

Abstract
The valosin-containing protein (p97) is a ubiquitin-dependent ATPase that plays central roles in ubiquitin proteasome system (UPS)-mediated protein degradation pathways. p97 has been recently identified as a putative substrate of active Caspase-6 (Casp6) in primary human neurons. Since Casp6 is activated in mild cognitive impairment (MCI) and Alzheimer's disease (AD) patients' brains, the targeting of p97 by Casp6 may represent an important step that leads to UPS impairment in AD. Here, we show that p97 is a Casp6 substrate in vitro and in vivo. Casp6 cleavage of recombinant p97 generated two N-terminal fragments of 28 and 20 kDa, which were not generated by the other two effector caspases, Caspase-3 and Caspase-7. ATP binding to the D1 ATPase ring of p97 reduced the susceptibility of the N-domain to caspase-mediated proteolysis. Mass spectrometric analysis identified VAPD179 as a Casp6 cleavage site within p97's N-domain. An anti-neoepitope serum immunohistochemically detected p97 cleaved at VAPD179 in the cytoplasm of the cell soma and neurites of hippocampal neurons in MCI and AD. Overexpression of p97 (1-179) fragment, representing p97 cleaved at D179, impaired the degradation of model substrates in the ubiquitin-fusion degradation and the N-end rule pathways, and destabilized endogenous p97. Collectively, these results show that p97 is cleaved by Casp6 in AD and suggest p97 cleavage as an important mechanism for UPS impairment.
Introduction
The accumulation and deposition of polyubiquitinated proteins into misfolded aggregates is a common characteristic of many neurodegenerative diseases (Schwartz and Ciechanover, 1999). In Alzheimer's disease (AD), ubiquitin is associated with neurofibrillary tangles and senile plaques in hippocampal brain tissue (Perry et al., 1987, 1989). Ubiquitin immunoreactivity is present in paired helical filaments (PHF) (Mori et al., 1987) and dystrophic neurites of aged and AD brains (Dickson et al., 1990a). Although Tau is a major target of ubiquitination (Mori et al., 1987), ubiquitin-immunopositive structures devoid of Tau indicate that other ubiquitinated proteins accumulate in AD brains (Shaw and Chau, 1988; Dickson et al., 1990b). The deposition of ubiquitinated proteins precedes tangle formation (García Gil et al., 2001) and is associated with the induction of the unfolded protein response (Hoozemans et al., 2009).
The early accumulation and deposition of ubiquitinated proteins in AD suggests dysfunction in ubiquitin proteasome system (UPS)-mediated protein degradation. Expression of a C-terminal frame-shift mutant of ubiquitin lacking the critical Gly76 residue necessary for polyubiquitination (van Leeuwen et al., 1998) impairs UPS-mediated protein degradation (Lindsten et al., 2002). Proteasome function is also compromised by the oxidation of the neuronal ubiquitin C-terminal hydrolase, UCH-L1 (Choi et al., 2004), which protects synapses against β-amyloid-induced cytotoxicity (Gong et al., 2006). Moreover, PHF may directly impair proteasome function by physical interaction (Keck et al., 2003). Collectively, these findings imply that alterations in protein translation, oxidative stress, and PHF formation may induce proteasome dysfunction in AD.
The cysteine protease, Caspase-6 (Casp6), is a putative effector of neurodegeneration in AD (LeBlanc, 2005; Albrecht et al., 2007). Neurotrophic factor deprivation in primary neurons activates Casp6 (LeBlanc et al., 1999; Nikolaev et al., 2009) and results in axonal degeneration independent of cell death (Guo et al., 2004; Nikolaev et al., 2009). This critical role of active Casp6 in mediating axonopathy in AD neurons prompted investigation of other consequences of Casp6 activation. In a proteomic screen, we identified p97, a chaperone-like ATPase and a well known regulator of protein ubiquitination, as a candidate substrate of Casp6 in primary human neurons (Klaiman et al., 2008). Interestingly, p97 is an important effector of protein ubiquitination (Rumpf and Jentsch, 2006) and disaggregation (Shcherbik and Haines, 2007). p97 facilitates UPS-mediated degradation in several pathways, including the ubiquitin fusion degradation (UFD) (Koegl et al., 1999; Richly et al., 2005), the N-end rule (Wójcik et al., 2006), and endoplasmic reticulum-associated degradation (ERAD) (Ye et al., 2001). Within these pathways, p97 forms complexes with ubiquitin-binding cofactors and E3 ubiquitin ligases and ensures the efficient ubiquitination and degradation of proteasome substrates (Halawani and Latterich, 2006; Jentsch and Rumpf, 2007). Moreover, mutations in p97 are linked to a familial form of frontotemporal dementia (Watts et al., 2004) that is associated with an accumulation of intranuclear ubiquitinated proteins (Forman et al., 2006). In this report, we investigated whether p97 is a substrate of Casp6 in vitro and in vivo. Our results implicate Casp6 in the proteolytic processing of p97 and suggest a novel mechanism for UPS impairment in AD.
Materials and Methods
Protein expression vectors.
Human p97 cDNA in pBluescript SK+ vector was kindly provided by Dr. Fred Indig (National Institute on Aging, National Institutes of Health, Baltimore, MD). p97ND1 (residues 1-480) and p97D2C (residues 481-806) were subcloned into the pTrcHis2A vectors (Invitrogen) to allow for C-terminal polyhistidine tagging. The p97ND1 fragment was amplified by PCR using the primers 5′-AGCTCGAGAATGGCCTCTGGAGCCGAT-3′ and 5′-CCATATGTACCTTATCCAAT ATCTTCCCAGGT-3′, which contained the restriction sites XhoI and KpnI, respectively. Similarly, p97D2C fragment was amplified using the primers 5′-AGCTCGAGAGGTGGCCTGGAGGATGTC-3′ and 5′-ATATGGTACCAGGCCATAC AGGTCATCGTC-3′, containing the restriction sites XhoI and KpnI, respectively. Subsequent to restriction digest, both PCR products were ligated into pTrcHis2A vectors. Full-length p97WT in pTrcHis2C was generated as previously described (Halawani et al., 2009).
Mammalian p97WT expression vectors were generated by cloning full-length murine p97WT cDNA into the p3X-FLAG-CMV10 vector for N-terminal tagging or p3X-FLAG-CMV14 vector for C-terminal tagging with the FLAG peptide (Sigma). Full-length p97 was amplified using the primers 5′-CTTGCGGCCGCGGCCTCTGGAGCCGATTCAAAA-3′ and 5′-GGGGATCCTTAGCCATACAGGTCATCGTCATT-3′, containing the restriction sites NotI and BamHI for cloning into the p3X-FLAG-CMV10. Similarly, primers 5′-CTTGCGGCCGCGGCCGCCACCATGGCCTCTGGAGCCGAT-3′ and 5′-GGGGATCCGCCATACAGTCAT CGTCATT-3′, with the restriction sites NotI and BamHI, were used for cloning into the p3X-FLAG-CMV14. Subsequent to restriction digest, the inserts were ligated into the appropriate vector. FLAG-tagged p97K524A was generated as described for p97WT except that the cDNA was supplied in the pQE9 vector (kind gift from Dr. Tom Rapoport, Harvard University, Boston, MA). The FLAG-p97 (1-179) vector was constructed using site-directed mutagenesis of the p3X-FLAG-p97WT-CMV10 as described above. Briefly, the codon 538ACA540 was changed to 538TAA540 using the primers 5′-TACTGTATTGTTGCTCCAGACTAAGTGATCCACTGT-3′ and 5′-GTCTGGAGCA ACAATACAGTAAGGGCTGGGATC-3′. All constructs were verified by sequencing at the McGill University and Genome Quebec Innovation Center Platform (Montreal, Quebec, Canada).
Caspase expression constructs pET23b-Casp3-His (Addgene plasmid 11821), pET23b-Casp6-His (Addgene plasmid 11823), pET23b-Casp7-His (Addgene plasmid 11825), and pET15b-Casp8ΔDED (Addgene plasmid 11827) were kindly provided by Dr. Guy Salvesen (The Burnham Institute, La Jolla, CA).
Protein expression and purification.
p97 proteins were expressed and purified as previously described with minor modifications (Halawani et al., 2009). Briefly, p97WT, p97ND1, and p97D2C were expressed in Top10 Escherichia coli strain. Overnight starter cultures were diluted 50 times in 2× YT media (16 g/L Tryptone, 10 g/L yeast extracts, 5 g/L NaCl) and grown at 37°C to an OD at 600 nm 0.6 with vigorous shaking. Following induction with 300 μm isopropyl β-d-thiogalactoside (IPTG), cells were grown for 4–5 h and harvested by centrifugation at 4000 g for 20 min. Cell pellets were lysed in buffer containing (in mm) 20 HEPES pH 7.5, 300 NaCl, 5 MgCl2, 2.5 DTT, and 20 imidazole and supplemented with 1 mg/ml lysozyme and a complete set of protease inhibitors (Roche). Lysates were sonicated and cleared by centrifugation at 26,000 g of for 45 min. Cleared supernatants were filtered with 0.2 μm filters (Millipore) and loaded on a pre-equilibrated Ni2+ affinity column (GE Healthcare). Bound proteins were washed with 10× column volume of lysis buffer, followed by a second wash with a 10× column volume of lysis buffer with 35 mm imidazole. Proteins were eluted with a 35–500 mm imidazole gradient over 5× column volume. For the p97ND1 and p97D2C protein fragments, eluted proteins were dialyzed against buffer containing (in mm) 20 HEPES pH 7.5, 150 NaCl, 5 MgCl2, and 2.5 DTT, using Slide-A-lyzer dialysis cassettes with a molecular weight cutoff of 10 kDa (Fisher). p97WT was further purified on a precalibrated 25 ml of Superdex-200 size-exclusion chromatography column (GE Healthcare). Peak fractions corresponding to hexameric p97 were eluted as 1 ml of fractions in buffer containing (in mm) 20 HEPES pH 7.5, 150 NaCl, 5 MgCl2, and 2.5 DTT. Final protein concentrations were determined using the Bradford assay (Bio-Rad). Purified proteins were snap frozen and stored at −80°C. All purifications were performed using the ÄKTA FPLC system (GE Healthcare).
Caspase protein expression was conducted using the BL21(DE3)pLysS E. coli stain using the following IPTG induction conditions: 200 μm IPTG was used for Casp3 and expression was allowed for 4–6 h at 30°C, Casp6 was induced with 50 μm IPTG and expressed overnight at 16°C, 200 μm IPTG was used for Casp7 expression at 30°C for 12–16 h, and Casp8 was induced with 200 μm IPTG for 4–6 h at 30°C. Cell harvesting, lysis, and Ni2+ affinity purification were performed as described above for p97 using the following buffer conditions: lysis buffer (50 mm Tris-HCl pH 8.0, 100 mm NaCl, and 10 mm imidazole supplemented with 1 mg/ml lysozyme), wash buffer (50 mm Tris-HCl pH 8.0, 500 mm NaCl, 10 mm imidazole), elution buffer (50 mm Tris-HCl pH 8.0, 100 mm NaCl using 10–500 mm imidazole gradient). Eluted proteins were dialyzed against buffer containing 50 mm Tris-HCl pH 8.0 and 100 mm NaCl as described above. Protein concentrations were determined using the Bradford assay (Bio-Rad). Caspase activity was verified using the appropriate fluorogenic substrates as previously described (LeBlanc et al., 1999).
In vitro p97 protein cleavage assay.
p97ND1, p97D2C, and p97WT were digested in HEPES buffer [0.1 m HEPES, pH 7.5, 10% sucrose, 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 10 mm DTT] or Stennicke's buffer (20 mm PIPES pH 7.4, 30 mm NaCl, 1 mm EDTA, 0.1% CHAPS, 10% sucrose, and 10 mm DTT) as indicated in the figure legends. Reactions were incubated at 37°C in 50 μl of volume using the indicated concentrations of p97 and caspase. Reactions assessing the effect of ATP on p97 proteolysis were also supplemented with 5 mm MgCl2, and p97 was preincubated with ATP for 1 h at 37°C before the addition of Casp3, Casp6, or Casp7. p97 proteolysis was assessed using SDS-PAGE followed by either Coomassie staining or Western blot analysis. Western blotting was performed with a monoclonal anti-p97 antibody raised using a p97 fragment containing residues 9–130 (BD Transduction Laboratories) or a polyclonal anti-p97 antibody raised against the C-terminal residues 786–806 (a generous gift from Dr. Maryam Taheri, McGill University, Montreal, Quebec, Canada). The monoclonal antibody was used at a dilution of 1:1000, whereas the polyclonal antibodies were used at a dilution of 1:3000. Following incubation with the appropriate HRP-conjugated secondary antibodies, the blots were visualized using the Storm 840 scanner or by standard autoradiography films (both from GE Healthcare).
Caspase activity assay.
Caspase activity assays were conducted using the fluorogenic substrates Ac-DEVD-AFC (Biomol International) for Casp3 and Casp7, and Ac-VEID-AFC (Biomol International) for Casp6. Ten nanomolar of hexameric p97 were incubated with 10 nm recombinant active Casp3, Casp6, or Casp7 in 50 μl of HEPES buffer (0.1 m HEPES, pH 7.5, 10% sucrose, 0.1% CHAPS, 10 mm DTT) supplemented with 5 mm MgCl2. For all reactions, 1 μm of either Ac-DEVD-AFC or Ac-VEID-AFC was used in the presence or absence of 2 mm ATP. For controls, fluorogenic substrates were incubated with either the caspase proteins or p97 in the presence or absence of 2 mm ATP. Quantification of AFC release was performed based on a free 7-amino-4-(trifluoromethyl)coumarin (AFC) (MP Biomedicals) standard curve as previously described (Zhang et al., 2000).
Identification of the Casp6 cleavage sites in p97 using liquid chromatography tandem mass spectroscopy analysis.
One microgram of p97ND1 or D2C fragments was digested with 320 ng of active recombinant Casp6 as indicated above. In parallel, similar digestion reactions were performed as controls with heat-inactivated Casp6. Ten replicates of each sample or control were subsequently pooled and prepared for tandem mass spectroscopy (MS/MS) analysis. Briefly, the CHAPS component of the reaction buffer was first removed using a nonionic detergent exchange cartridge (Michrom Bioresources) according to the manufacturer's instructions. The eluted sample was dried in a speed vacuum and resolubilized in 20 μl of 1 m triethylammonium bicarbonate, 2 μl of 2% SDS, and 2 μl of 50 mm tris[2-carboxyethyl]phosphine for 1 h at 60°C. Following treatment with 1 μl of 84 mm iodoacetamide, trypsin was added and samples were further incubated overnight in a thermomixer at 37°C and 700 rpm. On the following day, samples were dried and resolubilized in buffer containing 0.1% trifluoroacetic acid and 2% acetonitrile in HPLC-grade water and loaded on an SDS removal column (Michrom Bioresources). The flow-through containing the peptides was subsequently speed vacuumed to dryness and submitted for liquid chromatography (LC)-MS/MS analysis. LC-MS/MS and peptide identification was conducted at the proteomics core facility at the Institute for Research in Immunology and Cancer (University of Montreal, Montreal, Quebec, Canada).
Generation and characterization of Casp6 cleaved p97 anti-neoepitope antiserum.
Casp6-cleaved p97 anti-neoepitope antiserum (anti-p97D179) was generated using the six amino acid peptide 174CIVAPD179, N-terminally conjugated to the keyhole limpet hemocyanin protein. Before peptide synthesis, a BLAST search was conducted against the human genome to ensure uniqueness of this peptide sequence to human p97. All steps involved in antiserum production, including peptide synthesis, conjugation, immunization, bleed production, and antisera collection, were performed by Sigma-Genosys. The antiserum bound the 174CIVAPD179 peptide at a dilution of 1:300,000 in an ELISA.
The anti-p97D179 antiserum was characterized using Western blotting and immunoprecipitation analyses in protein extracts from mouse neuroblastoma N2a cells overexpressing either FLAG vector alone, FLAG-tagged full-length p97WT, or FLAG-tagged p97(1-179). Transfections were performed on 500,000 N2a cells using 4 μg of p3X-FLAG-CMV vector, FLAG-p97WT, or FLAG-p97(1-179) and 8 μg of polyethylenimine (Polysciences). Twenty-four hours posttransfection, cells were washed two times with cold PBS and lysed in RIPA buffer containing protease inhibitors [0.1 μg/ml N-α-p-tosyl-l-lysine chloromethyl ketone (TLCK), 0.5 μg/ml leupeptin, 38 μg/ml 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), and 0.1 μg/ml pepstatin A). The lysates were cleared by centrifugation (16,000 g of for 20 min] and loaded on an SDS-PAGE gel for analysis by Western blotting. Briefly, membranes were blocked with 5% nonfat milk in TBST for 1 h at room temperature and probed with an anti-FLAG-M2 antibody (Sigma-Aldrich-) at a dilution of 1:1000, an anti-p97N antibody (BD Biosciences Pharmingen) at a dilution of 1:1000, or the anti-p97D179 antiserum at a dilution of 1:20,000 in blocking solution. The appropriate HRP-conjugated secondary antibodies were applied at a dilution of 1:5000. Detection was performed using enhanced chemiluminescence plus and the Storm 840 scanner system (GE Healthcare). As a loading control, blots were further probed with a monoclonal anti-β actin antibody at a dilution of 1:5000 (Sigma-Aldrich) and developed as described above.
Immunoprecipitation was performed on protein extracts from N2a cells overexpressing p3X-FLAG-CMV vector, p97WT-FLAG, or FLAG-p97(1-179). Briefly, cells were transfected and lysed as described above. Approximately 300 μg of protein extracts were precleared with 20 μl of 20% protein A Sepharose slurry (GE Healthcare) for 1 h at 4°C. Protein A Sepharose beads were pelleted by centrifugation at 16,000 g of for 1 min. The cleared supernatants were transferred to clean tubes and further incubated with 20 μl of 20% protein A Sepharose slurry alone or with either preimmune serum or the anti-p97D179 antiserum at a dilution of 1:2000 overnight. Following five washes with 1 ml of RIPA buffer, the Sepharose beads were boiled in 2× SDS loading buffer containing 50 mm DTT and analyzed by SDS-PAGE followed by Western blotting. Membrane blocking was performed as described above. Polyvinylidene fluoride membranes were incubated with an anti-FLAG-M2 antibody at a dilution of 1:1000 for 1 h at room temperature. Following incubation with an HRP-conjugated secondary antibody at a dilution of 1:10,000, detection was performed with ECL plus and the Storm 840 scanner system (GE Healthcare).
Immunohistochemistry on human brain tissue.
Human brain tissue from persons with no cognitive impairment, mild cognitive impairment, or AD was obtained from participants in the Religious Orders Study (Bennett et al., 2002, 2005). Mini-mental score examination of the cases used in this study were previously reported (Albrecht et al., 2007). Pathological scoring of anti-p97D179 hippocampal immunoreactivity was performed blinded according to the following criteria: a score of 0, 1, 2, or 3 represented absent, rare, abundant, or very abundant immunopositive neurons in the hippocampus, respectively. Immunohistochemical detection of p97D179 (1/1000), p20Casp6 (1277 at 1/1000), and TauΔCasp6 (1/12,000) (Albrecht et al., 2007) was performed on 4% paraformaldehyde-fixed and paraffin-embedded human brain sections of 4 μm thickness. Antigen retrieval was performed by autoclaving the tissue for 15 min in 8 mm sodium citrate buffer pH 6.0. Immunohistochemical analysis was conducted manually using a diaminobenzidine I-VIEW detection kit (Ventana Medical Systems). Tissue was blocked for 20 min and incubated with antiserum. Immunoreactivity was detected with a secondary biotinylated IgG antibody for 30 min and then streptavidin-HRP for 30 min. The immunoreactivity was revealed with diaminobenzidine and the tissue section was counterstained with hematoxylin (Vector Laboratories). Colocalization of active Casp6 and p97(1-179) was performed on one AD case kindly provided by Dr. Catherine Bergeron (University of Toronto, Toronto, Ontario, Canada) using a double-labeling approach as described previously (Albrecht et al., 2007).
Protein expression analysis.
N2a neuroblastoma cells were plated at a density of 500,000 cells/well for 24 h in MEM (Invitrogen) supplemented with 10% fetal bovine serum (Fisher). Cells were transfected in 6-well plates using 3.5 μg of p3X-FLAG-CMV vector or p97 vectors p97WT-FLAG, p97K524A-FLAG, FLAG-p97(1-179), and 0.5 μg of either the ubiquitin fusion degradation reporter UbG76V-GFP (Addgene plasmid 11941) or the N-end rule reporter Ub-R-GFP (Addgene plasmid 11939), and 20 μg of polyethylenimine (Polysciences). After 48 h, cells were trypsinized, washed in PBS, and lysed in RIPA buffer containing protease inhibitors (0.1 μg/ml TLCK, 0.5 μg/ml leupeptin, 38 μg/ml AEBSF, and 0.1 μg/ml pepstatin A). Protein concentrations were quantified with the BCA protein assay (Fischer). Thirty micrograms of protein were loaded on SDS-PAGE gels and analyzed by Western blotting using either a monoclonal anti-FLAG antibody (Sigma) or a polyclonal anti-C terminal antiserum for p97 (a kind gift from Dr. Maryam Taheri, McGill University, Montreal, Quebec, Canada).
Fluorescence microscopy.
N2a cells were cotransfected with 5 μg of p3X-FLAG-CMV vector, p97WT-FLAG, p97K524A-FLAG, or FLAG-p97(1-179); and 750 ng of UbG76V-GFP or Ub-R-GFP using 10 μg of polyethylenimine (Polysciences). GFP fluorescence was visualized using and a Nikon Eclipse, TE2000-U, inverted fluorescence microscope under 20× magnification.
Flow cytometry for detection of UPS model substrates.
Transfections of N2a neuroblastoma cells were performed as described above. For analysis of cell fluorescence, cells were trypsinized, washed with PBS, and fixed with 1% paraformaldehyde for 30 min at room temperature. Flow cytometry was performed on at least 20,000 cells using the FACSCalibur flow cytometer (BD Biosciences). The data were analyzed using the CellQuest software (BD Biosciences).
Statistical analysis.
All statistics were conducted using the Stat View 5.0 software (SAS Institute). The variance was analyzed from three replicates using a one-way ANOVA followed by the Scheffé post hoc test. Significance was determined at a p value <0.05.
Results
p97 is cleaved by Casp6 in vitro
Fifteen putative Casp6 cleavage sites were identified based on the presence of consensus sites (Thornberry et al., 2000) in p97's primary amino acid sequence (Fig. 1A). To examine whether p97 is indeed cleaved by Casp6, p97ND1 (residues 1-480) or p97D2C (residues 481-806) fragments were incubated with active Casp6. The 54 kDa p97ND1 fragment (p54) generated 50 kDa (p50), 38 kDa (p38), 28 kDa (p28), and 20 kDa (p20) cleavage products, which were immunoreactive with a monoclonal antibody raised against the N-domain of p97 (Fig. 1B, top panel). Similarly, the 43 kDa p97D2C (p43) fragment resulted in a 30 kDa (p30) cleavage product, which was immunoreactive with a polyclonal antiserum against the C-terminal domain of p97 (Fig. 1C, top panel). p97ND1 and D2C cleavage was dependent on Casp6 concentration (Fig. 1B,C, top panels) and inhibited by Casp6 peptide inhibitor, Z-VEID-FMK (Fig. 1B,C, lower panels). These results indicate that p97 indeed contains caspase consensus sequences that are recognized and cleaved by Casp6 in vitro.
Figure 1.
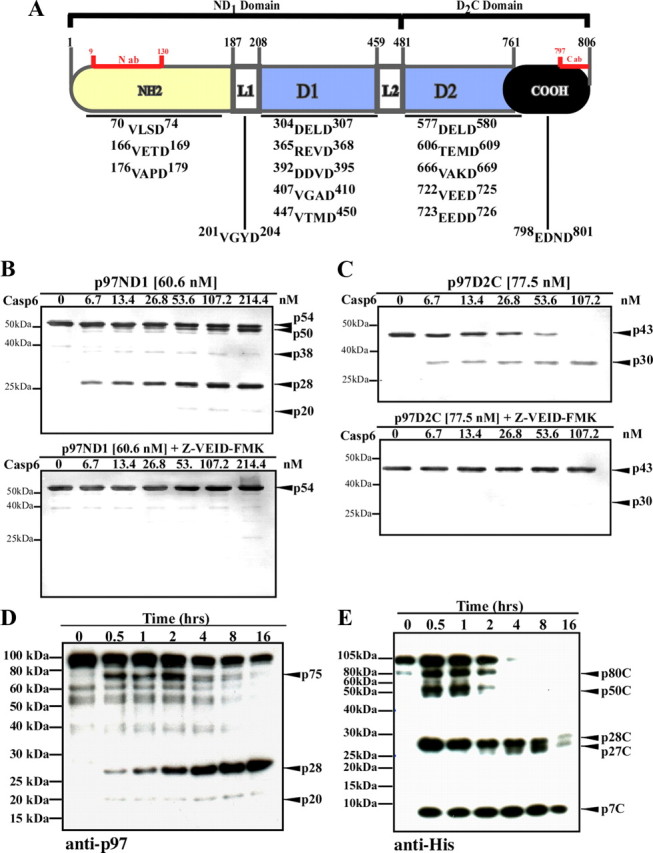
p97 cleavage by Casp6 in vitro. A, Schematic representation of p97 structure domains with the localization of the antibody binding sites and putative Casp6 cleavage sites indicated. ab, Antibody; N, N-terminal domain; L1, linker-1; D1, D1 AAA+ domain; L2, linker-2; D2, D2 AAA+ domain; C, C-terminal domain. B, C, Western blot analysis of p97ND1 (B) or p97D2C (C) incubated with Casp6 using a monoclonal anti-N-terminal antibody (B) or a polyclonal anti-C terminal antibody (C) for p97. Upper panels, Digestion reaction in the absence of inhibitor. Lower panels, Digestion reaction in the presence of the inhibitor, Z-VEID-FMK at 10 μm concentration. D, E, Western blot analysis of 100 nm hexameric p97 incubated with 300 nm active Casp6, using a monoclonal anti-N-terminal antibody for p97 (D) or a monoclonal anti-His tag antibody (E).
Since p97 physiologically exists as a hexamer, and hexameric p97 is partially resistant to proteolytic processing (Wang et al., 2003; Halawani et al., 2009), we investigated whether assembled p97 is also a substrate of Casp6. Purified hexameric p97 contained minor C-terminal truncations commonly observed (Halawani et al., 2009) and readily detectable with an anti-N-terminal antibody for p97 (Fig. 1D). Casp6 processed hexameric p97 within 30 min of incubation, generating 75 kDa (p75), 28 kDa (p28), and 20 kDa (p20) N-terminal cleavage products (Fig. 1D). After 30 min of incubation, p75 was the predominant cleavage product, followed by p28. Within 4 h of incubation, the quantity of the p75 fragment decreased and p28 became the major N-terminal cleavage product. We next used an antibody against the C-terminal polyhistidine-tag for p97. After 30 min of incubation, this antibody detected multiple C-terminal cleavage products for p97: p80C, p50C, p28C, p27C, and p7C (Fig. 1E). These results indicate that full-length hexameric p97 is a substrate of Casp6 in vitro.
N-terminal processing of p97 by Casp6 is distinct from cleavage by effectors Casp3 and Casp7
To determine whether p97 is also a substrate of other caspases, full-length hexameric p97 was incubated with effectors Casp3 and Casp7 and initiator Casp8. Casp3 and Casp7 both partially processed p97 into p75 and p40 N-terminal fragments (Fig. 2A,B). Furthermore, the p40 fragment appeared as a doublet when cleaved by Casp3 (Fig. 2A) compared with Casp7 (Fig. 2B). Unlike Casp3 and Casp7, Casp8 did not cleave p97 (Fig. 2C). Interestingly, only cleavage by Casp6 generated the low molecular weight N-terminal cleavage products, p28 and p20 (Fig. 2D). These findings suggest that Casp6 consensus sites in p97 are distinct from those targeted by effector Casp3 and Casp7.
Figure 2.

p97 cleavage by effector Casp3 and Casp7 and initiator Casp8 in vitro. Western blot analysis of hexameric p97 (100 nm) treated with active Casp3 (A), Casp7 (B), Casp8 (C), or Casp6 (D) for 4 h, with a monoclonal anti-N-terminal p97 antibody.
ATP-dependent conformational changes in p97 reduce the accessibility of the Casp6 cleavage site
Given that (1) Casp6 uniquely generates the N-terminal p28 fragment, (2) the D1 domain is responsible for forming the p97 homohexamer, and (3) the D1 nucleotide binding state regulates the N-terminal domain conformation (Davies et al., 2005), we assessed the role of nucleotide binding on p97ND1 susceptibility to Casp6-mediated cleavage. In the absence of ATP, p97ND1 cleavage by Casp6 generated the predominant cleavage product p28 (Figs. 1B and 3A). In the presence of ATP, Casp6 generated the p50 product more readily, indicating that p97ND1 was preferentially cleaved at the C terminus. To investigate the effect of Casp6 concentration on p97ND1 processing, p97ND1 was incubated with increasing concentrations of Casp6 in the presence or absence of ATP (Fig. 3B). Consistent with Figure 3A, generation of p28 and p50 cleavage products was dependent on Casp6 concentration in the absence of ATP. However, only p50 was generated in the presence of ATP, indicating that the N-domain of p97 remained resistant to Casp6 processing. Furthermore, p50 generation was dependent on the ATP concentration (Fig. 3C), indicating that ATP binding enhances p97ND1 processing at the C-terminal end. These results strongly suggest that the ATP binding state of the D1 ring influences p97 N-terminal susceptibility to Casp6-mediated processing.
Figure 3.
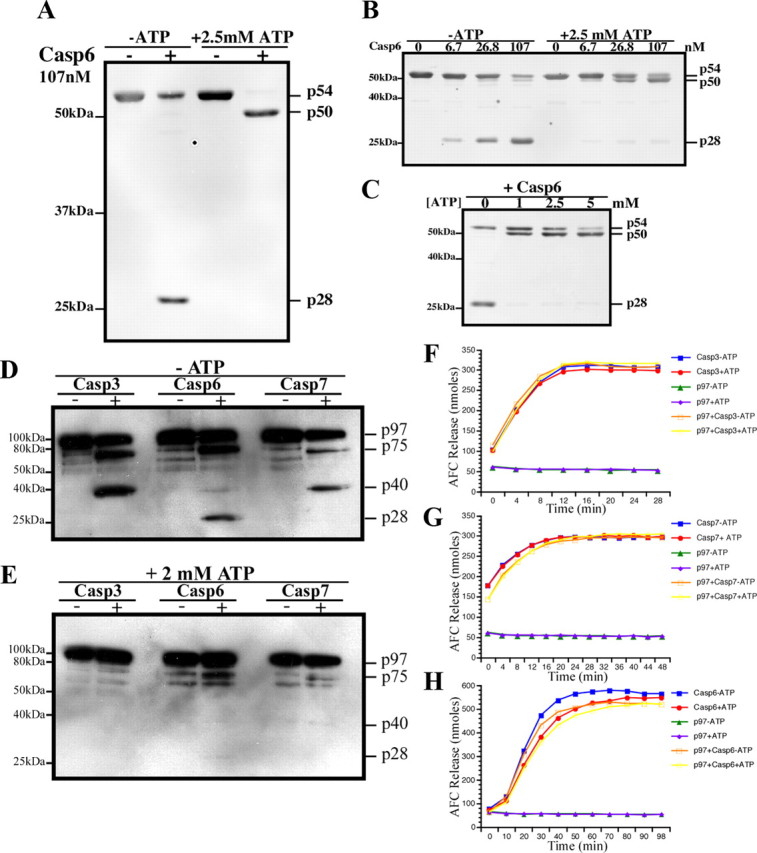
ATP-dependent modulation of p97 cleavage by caspases. A–C, Western blot analyses of p97 ND1 domain cleaved with Casp6 with a monoclonal anti-N-terminal p97 antibody. D, E, Western blot analysis of full-length p97 cleaved with Casp3, Casp6, and Casp7 without (D) or with ATP (E). F–H, Casp3 (F), Casp7 (G), and Casp6 (H) activity assays in the presence or absence of ATP and p97.
We next investigated whether nucleotide binding also modulates full-length p97 cleavage by caspases. In the absence of ATP, cleavage of p97 by Casp3 and Casp7 generated p75 and p40, whereas cleavage by Casp6 generated p75 and p28, but not p40 (Figs. 2 and 3D). In the presence of ATP, p97 cleavage by all caspases was drastically reduced, but not abolished (Fig. 3E). Several possibilities could account for the reduced processing of p97, such as: (1) ATP may inhibit caspase activity, (2) p97 may inhibit caspase activity in the presence of ATP, and (3) ATP-binding to p97 may modulate the accessibility of the caspase cleavage site. Casp3 or Casp7 DEVDase activity was neither affected by ATP alone nor by ATP in the presence of p97 (Fig. 3F,G). Incubation of active Casp6 with ATP alone, p97 alone, or p97 in the presence of ATP only slightly reduced its VEIDase activity (Fig. 3H). Since p97 processing by all caspases was reduced in the presence of ATP, these data indicate that ATP-driven conformational changes in p97 likely determine the accessibility of the Casp6 cleavage site.
Identification of the Casp6 cleavage site in p97
Considering the role of Casp6 in mediating neurodegeneration in AD, we sought to identify the Casp6 cleavage sites in p97 using LC-MS/MS. Digestion of recombinant p97ND1 fragment with active Casp6 resulted in the identification of four semitryptic peptides of a total of 50 sequenced peptides (Table 1). All semitryptic peptides contained an aspartic acid residue at the C terminus (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Two peptides contained the consensus sites 176VAPD179 and 304DELD307, which were previously predicted (Fig. 1A). Two additional peptides contained the unique cleavage sites 435ETID438 and 475TWED478. Processing of the p97D2C fragment with active Casp6 yielded only a single semitryptic peptide of a total of 41 sequenced peptides (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Unlike the ND1 semitryptic peptides, the D2C-derived peptide contained an aspartic acid residue at the N-terminal end. Analysis of the sequence immediately preceding the peptide revealed the predicted cleavage site 722VEED725 (Table 1). As expected, no semitryptic peptides were identified in p97ND1 or D2C fragments treated with heat-inactivated Casp6 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). All together, these results indicate that p97 contains at least five functional Casp6 cleavage sites.
Table 1.
Semitryptic peptides of Casp6 cleaved p97 identified using mass spectrometry
| Identified semitryptic peptide | Consensus site | p97 domain localization | Theoretical molecular weight from the N domain (kDa) |
|---|---|---|---|
| 165VVETDPSPYCIVAPD179 | VAPD179 | N domain | 20.0 |
| 296NAPAIIFIDELD307 | DELD307 | D1 AAA + ring | 34.1 |
| 426KMDLIDLEDETID438 | ETID438 | D1 AAA + ring | 48.7 |
| 466ETVVEVPQVTWED478 | TWED478 | D1-D2 linker | 53.2 |
| 714QTNPSAMEVEEDDPVPEI R732 | VEED732 | D2 AAA + ring | 80.4 |
p97 is cleaved at VAPD179 in AD
To confirm p97 cleavage in AD, we generated a neoepitope antiserum recognizing p97 cleavage at D179 (anti-p97D179), the only N-terminal Casp6 cleavage site consistently identified by mass spectrometry (Table 1). We assessed the specificity of the anti-p97D179 antiserum using cell extracts expressing vector only, FLAG-tagged full-length p97 (p97WT), or a FLAG-tagged p97(1-179) fragment, representing p97 cleaved at D179. Overexpression of p97WT and p97(1-179) was confirmed using an anti-FLAG antibody (Fig. 4A) and an anti-N-terminal antibody for p97 (Fig. 4B). Immunoblotting with anti-p97D179 specifically recognized p97(1-179) and not p97WT (Fig. 4C). Furthermore, the anti-p97D179 antiserum specifically immunoprecipitated p97(1-179), but not full-length p97WT from N2a protein extracts (Fig. 4D). p97(1-179) could not be immunoprecipitated with preimmune rabbit serum (Fig. 4D). A nonspecific band, likely corresponding to IgG, appeared in all extracts immunoprecipitated with either the anti-p97D179 antiserum or the preimmune serum. Thus, the anti-p97D179 antiserum specifically recognizes p97 cleavage at D179 and shows no immunoreactivity against full-length p97.
Figure 4.

Specificity of the anti-p97D179 neoepitope antiserum. A–C, Upper panels, Western blot analysis of N2a cell protein extracts overexpressing FLAG vector, p97WT-FLAG, or FLAG-p97(1-179), using an anti-FLAG antibody (A), anti-p97N-terminal antibody (B), and the anti-p97D179 antiserum (C). Lower panels, Western blot analysis with an anti-β-actin antibody on the blots probed in A–C. D, Western blot (WB) analysis with an anti-FLAG antibody of p97 or p97(1-179) immunoprecipitation using the anti-p97D179 antiserum or preimmune serum with proteins extracts of cells expressing FLAG vector, p97WT-FLAG, or FLAG-p97(1-179).
To determine whether p97 is cleaved at D179 in AD brains, we performed immunohistological staining on hippocampal tissue sections from four persons with no cognitive impairment (NCI), five with mild cognitive impairment (MCI), and 15 with AD with the anti-p97D179 antiserum (Table 2 and Fig. 5A). The results showed negative or rare immunopositive neurons in the hippocampus of most NCI and MCI cases. One case of MCI showed very abundant immunopositive neurons, consistent with the previous detection of active Casp6 in some aged NCI brains (Albrecht et al., 2007) and the presence of AD pathology in persons with MCI and NCI (Bennett et al., 2005). Of the 15 AD cases, immunopositive neurons were rare or absent in mild AD according to the Mini-Mental State Examination (MMSE) score (20–24) and increased to abundant and very abundant levels in the more severe cases of AD (MMSE score ≤16), except in two cases. Again, these two cases were the same previously found to have low levels of senile plaques and neurofibrillary tangles (Albrecht et al., 2007). Furthermore, p97D179 immunoreactivity localized to the same hippocampal region where active Casp6 and TauΔCasp6 were detected (Fig. 5B). Interestingly, the immunoreactivity of the anti-p97D179 antiserum in neurons had a granular appearance and was localized to the cytoplasm of the neuronal cell body and to large neurites (Fig. 5C). Furthermore, AD hippocampal neurons displayed strong immunoreactivity with the anti-p97D179 antiserum and not the preimmune rabbit serum (Fig. 5D). Neuronal cells labeled with the anti-p97D179 antiserum were also immunopositive for active Casp6, as p97D179 and active Casp6 epitopes colocalized in the cytoplasm of hippocampal neurons (Fig. 5E). Collectively, these results provide evidence for p97 cleavage at D179 in hippocampal AD tissue and implicate Casp6 activation in p97 processing.
Table 2.
Abundance of anti-p97D179 immunopositive neurons in the hippocampus of NCI, MCI, and AD individuals
| Pathology score | MMSE score | |
|---|---|---|
| NCI (n = 4) | 0 | 30 |
| 0 | 29 | |
| 0 | 28 | |
| MCI (n = 4) | 0 | 29 |
| 0 | 29 | |
| 0 | 28 | |
| 3 | 26 | |
| AD (n = 15) | 1 | 24 |
| 1 | 24 | |
| 1 | 22 | |
| 1 | 21 | |
| 0 | 20 | |
| 1 | 16 | |
| 3 | 15 | |
| 0 | 15 | |
| 2 | 12 | |
| 3 | 10 | |
| 2 | 9 | |
| 2 | 7 | |
| 3 | 2 | |
| 3 | 1 |
Scores: 0, absent; 1, rare; 2, abundant; and 3, very abundant amounts of anti-p97D179 immunopositive neurons in the hippocampus.
Figure 5.

Assessment of p97 cleavage in AD. A, Micrograph of p97D179 neoepitope antiserum (1/1000) immunostaining developed with diaminobenzidine in the dentate gyrus of one representative case of NCI, MCI, and AD hippocampal tissue. B, Micrographs of the CA2/CA3 area of the hippocampus of consecutive tissue sections from an AD individual stained with anti-p97D179, anti-p20Casp6, and anti-Tau cleaved by Casp6 (TauΔCasp6). All micrographs were taken from the same areas for comparison. C, Magnified micrographs of a neuron showing the granularity of the p97D179-positive staining in the cytoplasm and neurite of neurons (+N) and a few neurons that were p97D179-negative (−N). D, Immunostaining of AD hippocampal tissue sections with anti-p97D179 antiserum or no primary antiserum. Lower panels show enlarged area of the CA2. E, Micrographs of hippocampal neurons coimmunostained with the anti-p97D179 antiserum (Fast Red) and anti-p20Casp6 antiserum (DAB).
Overexpression of p97(1-179) fragment compromises UPS-mediated protein degradation
We next assessed the effect of p97(1-179) expression on UPS-dependent protein degradation pathways. We used two previously described UPS reporter constructs (Dantuma et al., 2000), UbG76V-GFP and Ub-R-GFP, in assessing the integrity of the UFD and the N-end rule pathway, respectively. In principle, both UbG76V-GFP and Ub-R-GFP are short-lived proteasome substrates, which accumulate quantitatively in response to proteasome inhibition. Transient coexpression of UbG76V-GFP with vector only or p97WT resulted in a weak baseline expression of UbG76V-GFP (Fig. 6A). In contrast, coexpression of an ATPase-deficient mutant of p97, p97K524A, led to a marked accumulation of UbG76V-GFP (Fig. 6A), indicating that UbG76V-GFP degradation is p97-dependent. Similarly, p97(1-179) expression resulted in the stabilization of UbG76V-GFP (Fig. 6A). Expression of p97WT, p97K524A, and p97(1-179) was verified with an anti-FLAG antibody (Fig. 6B). Interestingly, immunoblotting with an anti-C-terminal antibody for full-length p97 revealed two cleavage products of 55 and 40 kDa, which were only present in cells expressing p97(1-179), but not p97WT or p97K524A (Fig. 6C). Analysis of the effect of p97(1-179) expression on the N-end rule pathway, using the Ub-R-GFP reporter construct, showed similar results (Fig. 6D–F). These results suggest that p97(1-179) expression impairs the UFD and N-end rule protein degradation pathways and destabilize endogenous p97 protein levels.
Figure 6.
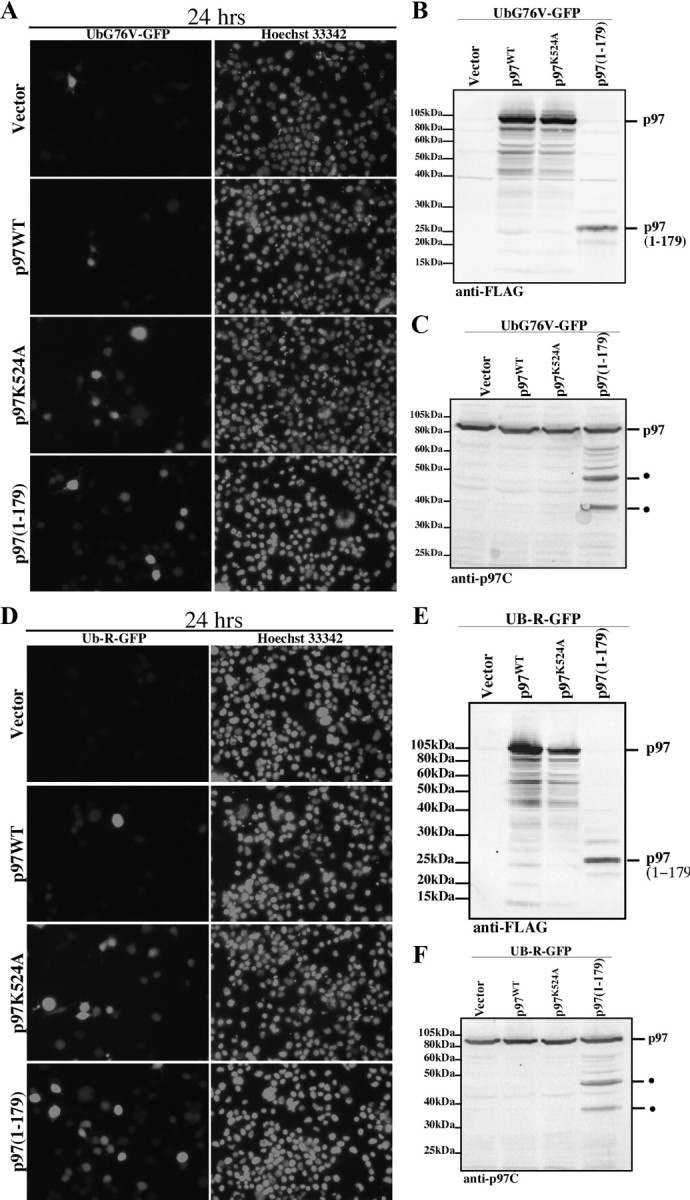
Effect of p97(1-179) expression on UPS reporter expression in N2a cells. GFP and Hoescht nuclear fluorescence of N2a cells coexpressing vector, p97WT-FLAG, p97K524A-FLAG, or FLAG-p97(1-179) with UbG76V-GFP (A) or Ub-R-GFP (D). Western blot analysis of N2a cell protein extracts overexpressing FLAG vector, p97WT-FLAG, p97K524A-FLAG, or FLAG-p97(1-179) with UbG76V-GFP (B, C) or Ub-R-GFP (E, F) using an anti-FLAG antibody (C, D) or an anti-p97 C-terminal antibody (E, F).
We next used flow cytometry to quantify the extent of UPS reporter accumulation in response to p97(1-179) expression. Baseline UbG76V-GFP expression, as evident in the number of GFP-positive cells and their mean GFP fluorescence intensity, was determined using vector cotransfected cells (Fig. 7A). Two populations were evident: a large cell population expressing low GFP fluorescence (P1) and a small population of high-GFP expressers (P2) (Fig. 7A). Compare to baseline GFP expression, overexpression of p97WT (Fig. 7B) or p97K524A (Fig. 7C) showed similar distributions of the P1 and P2 populations. In contrast, p97(1-179) expression led to a marked increase in the P2 population (Fig. 7D). Quantification of the number of GFP-positive cells in the low or high GFP-expressing population showed no significant difference between vector, p97WT, or p97K524A-expressing cells (Fig. 7E,F). However, p97(1-179) expression resulted in a significant decrease in the number of GFP-positive cells in population P1 and a corresponding significant increase in population P2. These results indicate that p97(1-179) expression leads to the stabilization, and thereby the accumulation of UbG76V-GFP in the cell population where they are normally rapidly degraded.
Figure 7.

Quantitative analysis of UPS reporter expression in N2a cells. A–D, Fluorescence activated cell sorting histograms of N2a cells coexpressing UbG76V-GFP and FLAG vector (A), p97WT-FLAG (B), p97K524A-FLAG (C), or FLAG-p97(1-179) (D) for 24 h. Data are representative of three independent experiments. E–H, The number of GFP-positive cells of P1 (E) or P2 (F) populations and the mean GFP fluorescence intensity of the P1 (G) or P2 (H) at 24 h. Data represents the average of three independent experiments. Error bars represent the SD between the three independent experiments. * indicates statistical significance at p ≤ 0.05 compared p97WT (Scheffé).
We next assessed the mean fluorescence intensity, reflecting the average accumulation of GFP per cell, of both the P1 and P2 populations. Compared with vector cotransfected cells, p97WT expression had no effect on the mean fluorescence intensity of the P1 (Fig. 7G) or P2 population (Fig. 7H). However, expression of p97K524A led to a significant increase in the mean fluorescence intensity of the P2 population, but not P1. These results suggest that p97 ATPase activity is necessary for the clearance of UbG76V-GFP in the high expresser population. In contrast, p97(1-179) expression had no significant effect on the mean fluorescence intensity of either the P1 or P2 populations. Furthermore, p97(1-179) expression had a similar effect on the degradation of the N-end rule pathway reporter, Ub-R-GFP (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Discussion
The accumulation of ubiquitinated proteins in hippocampal neurons is an important feature of neurodegeneration in AD and suggests impairment in UPS-mediated protein degradation. In AD, neurodegeneration is accompanied by Casp6 activation (Guo et al., 2004; Albrecht et al., 2007, 2009) and Casp6 cleavage of cytoskeletal proteins, which are critical for axonal function (Guo et al., 2004; Klaiman et al., 2008). In this study, we add the ubiquitin-dependent ATPase, p97, to the repertoire of Casp6 substrates in AD and propose Casp6-mediated processing of p97 as a novel mechanism of UPS impairment in AD.
Two lines of evidence support that p97 is a substrate of Casp6. First, recombinant p97 was readily cleaved by active Casp6 as shown by immunodetection of the cleaved fragments and mass spectrometric identification of the cleavage sites. p97 was previously reported as a putative Casp6 substrate in human primary neurons (Klaiman et al., 2008). However, definitive identification of p97 as a Casp6 substrate was complicated by the activation of Casp3 and Casp7 in these experiments (Klaiman et al., 2008), which theoretically could also cleave p97. Our results now conclusively confirm that p97 is a Casp6 substrate. Second, p97 processing at the Casp6 consensus site, VAPD179, was demonstrated in the hippocampus of AD brain regions also displaying abundant activation of Casp6. Active Casp6 is present in characteristic pathologic features of both sporadic and familial AD: neuritic plaques, neuropil threads, and neurofibrillary tangles (Guo et al., 2004; Albrecht et al., 2007, 2009). Casp6 activation positively correlates with cognitive decline in aged individuals and occurs at all stages of AD (Albrecht et al., 2007). Accordingly, since p97 is a Casp6 substrate, its processing should be evident in neuronal populations displaying Casp6 activation. Indeed, p97 was colocalized with active Casp6 in AD hippocampal neurons and p97D179 immunoreactivity was evident in MCI and AD hippocampal regions, which also stain positive for active Casp6 and Tau cleaved by Casp6.
We also show that p97D179 impairs UPS-mediated protein degradation because overexpression of p97(1-179) in cells leads to the accumulation of well characterized reporters of the ubiquitin fusion degradation and the N-end rule pathways (Dantuma et al., 2000). Although it is unclear how UPS function is impaired at the mechanistic level, p97(1-179) contains a protein-binding fold that is known to interact with ubiquitin (Meyer et al., 2000), ubiquitin-binding proteins (Meyer et al., 2002), and p97 cofactors in membrane fusion (Kondo et al., 1997) and ERAD (Ye et al., 2001). Therefore, p97(1-179) may competitively displace the binding of endogenous p97 to these regulatory proteins. p97 also uses ubiquitin as a recognition signal to segregate membrane tethered protein complexes (Hitchcock et al., 2001; Flierman et al., 2003; Ye et al., 2003; Shcherbik and Haines, 2007), and may thereby mediate the disassembly or disaggregation of ubiquitinated proteins. The competitive binding of p97(1-179) to ubiquitinated proteins may interfere with p97 recruitment and subsequent disaggregation of these complexes. Furthermore, reduction in p97 protein levels as a result of proteolysis in p97(1-179)-expressing cells could also contribute to UPS impairment. Consistently, knockdown of p97 by RNA interference impairs proteasome function in both the ubiquitin-fusion degradation and N-end rule pathways (Wójcik et al., 2006). Thus, in AD, Casp6 may exacerbate the accumulation of ubiquitinated proteins through the proteolytic processing of p97 and the generation of an N-terminal fragment of p97. p97D179 immunostaining in AD neurons was predominantly cytosolic and granular in appearance, which is reminiscent of ubiquitin immunostaining in AD (Hoozemans et al., 2009).
Our study shows that p97 is readily cleaved in the absence of ATP and that ATP binding to p97 protects it from proteolysis. Given that p97 undergoes drastic conformational changes in response to nucleotide binding (Dreveny et al., 2004), our data indicate that ATP-driven conformational changes could alter either the structure or the accessibility of the Casp6 cleavage site within the N-domain. Interestingly, mitochondrial dysfunction and ATP depletion have been implicated in neurodegeneration in AD (Wang et al., 2009a,b; Yao et al., 2009). Accordingly, it remains to be established whether cytosolic ATP depletion may increase the proteolytic susceptibility of p97 to Casp6-mediated proteolysis in vivo. In cells, p97 conformational state is likely influenced by multiple factors, including adaptor protein binding and posttranslational modifications such as ubiquitination (Hitchcock et al., 2001) and phosphorylation (Klein et al., 2005). Other studies have demonstrated that phosphorylation could modulate the susceptibility of a substrate to caspase-mediated processing, either positively or negatively. For example, Bid phosphorylation by casein kinases I and II decrease its sensitivity to Casp8-mediated cleavage (Desagher et al., 2001). Similarly, mutant Huntingtin phosphorylation by cyclin-dependent kinase 5 reduced its cleavage by Casp3 (Luo et al., 2005). In other cases, phosphorylation, such as mitogen-activated protein kinase-mediated phosphorylation of the androgen receptor, enhances substrate cleavage by caspases (LaFevre-Bernt and Ellerby, 2003).
Collectively, our results identify p97 as a substrate of Casp6 in AD and suggest that p97 proteolysis is a novel mechanism of UPS impairment in neurodegeneration. These findings further reinforce the role of Casp6 in affecting multiple neurodegenerative pathways in AD. Considering that Casp6 activation is also linked to increased amyloid β production (LeBlanc, 1995, 1999), tau protein cleavage (Guo et al., 2004), α-tubulin cleavage (Klaiman et al., 2008), and synaptic protein degradation (Guo et al., 2004; Albrecht et al., 2007, 2009), the additional identification of p97 as a Casp6 substrate also links Casp6 to polyubiquitinated protein accumulation in AD.
Footnotes
Part of this work was supported by grants from the Human Frontier Science Program, Genome Canada, and Silicon Kinetics-Genocean to M.L., National Institute on Aging Grant P30AG10161 to D.A.B., and Canadian Institute of Health Research Grant MOP-81146 to A.L.B. We gratefully acknowledge Jennifer Hammond for assistance with the immunohistochemistry, Dr. Steffen Albrecht for assisting in the interpretation of the immunohistochemical data, and Dr. Guy Klaiman for initial guidance with the purification of Casp6. We thank Dr. Guy Salvesen for providing the caspase recombinant expression constructs, Dr. Nico Dantuma for providing the UPS-GFP model cDNA constructs, and Dr. John Bergeron and Dr. Maryam Taheri for generously providing the polyclonal antibody against the C-terminal domain of mammalian p97. We are grateful to Dr. Julie Jodoin, Dr. Jan Schnitzer, Andrea Lee, and Jasmine Ramcharitar for critically reading the manuscript.
References
- Albrecht S, Bourdeau M, Bennett D, Mufson EJ, Bhattacharjee M, LeBlanc AC. Activation of caspase-6 in aging and mild cognitive impairment. Am J Pathol. 2007;170:1200–1209. doi: 10.2353/ajpath.2007.060974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht S, Bogdanovic N, Ghetti B, Winblad B, LeBlanc AC. Caspase-6 activation in familial Alzheimer disease brains carrying amyloid precursor protein or Presenilin I or Presenilin II mutations. J Neuropathol Exp Neurol. 2009;68:1282–1293. doi: 10.1097/NEN.0b013e3181c1da10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J. Natural history of mild cognitive impairment in older persons. Neurology. 2002;59:198–205. doi: 10.1212/wnl.59.2.198. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS. Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology. 2005;64:834–841. doi: 10.1212/01.WNL.0000152982.47274.9E. [DOI] [PubMed] [Google Scholar]
- Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson's and Alzheimer's diseases. J Biol Chem. 2004;279:13256–13264. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol. 2000;18:538–543. doi: 10.1038/75406. [DOI] [PubMed] [Google Scholar]
- Davies JM, Tsuruta H, May AP, Weis WI. Conformational changes of p97 during nucleotide hydrolysis determined by small-angle X-ray scattering. Structure. 2005;13:183–195. doi: 10.1016/j.str.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Desagher S, Osen-Sand A, Montessuit S, Magnenat E, Vilbois F, Hochmann A, Journot L, Antonsson B, Martinou JC. Phosphorylation of bid by casein kinases I and II regulates its cleavage by caspase 8. Mol Cell. 2001;8:601–611. doi: 10.1016/s1097-2765(01)00335-5. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Wertkin A, Kress Y, Ksiezak-Reding H, Yen SH. Ubiquitin immunoreactive structures in normal human brains: distribution and developmental aspects. Lab Invest. 1990a;63:87–99. [PubMed] [Google Scholar]
- Dickson DW, Wertkin A, Mattiace LA, Fier E, Kress Y, Davies P, Yen SH. Ubiquitin immunoelectron microscopy of dystrophic neurites in cerebellar senile plaques of Alzheimer's disease. Acta Neuropathol. 1990b;79:486–493. doi: 10.1007/BF00296107. [DOI] [PubMed] [Google Scholar]
- Dreveny I, Kondo H, Uchiyama K, Shaw A, Zhang X, Freemont PS. Structural basis of the interaction between the AAA ATPase p97/VCP and its adaptor protein p47. EMBO J. 2004;23:1030–1039. doi: 10.1038/sj.emboj.7600139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flierman D, Ye Y, Dai M, Chau V, Rapoport TA. Polyubiquitin serves as a recognition signal, rather than a ratcheting molecule, during retrotranslocation of proteins across the endoplasmic reticulum membrane. J Biol Chem. 2003;278:34774–34782. doi: 10.1074/jbc.M303360200. [DOI] [PubMed] [Google Scholar]
- Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, Jhaveri BS, Karlawish JH, Pestronk A, Smith TW, Tu PH, Watts GD, Markesbery WR, Smith CD, Kimonis VE. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol. 2006;65:571–581. doi: 10.1097/00005072-200606000-00005. [DOI] [PubMed] [Google Scholar]
- García Gil ML, Morán MA, Gómez-Ramos P. Ubiquitinated granular structures and initial neurofibrillary changes in the human brain. J Neurol Sci. 2001;192:27–34. doi: 10.1016/s0022-510x(01)00587-1. [DOI] [PubMed] [Google Scholar]
- Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell. 2006;126:775–788. doi: 10.1016/j.cell.2006.06.046. [DOI] [PubMed] [Google Scholar]
- Guo H, Albrecht S, Bourdeau M, Petzke T, Bergeron C, LeBlanc AC. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer's disease. Am J Pathol. 2004;165:523–531. doi: 10.1016/S0002-9440(10)63317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halawani D, Latterich M. p97: the cell's molecular purgatory? Mol Cell. 2006;22:713–717. doi: 10.1016/j.molcel.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Halawani D, LeBlanc AC, Rouiller I, Michnick SW, Servant MJ, Latterich M. Hereditary inclusion body myopathy-linked p97/VCP mutations in the NH2 domain and the D1 ring modulate p97/VCP ATPase activity and D2 ring conformation. Mol Cell Biol. 2009;29:4484–4494. doi: 10.1128/MCB.00252-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock AL, Krebber H, Frietze S, Lin A, Latterich M, Silver PA. The conserved npl4 protein complex mediates proteasome-dependent membrane-bound transcription factor activation. Mol Biol Cell. 2001;12:3226–3241. doi: 10.1091/mbc.12.10.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer's disease hippocampus. Am J Pathol. 2009;174:1241–1251. doi: 10.2353/ajpath.2009.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch S, Rumpf S. Cdc48 (p97): a “molecular gearbox” in the ubiquitin pathway? Trends Biochem Sci. 2007;32:6–11. doi: 10.1016/j.tibs.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Keck S, Nitsch R, Grune T, Ullrich O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer's disease. J Neurochem. 2003;85:115–122. doi: 10.1046/j.1471-4159.2003.01642.x. [DOI] [PubMed] [Google Scholar]
- Klaiman G, Petzke TL, Hammond J, LeBlanc AC. Targets of caspase-6 activity in human neurons and Alzheimer disease. Mol Cell Proteomics. 2008;7:1541–1555. doi: 10.1074/mcp.M800007-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JB, Barati MT, Wu R, Gozal D, Sachleben LR, Jr, Kausar H, Trent JO, Gozal E, Rane MJ. Akt-mediated valosin-containing protein 97 phosphorylation regulates its association with ubiquitinated proteins. J Biol Chem. 2005;280:31870–31881. doi: 10.1074/jbc.M501802200. [DOI] [PubMed] [Google Scholar]
- Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell. 1999;96:635–644. doi: 10.1016/s0092-8674(00)80574-7. [DOI] [PubMed] [Google Scholar]
- Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, Warren G. p47 is a cofactor for p97-mediated membrane fusion. Nature. 1997;388:75–78. doi: 10.1038/40411. [DOI] [PubMed] [Google Scholar]
- LaFevre-Bernt MA, Ellerby LM. Kennedy's disease: phosphorylation of the polyglutamine-expanded form of androgen receptor regulates its cleavage by caspase-3 and enhances cell death. J Biol Chem. 2003;278:34918–34924. doi: 10.1074/jbc.M302841200. [DOI] [PubMed] [Google Scholar]
- LeBlanc A. Increased production of 4 kDa amyloid beta peptide in serum deprived human primary neuron cultures: possible involvement of apoptosis. J Neurosci. 1995;15:7837–7846. doi: 10.1523/JNEUROSCI.15-12-07837.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc AC. The role of apoptotic pathways in Alzheimer's disease neurodegeneration and cell death. Curr Alzheimer Res. 2005;2:389–402. doi: 10.2174/156720505774330573. [DOI] [PubMed] [Google Scholar]
- LeBlanc A, Liu H, Goodyer C, Bergeron C, Hammond J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer's disease. J Biol Chem. 1999;274:23426–23436. doi: 10.1074/jbc.274.33.23426. [DOI] [PubMed] [Google Scholar]
- Lindsten K, de Vrij FM, Verhoef LG, Fischer DF, van Leeuwen FW, Hol EM, Masucci MG, Dantuma NP. Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J Cell Biol. 2002;157:417–427. doi: 10.1083/jcb.200111034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Vacher C, Davies JE, Rubinsztein DC. Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J Cell Biol. 2005;169:647–656. doi: 10.1083/jcb.200412071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 2000;19:2181–2192. doi: 10.1093/emboj/19.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HH, Wang Y, Warren G. Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1-Npl4. EMBO J. 2002;21:5645–5652. doi: 10.1093/emboj/cdf579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Kondo J, Ihara Y. Ubiquitin is a component of paired helical filaments in Alzheimer's disease. Science. 1987;235:1641–1644. doi: 10.1126/science.3029875. [DOI] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Perry G, Friedman R, Shaw G, Chau V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci U S A. 1987;84:3033–3036. doi: 10.1073/pnas.84.9.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry G, Mulvihill P, Fried VA, Smith HT, Grundke-Iqbal I, Iqbal K. Immunochemical properties of ubiquitin conjugates in the paired helical filaments of Alzheimer disease. J Neurochem. 1989;52:1523–1528. doi: 10.1111/j.1471-4159.1989.tb09203.x. [DOI] [PubMed] [Google Scholar]
- Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005;120:73–84. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Rumpf S, Jentsch S. Functional division of substrate processing cofactors of the ubiquitin-selective Cdc48 chaperone. Mol Cell. 2006;21:261–269. doi: 10.1016/j.molcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Schwartz AL, Ciechanover A. The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu Rev Med. 1999;50:57–74. doi: 10.1146/annurev.med.50.1.57. [DOI] [PubMed] [Google Scholar]
- Shaw G, Chau V. Ubiquitin and microtubule-associated protein tau immunoreactivity each define distinct structures with differing distributions and solubility properties in Alzheimer brain. Proc Natl Acad Sci U S A. 1988;85:2854–2858. doi: 10.1073/pnas.85.8.2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbik N, Haines DS. Cdc48p(Npl4p/Ufd1p) binds and segregates membrane-anchored/tethered complexes via a polyubiquitin signal present on the anchors. Mol Cell. 2007;25:385–397. doi: 10.1016/j.molcel.2007.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry NA, Chapman KT, Nicholson DW. Determination of caspase specificities using a peptide combinatorial library. Methods Enzymol. 2000;322:100–110. doi: 10.1016/s0076-6879(00)22011-9. [DOI] [PubMed] [Google Scholar]
- van Leeuwen FW, de Kleijn DP, van den Hurk HH, Neubauer A, Sonnemans MA, Sluijs JA, Köycü S, Ramdjielal RD, Salehi A, Martens GJ, Grosveld FG, Peter J, Burbach H, Hol EM. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer's and Down patients. Science. 1998;279:242–247. doi: 10.1126/science.279.5348.242. [DOI] [PubMed] [Google Scholar]
- Wang Q, Song C, Yang X, Li CC. D1 ring is stable and nucleotide-independent, whereas D2 ring undergoes major conformational changes during the ATPase cycle of p97-VCP. J Biol Chem. 2003;278:32784–32793. doi: 10.1074/jbc.M303869200. [DOI] [PubMed] [Google Scholar]
- Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J Neurochem. 2009a;109(Suppl 1):153–159. doi: 10.1111/j.1471-4159.2009.05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009b;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- Wójcik C, Rowicka M, Kudlicki A, Nowis D, McConnell E, Kujawa M, DeMartino GN. Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol Biol Cell. 2006;17:4606–4618. doi: 10.1091/mbc.E06-05-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol. 2003;162:71–84. doi: 10.1083/jcb.200302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Goodyer C, LeBlanc A. Selective and protracted apoptosis in human primary neurons microinjected with active caspase-3, -6, -7, and -8. J Neurosci. 2000;20:8384–8389. doi: 10.1523/JNEUROSCI.20-22-08384.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]