX regimen, capecitabine Oxaliplatin | II | CINJ, National Cancer Institute (NCI) |
| NCT00809237 | Non-small cell lung cancer | HCQ, gefitinib | I/II | National University Hospital-Singapore, Massachusetts General Hospital, AstraZeneca |
| NCT00909831 | Unspecified; adult solid tumor, protocol specific | HCQ, temsirolimus | I | University of Pennsylvania, NCI |
| NCT00714181 | Unspecified; adult solid tumor, protocol specific | HCQ, temozolomide | I | University of Pennsylvania, NCI |
| NCT00786682 | Prostate cancer | HCQ, docetaxel | II | UMDNJ, NCI |
| NCT01206530 | Rectal, colon, metastasis, adenocarcinoma | HCQ, oxaliplatin, leucovorin, 5-FU, bevacizumab | I/II | Abramson Cancer Center of the University of Pennsylvania |
| NCT00726596 | Prostate cancer | HCQ | II | UMDNJ, NCI |
| NCT01013737 | Advanced solid tumors | HCQ, vorinostat | I | The University of Texas Health Sciences Center at San Antonio, Merck |
| NCT00977470 | Non-small cell lung cancer | HCQ, erlotinib | II | Massachusetts General Hospital, Dana Farber Cancer Institute, Beth Israel Deaconess Medical Center, Genentech |
| NCT00486603 | Brain and central nervous system tumors | HCQ, temozolomide | I/II | NCI |
| NCT01128296 | Pancreatic cancer | HCQ, gemcitabine | I | University of Pittsburgh, NIH |
| NCT00568880 | Multiple myeloma and plasma cell neoplasm | HCQ, bortezomib | I/II | University of Pennsylvania, NCI |
| NCT01144169 | Renal cell carcinoma | HCQ | I | NIH |
| NCT01292408 | Breast cancer | HCQ | II | Radboud University |
| NCT00962845 | Melanoma | HCQ | I | CINJ, NCI |
| NCT01227135 | Leukemia | HCQ, imatinib mesylate | II | University of Glasgow |
| NCT00771056 | B-cell chronic lymphocytic leukemia | HCQ | II | North Shore Long Island Jewish Health System |
| NCT00969306 | Small cell lung cancer | CQ, A-CQ 100 | I/II | Maastricht Radiation Oncology, Maastricht University Medical Center, NCI |
| NCT01023477 | Ductal carcinoma in situ (DCIS) | CQ tamoxifen | I/II | Inova Health Care Services George Mason University, Department of Defense, US Army Medical Research and Material Command |
B. Other Autophagy Inhibitors
Heat shock proteins (HSPs) function as molecular chaperones to regulate protein folding, intracellular protein trafficking, and re-folding of misfolded proteins. Elevated levels of these chaperones have been found in a number of cancers including gastric, breast, endometrial, ovarian, colon, lung, and prostate.209 While not considered bona fide oncoproteins, overexpression of Hsp70 contributes to oncogenesis in vitro210 and in vivo.211 Pharmacological inhibition of Hsp70 by the small molecule inhibitor 2-phenylethynesulfonamide (PES) is selectively cytotoxic to tumor cells, due to the fact that they overexpress Hsp70 and generally experience increased proteotoxic stress.212 PES-mediated toxicity is associated with protein aggregation and impairment of lysosomal function, resulting in inhibition of autophagy. Notably, like CQ, PES is effective at inhibiting lymphomagenesis in a preclinical mouse model of Burkitt’s lymphoma.212
IX. AUTOPHAGY INDUCERS IN CANCER THERAPY
While the general consensus is that autophagy inhibition is an effective strategy for cancer therapy, some drugs that are being used in the clinic induce autophagy. In most cases, however, it has not been proven that these drugs induce death via the autophagy pathway. Indeed, for many of these drugs it is hypothesized that combining them with autophagy inhibitors may improve efficacy. A list of chemotherapeutic agents that induce autophagy is provided in Table 3.
TABLE 3.
Clinical Trials Utilizing Drugs That Impact Autophagy”
| Agent | Target |
|---|---|
| Tamoxifen | Estrogen receptor |
| Temozolomide | DNA |
| γ-irradiation | DNA |
| SAHA | HDAC |
| Hyperthermia | Unknown |
| Arsenic trioxide | Mitochondria |
| Resveratrol | Multiple |
| Rapamycin | mTOR |
| Temsirolimus | mTOR |
| Everolimus | mTOR |
| 2-deoxy-D-glucose | Glycolysis |
| Endostatin | Angiogenesis |
| Sorafenib | TKI, VEGFR |
| Lonafarnib | Farnesyltransferase |
Rapamycin is a macrolide antibiotic that inhibits the mTOR complex by binding to FK-binding protein 12. Although initially approved by the US Food and Drug Administration as an immunosuppressant, rapamycin and its derivatives are currently being used in clinical trials for the treatment of various malignancies. Initially, the effectiveness of mTOR inhibitors was solely attributed to inhibition of the PI3K-Akt-mTOR axis; however, some reports suggest that the autophagy pathway may be involved in cell death induced by mTOR inhibitors. Specifically, the mTOR inhibitor RAD001 was found to sensitize prostate tumor cells to cell death by radiation, and this increase in cell death was associated with the induction of autophagy.213
Arsenic trioxide is an efficient agent for the treatment of newly diagnosed or relapsed acute promyelocytic leukemia (APL) and it can be used as a single agent with minimal myelotoxicity.214 Treatment of tumor cells with this drug leads to autophagy.215 In malignant glioma models, arsenic trioxide induced autophagy followed by non-apoptotic cell death.216,217 Recently, a study investigated the synergistic effects of ionizing radiation and arsenic trioxide in human osteosarcoma models. The combination of radiation with arsenic trioxide increased G2/M arrest with subsequent cell death.218 It should be noted that it is not clear if cell death by arsenic trioxide occurs via, or is accompanied by, autophagy.
2-Deoxy-glucose (2-DG) is an agent commonly used in cancer imaging that resembles glucose but cannot undergo glycolysis in the cell. Recently it was found that 2-DG induces autophagy in prostate cancer cell lines and in the cells of patients treated with this compound219,220; in tumor cell lines this autophagy was dependent on Beclin-1. Interestingly, rather than enhancing cell death by 2-DG, autophagy was found to function as a survival mechanism in treated cells. Therefore, the use of 2-DG for cancer therapy, possibly in combination with autophagy inhibitors, is currently being investigated.
X. SUMMARY
Autophagy is a critical pathway for the survival of tumor cells. However, whether some tumor types are more sensitive to autophagy inhibition remains to be determined. It will be especially important to determine if autophagy inhibitors are more effective when tumor cells suffer particular oncogenic insults; for example, if autophagy inhibition is cytotoxic for Ras-transformed tumor cells, but not Myc-transformed cells. The search for genes or single nucleotide polymorphisms that impact the sensitivity of tumor cells to autophagy inhibitors should start with the NCI 60 panel of cell lines; such data could provide researchers with clues as to which cancer(s) could and should be treated with autophagy inhibitors. In addition, it will be important to determine if autophagy inhibitors synergize with all, or some, conventional cytotoxic anti-cancer drugs.
The role of autophagy in tumor cell death remains unclear; specifically, at present it is not clear if autophagy accompanies, or induces, cell death by anti-cancer agents. And if tumor cells treated with autophagy inducers like mTOR inhibitors do not require autophagy for cell death, it will be important to determine what pathway they die from: necrosis, necroptosis, or some other pathway. Addressing the issue of whether cells die from autophagy will require the use of more sophisticated markers of autophagy than the mere presence of autophagosomes. Finally, there is dire need for new inhibitors of autophagy; the side effects associated with CQ may prohibit its use. Because autophagy pathways use several enzymes unique to this process, including ULK1, PI3K III, and Atg4, it seems likely that identifying inhibitors of these enzymes will prove productive for future anti-cancer therapies.
Acknowledgments
This work was supported by funding from the National Institutes of Health (R01 CA 139319 and R01 CA102184).
Footnotes
The authors declare that they have no financial interests in this work.
References
- 1.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–21. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333(1–2):169–74. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- 4.Orsi A, Polson HE, Tooze SA. Membrane trafficking events that partake in autophagy. Curr Opin Cell Biol. 2010;22(2):150–6. doi: 10.1016/j.ceb.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 5.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2(4):330–5. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141(4):656–67. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rambold AS, Lippincott-Schwartz J. Starved cells use mitochondria for autophagosome biogenesis. Cell Cycle. 2010;9(18):3633–4. doi: 10.4161/cc.9.18.13170. [DOI] [PubMed] [Google Scholar]
- 8.Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23(18):3151–71. doi: 10.1038/sj.onc.1207542. [DOI] [PubMed] [Google Scholar]
- 9.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150(6):1507–13. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott SV, Nice DC, 3rd, Nau JJ, Weisman LS, Kamada Y, Keizer-Gunnink I, Funakoshi T, Veenhuis M, Ohsumi Y, Klionsky DJ. Apg13p and Vac8p are part of a complex of phosphoproteins that are required for cytoplasm to vacuole targeting. J Biol Chem. 2000;275(33):25840–9. doi: 10.1074/jbc.M002813200. [DOI] [PubMed] [Google Scholar]
- 11.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10(3):457–68. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 12.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, Guan JL, Oshiro N, Mizushima N. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin-1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19(12):5360–72. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8(7):688–99. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 17.Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2008;105(49):19211–6. doi: 10.1073/pnas.0810452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mulé JJ, Pledger WJ, Wang HG. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9(10):1142–51. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152(3):519–30. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395(6700):395–8. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 21.Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem. 1998;273(51):33889–92. doi: 10.1074/jbc.273.51.33889. [DOI] [PubMed] [Google Scholar]
- 22.Shintani T, Mizushima N, Ogawa Y, Matsuura A, Noda T, Ohsumi Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J. 1999;18(19):5234–41. doi: 10.1093/emboj/18.19.5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanida I, Mizushima N, Kiyooka M, Ohsumi M, Ueno T, Ohsumi Y, Kominami E. Apg7p/Cvt2p: A novel protein-activating enzyme essential for autophagy. Mol Biol Cell. 1999;10(5):1367–79. doi: 10.1091/mbc.10.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116(Pt 9):1679–88. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- 25.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408(6811):488–92. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 26.Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, Ohsumi M, Takao T, Noda T, Ohsumi Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000;151(2):263–76. doi: 10.1083/jcb.151.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152(4):657–68. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282(52):37298–302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 29.Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, Sawada N, Yamada A, Mizushima N, Uchiyama Y, Kominami E, Tanaka K, Komatsu M. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell. 2008;19(11):4762–75. doi: 10.1091/mbc.E08-03-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, Hailey DW, Lippincott-Schwartz J, Tooze SA. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119(Pt 18):3888–900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 31.Reggiori F, Tucker KA, Stromhaug PE, Klionsky DJ. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell. 2004;6(1):79–90. doi: 10.1016/s1534-5807(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 32.Ropolo A, Grasso D, Pardo R, Sacchetti ML, Archange C, Lo Re A, Seux M, Nowak J, Gonzalez CD, Iovanna JL, Vaccaro MI. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J Biol Chem. 2007;282(51):37124–33. doi: 10.1074/jbc.M706956200. [DOI] [PubMed] [Google Scholar]
- 33.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–14. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280(48):40282–92. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 35.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–63. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 36.Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Øvervatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33(4):505–16. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 37.Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D, Rusten TE, Stenmark H, Brech A. Ref(2) P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol. 2008;180(6):1065–71. doi: 10.1083/jcb.200711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 39.Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A. 2008;105(52):20567–74. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12(9):836–41. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 41.Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, Noda T, Ohsumi Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol. 1999;147(2):435–46. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Darsow T, Rieder SE, Emr SD. A multispecificity syntaxin homologue, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J Cell Biol. 1997;138(3):517–29. doi: 10.1083/jcb.138.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ishihara N, Hamasaki M, Yokota S, Suzuki K, Kamada Y, Kihara A, Yoshimori T, Noda T, Ohsumi Y. Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol Biol Cell. 2001;12(11):3690–702. doi: 10.1091/mbc.12.11.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117(Pt 20):4837–48. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 45.Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117(Pt 13):2687–97. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- 46.Atlashkin V, Kreykenbohm V, Eskelinen EL, Wenzel D, Fayyazi A, Fischer von Mollard G. Deletion of the SNARE vti1b in mice results in the loss of a single SNARE partner, syntaxin 8. Mol Cell Biol. 2003;23(15):5198–207. doi: 10.1128/MCB.23.15.5198-5207.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jahreiss L, Menzies FM, Rubinsztein DC. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 2008;9(4):574–87. doi: 10.1111/j.1600-0854.2008.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25(3):1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, Ido Y. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab. 2010;298(4):E751–60. doi: 10.1152/ajpendo.00745.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neufeld TP. TOR-dependent control of autophagy: biting the hand that feeds. Curr Opin Cell Biol. 2010;22(2):157–68. doi: 10.1016/j.ceb.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 52.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6(6):458–71. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 53.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9(2):218–24. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 54.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105(9):3374–9. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40(2):294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22(2):177–80. doi: 10.1016/j.ceb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 59.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29(10):2570–81. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, Lambin P, van der Kogel AJ, Koritzinsky M, Wouters BG. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120(1):127–41. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–75. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 63.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 64.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 65.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443(7113):780–6. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 66.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169(3):425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eskelinen EL. Fine structure of the autophagosome. Methods Mol Biol. 2008;445:11–28. doi: 10.1007/978-1-59745-157-4_2. [DOI] [PubMed] [Google Scholar]
- 68.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clavé C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di Sano F, Dice JF, Difiglia M, Dinesh-Kumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Dröge W, Dron M, Dunn WA, Jr, Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fésüs L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, González-Estévez C, Gorski S, Gottlieb RA, Häussinger D, He YW, Heidenreich K, Hill JA, Høyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jäättelä M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, Komatsu M, Kominami E, Kondo S, Kovács AL, Kroemer G, Kuan CY, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei HY, Lenardo MJ, Levine B, Lieberman A, Lim KL, Lin FC, Liou W, Liu LF, Lopez-Berestein G, López-Otín C, Lu B, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mautner J, Meijer AJ, Meléndez A, Michels P, Miotto G, Mistiaen WP, Mizushima N, Mograbi B, Monastyrska I, Moore MN, Moreira PI, Moriyasu Y, Motyl T, Münz C, Murphy LO, Naqvi NI, Neufeld TP, Nishino I, Nixon RA, Noda T, Nürnberg B, Ogawa M, Oleinick NL, Olsen LJ, Ozpolat B, Paglin S, Palmer GE, Papassideri I, Parkes M, Perlmutter DH, Perry G, Piacentini M, Pinkas-Kramarski R, Prescott M, Proikas-Cezanne T, Raben N, Rami A, Reggiori F, Rohrer B, Rubinsztein DC, Ryan KM, Sadoshima J, Sakagami H, Sakai Y, Sandri M, Sasakawa C, Sass M, Schneider C, Seglen PO, Seleverstov O, Settleman J, Shacka JJ, Shapiro IM, Sibirny A, Silva-Zacarin EC, Simon HU, Simone C, Simonsen A, Smith MA, Spanel-Borowski K, Srinivas V, Steeves M, Stenmark H, Stromhaug PE, Subauste CS, Sugimoto S, Sulzer D, Suzuki T, Swanson MS, Tabas I, Takeshita F, Talbot NJ, Tallóczy Z, Tanaka K, Tanaka K, Tanida I, Taylor GS, Taylor JP, Terman A, Tettamanti G, Thompson CB, Thumm M, Tolkovsky AM, Tooze SA, Truant R, Tumanovska LV, Uchiyama Y, Ueno T, Uzcátegui NL, van der Klei I, Vaquero EC, Vellai T, Vogel MW, Wang HG, Webster P, Wiley JW, Xi Z, Xiao G, Yahalom J, Yang JM, Yap G, Yin XM, Yoshimori T, Yu L, Yue Z, Yuzaki M, Zabirnyk O, Zheng X, Zhu X, Deter RL. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4(2):151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karim MR, Kanazawa T, Daigaku Y, Fujimura S, Miotto G, Kadowaki M. Cytosolic LC3 ratio as a sensitive index of macroautophagy in isolated rat hepatocytes and H4-II-E cells. Autophagy. 2007;3(6):553–60. doi: 10.4161/auto.4615. [DOI] [PubMed] [Google Scholar]
- 71.Tanida I, Ueno T, Kominami E. LC3 and autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- 72.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 73.Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, Klionsky DJ. In search of an “autophagomometer”. Autophagy. 2009;5(5):585–9. doi: 10.4161/auto.5.5.8823. [DOI] [PubMed] [Google Scholar]
- 74.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3(5):452–60. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 75.Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007;3(4):323–8. doi: 10.4161/auto.4012. [DOI] [PubMed] [Google Scholar]
- 76.Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26(31):8057–68. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pattingre S, Petiot A, Codogno P. Analyses of Galpha-interacting protein and activator of G-protein-signaling-3 functions in macroautophagy. Methods Enzymol. 2004;390:17–31. doi: 10.1016/S0076-6879(04)90002-X. [DOI] [PubMed] [Google Scholar]
- 78.Ueno T, Ishidoh K, Mineki R, Tanida I, Murayama K, Kadowaki M, Kominami E. Autolysosomal membrane-associated betaine homocysteine methyltransferase. Limited degradation fragment of a sequestered cytosolic enzyme monitoring autophagy. J Biol Chem. 1999;274(21):15222–9. doi: 10.1074/jbc.274.21.15222. [DOI] [PubMed] [Google Scholar]
- 79.Mercer CA, Kaliappan A, Dennis PB. Macroautophagy-dependent, intralysosomal cleavage of a betaine homocysteine methyltransferase fusion protein requires stable multimerization. Autophagy. 2008;4(2):185–94. doi: 10.4161/auto.5275. [DOI] [PubMed] [Google Scholar]
- 80.Dennis PB, Mercer CA. The GST-BHMT assay and related assays for autophagy. Methods Enzymol. 2009;452:97–118. doi: 10.1016/S0076-6879(08)03607-0. [DOI] [PubMed] [Google Scholar]
- 81.Bauvy C, Meijer AJ, Codogno P. Assaying of autophagic protein degradation. Methods Enzymol. 2009;452:47–61. doi: 10.1016/S0076-6879(08)03604-5. [DOI] [PubMed] [Google Scholar]
- 82.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 83.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72(11):8586–96. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kametaka S, Okano T, Ohsumi M, Ohsumi Y. Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J Biol Chem. 1998;273(35):22284–91. doi: 10.1074/jbc.273.35.22284. [DOI] [PubMed] [Google Scholar]
- 85.Yoshimoto K, Hanaoka H, Sato S, Kato T, Tabata S, Noda T, Ohsumi Y. Processing of ATG8s, ubiquitin-like proteins, and their deconjugation by ATG4s are essential for plant autophagy. Plant Cell. 2004;16(11):2967–83. doi: 10.1105/tpc.104.025395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301(5638):1387–91. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- 87.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005;1(1):46–52. doi: 10.4161/auto.1.1.1542. [DOI] [PubMed] [Google Scholar]
- 89.Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S, Gilliam TC, Levine B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59(1):59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- 90.Miracco C, Cosci E, Oliveri G, Luzi P, Pacenti L, Monciatti I, Mannucci S, De Nisi MC, Toscano M, Malagnino V, Falzarano SM, Pirtoli L, Tosi P. Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int J Oncol. 2007;30(2):429–36. [PubMed] [Google Scholar]
- 91.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100(25):15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, Jung JU. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10(7):776–87. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Noble CG, Dong JM, Manser E, Song H. Bcl-xL and UVRAG cause a monomer-dimer switch in Beclin1. J Biol Chem. 2008;283(38):26274–82. doi: 10.1074/jbc.M804723200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ionov Y, Nowak N, Perucho M, Markowitz S, Cowell JK. Manipulation of nonsense mediated decay identifies gene mutations in colon cancer Cells with microsatellite instability. Oncogene. 2004;23(3):639–45. doi: 10.1038/sj.onc.1207178. [DOI] [PubMed] [Google Scholar]
- 95.Kim MS, Jeong EG, Ahn CH, Kim SS, Lee SH, Yoo NJ. Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Hum Pathol. 2008;39(7):1059–63. doi: 10.1016/j.humpath.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 96.Bekri S, Adelaide J, Merscher S, Grosgeorge J, Caroli-Bosc F, Perucca-Lostanlen D, Kelley PM, Pébusque MJ, Theillet C, Birnbaum D, Gaudray P. Detailed map of a region commonly amplified at 11q13-->q14 in human breast carcinoma. Cytogenet Cell Genet. 1997;79(1–2):125–31. doi: 10.1159/000134699. [DOI] [PubMed] [Google Scholar]
- 97.Lee JW, Jeong EG, Soung YH, Nam SW, Lee JY, Yoo NJ, Lee SH. Decreased expression of tumour suppressor Bax-interacting factor-1 (Bif-1), a Bax activator, in gastric carcinomas. Pathology. 2006;38(4):312–5. doi: 10.1080/00313020600820880. [DOI] [PubMed] [Google Scholar]
- 98.Coppola D, Khalil F, Eschrich SA, Boulware D, Yeatman T, Wang HG. Down-regulation of Bax-interacting factor-1 in colorectal adenocarcinoma. Cancer. 2008;113(10):2665–70. doi: 10.1002/cncr.23892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Coppola D, Oliveri C, Sayegh Z, Boulware D, Takahashi Y, Pow-Sang J, Djeu JY, Wang HG. Bax-interacting factor-1 expression in prostate cancer. Clin Genitourin Cancer. 2008;6(2):117–21. doi: 10.3816/CGC.2008.n.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim SY, Oh YL, Kim KM, Jeong EG, Kim MS, Yoo NJ, Lee SH. Decreased expression of Bax-interacting factor-1 (Bif-1) in invasive urinary bladder and gallbladder cancers. Pathology. 2008;40(6):553–7. doi: 10.1080/00313020802320440. [DOI] [PubMed] [Google Scholar]
- 101.Balakrishnan A, von Neuhoff N, Rudolph C, Kamphues K, Schraders M, Groenen P, van Krieken JH, Callet-Bauchu E, Schlegelberger B, Steinemann D. Quantitative microsatellite analysis to delineate the commonly deleted region 1p22.3 in mantle cell lymphomas. Genes Chromosomes Cancer. 2006;45(10):883–92. doi: 10.1002/gcc.20352. [DOI] [PubMed] [Google Scholar]
- 102.Cuddeback SM, Yamaguchi H, Komatsu K, Miyashita T, Yamada M, Wu C, Singh S, Wang HG. Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. J Biol Chem. 2001;276(23):20559–65. doi: 10.1074/jbc.M101527200. [DOI] [PubMed] [Google Scholar]
- 103.Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11(4):468–76. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11(4):385–96. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 105.Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228(4706):1440–3. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 106.McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP, Korsmeyer SJ. Bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. 1989;57(1):79–88. doi: 10.1016/0092-8674(89)90174-8. [DOI] [PubMed] [Google Scholar]
- 107.Saeki K, Yuo A, Okuma E, Yazaki Y, Susin SA, Kroemer G. Bcl-2 down-regulation causes autophagy in a caspase-independent manner in human leukemic HL60 cells Bcl-2 down-regulation causes autophagy in a caspase-independent manner in human leukemic HL60 cells. Cell Death Differ. 2000;7(12):1263–9. doi: 10.1038/sj.cdd.4400759. [DOI] [PubMed] [Google Scholar]
- 108.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 anti-apoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 109.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26(10):2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–88. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem. 2009;284(5):2719–28. doi: 10.1074/jbc.M805920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zalckvar E, Berissi H, Eisenstein M, Kimchi A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy. 2009;5(5):720–2. doi: 10.4161/auto.5.5.8625. [DOI] [PubMed] [Google Scholar]
- 113.Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, Geneste O, Kroemer G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007;3(4):374–6. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 114.Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, Hirsch JA, Stein R, Pinkas-Kramarski R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy. 2007;3(6):561–8. doi: 10.4161/auto.4713. [DOI] [PubMed] [Google Scholar]
- 115.Liang C, EX, Jung JU. Downregulation of autophagy by herpesvirus Bcl-2 homologs. Autophagy. 2008;4(3):268–72. doi: 10.4161/auto.5210. [DOI] [PubMed] [Google Scholar]
- 116.Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64(12):4286–93. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- 117.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14(1):146–57. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 118.Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14(3):500–10. doi: 10.1038/sj.cdd.4402039. [DOI] [PubMed] [Google Scholar]
- 119.Rashmi R, Pillai SG, Vijayalingam S, Ryerse J, Chinnadurai G. BH3-only protein BIK induces caspase-independent cell death with autophagic features in Bcl-2 null cells. Oncogene. 2008;27(10):1366–75. doi: 10.1038/sj.onc.1210783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369(6483):756–8. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 121.Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva NR, Hall MN. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73(3):585–96. doi: 10.1016/0092-8674(93)90144-f. [DOI] [PubMed] [Google Scholar]
- 122.Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253(5022):905–9. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 123.Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15(7):807–26. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- 124.Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. EMBO J. 1997;16(12):3693–704. doi: 10.1093/emboj/16.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Meyuhas O. Synthesis of the translational apparatus is regulated at the translational level. Eur J Biochem. 2000;267(21):6321–30. doi: 10.1046/j.1432-1327.2000.01719.x. [DOI] [PubMed] [Google Scholar]
- 126.Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001;20(16):4370–9. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–34. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 129.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10(1):151–62. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 130.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121(2):179–93. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 131.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A. 2004;101(37):13489–94. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Stevens C, Lin Y, Harrison B, Burch L, Ridgway RA, Sansom O, Hupp T. Peptide combinatorial libraries identify TSC2 as a death-associated protein kinase (DAPK) death domain-binding protein and reveal a stimulatory role for DAPK in mTORC1 signaling. J Biol Chem. 2009;284(1):334–44. doi: 10.1074/jbc.M805165200. [DOI] [PubMed] [Google Scholar]
- 133.Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18(13):1533–8. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zeng X, Kinsella TJ. Mammalian target of rapamycin and S6 kinase 1 positively regulate 6-thioguanine-induced autophagy. Cancer Res. 2008;68(7):2384–90. doi: 10.1158/0008-5472.CAN-07-6163. [DOI] [PubMed] [Google Scholar]
- 136.Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7(2):167–78. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 137.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431(7005):200–5. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 138.Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14(18):1650–6. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 139.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275(2):992–8. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 140.Rusten TE, Lindmo K, Juhasz G, Sass M, Seglen PO, Brech A, Stenmark H. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev Cell. 2004;7(2):179–92. doi: 10.1016/j.devcel.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 141.Klionsky DJ, Meijer AJ, Codogno P. Autophagy and p70S6 kinase. Autophagy. 2005;1(1):59–60. doi: 10.4161/auto.1.1.1536. discussion -1. [DOI] [PubMed] [Google Scholar]
- 142.Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12(Suppl 2):1509–18. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- 143.Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243(1–2):240–6. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- 144.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 145.Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, Ogier-Denis E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276(38):35243–6. doi: 10.1074/jbc.C100319200. [DOI] [PubMed] [Google Scholar]
- 146.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102(23):8204–9. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, Carnuccio R, Kroemer G. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle. 2009;8(10):1571–6. doi: 10.4161/cc.8.10.8498. [DOI] [PubMed] [Google Scholar]
- 148.Budanov AV, Karin M. p53 target genes sestrin1 and ses-trin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134(3):451–60. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126(1):121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 150.Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D’Amelio M, Djavaheri-Mergny M, Cecconi F, Tavernarakis N, Kroemer G. A dual role of p53 in the control of autophagy. Autophagy. 2008;4(6):810–4. doi: 10.4161/auto.6486. [DOI] [PubMed] [Google Scholar]
- 151.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10(6):676–87. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A, Vicencio JM, Soussi T, Kroemer G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle. 2008;7(19):3056–61. doi: 10.4161/cc.7.19.6751. [DOI] [PubMed] [Google Scholar]
- 153.Humbey O, Pimkina J, Zilfou JT, Jarnik M, Dominguez-Brauer C, Burgess DJ, Eischen CM, Murphy ME. The ARF tumor suppressor can promote the progression of some tumors. Cancer Res. 2008;68(23):9608–13. doi: 10.1158/0008-5472.CAN-08-2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, Kimchi A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell. 2006;22(4):463–75. doi: 10.1016/j.molcel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 155.Abida WM, Gu W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008;68(2):352–7. doi: 10.1158/0008-5472.CAN-07-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pimkina J, Humbey O, Zilfou JT, Jarnik M, Murphy ME. ARF induces autophagy by virtue of interaction with Bcl-xl. J Biol Chem. 2009;284(5):2803–10. doi: 10.1074/jbc.M804705200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pimkina J, Murphy ME. ARF, autophagy and tumor suppression. Autophagy. 2009;5(3):397–9. doi: 10.4161/auto.5.3.7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol. 2002;157(3):455–68. doi: 10.1083/jcb.200109094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kang C, You YJ, Avery L. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev. 2007;21(17):2161–71. doi: 10.1101/gad.1573107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26(7):1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282(25):18573–83. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- 162.Gunn JM, Clark MG, Knowles SE, Hopgood MF, Ballard FJ. Reduced rates of proteolysis in transformed cells. Nature. 1977;266(5597):58–60. doi: 10.1038/266058a0. [DOI] [PubMed] [Google Scholar]
- 163.Kisen GO, Tessitore L, Costelli P, Gordon PB, Schwarze PE, Baccino FM, Seglen PO. Reduced autophagic activity in primary rat hepatocellular carcinoma and ascites hepatoma cells. Carcinogenesis. 1993;14(12):2501–5. doi: 10.1093/carcin/14.12.2501. [DOI] [PubMed] [Google Scholar]
- 164.Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Kim SS, Ahn CH, Yoo NJ, Lee SH. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol. 2009;217(5):702–6. doi: 10.1002/path.2509. [DOI] [PubMed] [Google Scholar]
- 165.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 166.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, Nelson DA, Jin S, White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, Stone CD, Brunt EM, Xavier RJ, Sleckman BP, Li E, Mizushima N, Stappenbeck TS, Virgin HW., 4th A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456(7219):259–63. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Granell S, Baldini G, Mohammad S, Nicolin V, Narducci P, Storrie B. Sequestration of mutated alpha1-antitrypsin into inclusion bodies is a cell-protective mechanism to maintain endoplasmic reticulum function. Mol Biol Cell. 2008;19(2):572–86. doi: 10.1091/mbc.E07-06-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Perlmutter DH. Autophagic disposal of the aggregation-prone protein that causes liver inflammation and carcinogenesis in alpha-1-antitrypsin deficiency. Cell Death Differ. 2009;16(1):39–45. doi: 10.1038/cdd.2008.103. [DOI] [PubMed] [Google Scholar]
- 170.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33(4):517–27. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21(11):1367–81. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21(13):1621–35. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavaré S, Arakawa S, Shimizu S, Watt FM, Narita M. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23(7):798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Xiao S, Chye ML. The Arabidopsis thaliana ACBP3 regulates leaf senescence by modulating phospholipid metabolism and ATG8 stability. Autophagy. 2010;6(6):802–4. doi: 10.1105/tpc.110.075333. [DOI] [PubMed] [Google Scholar]
- 175.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Autophagy mediates the process of cellular senescence characterizing bile duct damages in primary biliary cirrhosis. Lab Invest. 2010;90(6):835–43. doi: 10.1038/labinvest.2010.56. [DOI] [PubMed] [Google Scholar]
- 176.Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem. 2006;281(28):19179–87. doi: 10.1074/jbc.M513377200. [DOI] [PubMed] [Google Scholar]
- 177.Schin KS, Clever U. Lysosomal and free acid phosphatase in salivary glands of chironomus tentans. Science. 1965;150(699):1053–5. doi: 10.1126/science.150.3699.1053. [DOI] [PubMed] [Google Scholar]
- 178.Nopanitaya W, Misch DW. Developmental cytology of the midgut in the flesh-fly, Sarcophaga bullata (Parker) Tissue Cell. 1974;6(3):487–502. doi: 10.1016/0040-8166(74)90040-8. [DOI] [PubMed] [Google Scholar]
- 179.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131(6):1137–48. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.McPhee CK, Logan MA, Freeman MR, Baehrecke EH. Activation of autophagy during cell death requires the engulfment receptor Draper. Nature. 2010;465(7301):1093–6. doi: 10.1038/nature09127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12(1):25–31. [PubMed] [Google Scholar]
- 182.Yu WH, Kumar A, Peterhoff C, Shapiro Kulnane L, Uchiyama Y, Lamb BT, Cuervo AM, Nixon RA. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide overproduction and localization in Alzheimer’s disease. Int J Biochem Cell Biol. 2004;36(12):2531–40. doi: 10.1016/j.biocel.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 183.Yang DS, Kumar A, Stavrides P, Peterson J, Peterhoff CM, Pawlik M, Levy E, Cataldo AM, Nixon RA. Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer’s disease. Am J Pathol. 2008;173(3):665–81. doi: 10.2353/ajpath.2008.071176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278(27):25009–13. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 185.Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1(1):23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 186.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119(6):753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 187.Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306(5698):1037–40. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 188.Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M, Kreymborg K, Altenberend F, Brandenburg J, Kalbacher H, Brock R, Driessen C, Rammensee HG, Stevanovic S. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A. 2005;102(22):7922–7. doi: 10.1073/pnas.0501190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.English L, Chemali M, Duron J, Rondeau C, Laplante A, Gingras D, Alexander D, Leib D, Norbury C, Lippé R, Desjardins M. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol. 2009;10(5):480–7. doi: 10.1038/ni.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Li Y, Wang LX, Yang G, Hao F, Urba WJ, Hu HM. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008;68(17):6889–95. doi: 10.1158/0008-5472.CAN-08-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Münz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307(5709):593–6. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 192.Bae D, Lu S, Taglienti CA, Mercurio AM. Metabolic stress induces the lysosomal degradation of neuropilin-1 but not neuropilin-2. J Biol Chem. 2008;283(42):28074–80. doi: 10.1074/jbc.M804203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shinohara ET, Cao C, Niermann K, Mu Y, Zeng F, Hallahan DE, Lu B. Enhanced radiation damage of tumor vasculature by mTOR inhibitors. Oncogene. 2005;24(35):5414–22. doi: 10.1038/sj.onc.1208715. [DOI] [PubMed] [Google Scholar]
- 194.Kim KW, Hwang M, Moretti L, Jaboin JJ, Cha YI, Lu B. Autophagy upregulation by inhibitors of caspase-3 and mTOR enhances radiotherapy in a mouse model of lung cancer. Autophagy. 2008;4(5):659–68. doi: 10.4161/auto.6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22(2):165–78. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, Coller HA, Dipaola RS, Gelinas C, Rabinowitz JD, White E. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25(5):460–70. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120(2):237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 198.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–23. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 199.Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11(4):448–57. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- 200.Bursch W, Ellinger A, Kienzl H, Torok L, Pandey S, Sikorska M, Walker R, Hermann RS. Active cell death induced by the anti-estrogens tamoxifen and ICI 164–384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis. 1996;17(8):1595–607. doi: 10.1093/carcin/17.8.1595. [DOI] [PubMed] [Google Scholar]
- 201.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61(2):439–44. [PubMed] [Google Scholar]
- 202.Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68(5):1485–94. doi: 10.1158/0008-5472.CAN-07-0562. [DOI] [PubMed] [Google Scholar]
- 203.Glaumann H, Ahlberg J. Comparison of different autophagic vacuoles with regard to ultrastructure, enzymatic composition, and degradation capacity--formation of crinosomes. Exp Mol Pathol. 1987;47(3):346–62. doi: 10.1016/0014-4800(87)90018-9. [DOI] [PubMed] [Google Scholar]
- 204.Poole B, Ohkuma S. Effect of weak bases on the intraly-sosomal pH in mouse peritoneal macrophages. J Cell Biol. 1981;90(3):665–9. doi: 10.1083/jcb.90.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117(2):326–36. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Geser A, Brubaker G, Draper CC. Effect of a malaria suppression program on the incidence of African Burkitt’s lymphoma. Am J Epidemiol. 1989;129(4):740–52. doi: 10.1093/oxfordjournals.aje.a115189. [DOI] [PubMed] [Google Scholar]
- 207.Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, Houghton JA, Huang P, Giles FJ, Cleveland JL. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110(1):313–22. doi: 10.1182/blood-2006-10-050260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sotelo J, Briceno E, Lopez-Gonzalez MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2006;144(5):337–43. doi: 10.7326/0003-4819-144-5-200603070-00008. [DOI] [PubMed] [Google Scholar]
- 209.Ciocca DR, Calderwood SK. Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones. 2005;10(2):86–103. doi: 10.1379/CSC-99r.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Volloch VZ, Sherman MY. Oncogenic potential of Hsp72. Oncogene. 1999;18(24):3648–51. doi: 10.1038/sj.onc.1202525. [DOI] [PubMed] [Google Scholar]
- 211.Seo JS, Park YM, Kim JI, Shim EH, Kim CW, Jang JJ, Kim SH, Lee WH. T cell lymphoma in transgenic mice expressing the human Hsp70 gene. Biochem Biophys Res Commun. 1996;218(2):582–7. doi: 10.1006/bbrc.1996.0103. [DOI] [PubMed] [Google Scholar]
- 212.Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009;36(1):15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Cao C, Subhawong T, Albert JM, Kim KW, Geng L, Sekhar KR, Gi YJ, Lu B. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 2006;66(20):10040–7. doi: 10.1158/0008-5472.CAN-06-0802. [DOI] [PubMed] [Google Scholar]
- 214.Miller WH, Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62(14):3893–903. [PubMed] [Google Scholar]
- 215.Qian W, Liu J, Jin J, Ni W, Xu W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk Res. 2007;31(3):329–39. doi: 10.1016/j.leukres.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 216.Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003;63(9):2103–8. [PubMed] [Google Scholar]
- 217.Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene. 2005;24(6):980–91. doi: 10.1038/sj.onc.1208095. [DOI] [PubMed] [Google Scholar]
- 218.Chiu HW, Lin W, Ho SY, Wang YJ. Synergistic effects of arsenic trioxide and radiation in osteosarcoma cells through the induction of both autophagy and apoptosis. Radiat Res. 2011 Mar 9; doi: 10.1667/RR2380.1. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 219.DiPaola RS, Dvorzhinski D, Thalasila A, Garikapaty V, Doram D, May M, Bray K, Mathew R, Beaudoin B, Karp C, Stein M, Foran DJ, White E. Therapeutic starvation and autophagy in prostate cancer: a new paradigm for targeting metabolism in cancer therapy. Prostate. 2008;68(16):1743–52. doi: 10.1002/pros.20837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Stein M, Lin H, Jeyamohan C, Dvorzhinski D, Gounder M, Bray K, Eddy S, Goodin S, White E, Dipaola RS. Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate. 2010;70(13):1388–94. doi: 10.1002/pros.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]