

Abstract
Homogeneously glycosylated proteins are important targets for fundamental research and for biopharmaceutical development. The use of unnatural protein–glycan linkages bearing structural similarity to their native counterparts can accelerate the synthesis of glycoengineered proteins. Here we report an approach toward generating homogeneously glycosylated proteins that involves chemical attachment of aminooxy glycans to recombinantly produced proteins via oxime linkages. We employed the recently introduced aldehyde tag method to obtain a recombinant protein with the aldehyde-bearing formylglycine residue at a specific site. Complex aminooxy glycans were synthesized using a new route that features N-pentenoyl hydroxamates as key intermediates that can be readily elaborated chemically and enzymatically. We demonstrated the method by constructing site-specifically glycosylated variants of the human growth hormone.
Introduction
Precise molecular control of protein glycosylation would advance the development of biopharmaceuticals as well as fundamental studies of glycosylation-dependent biological processes.1−4 Biological methods on their own have failed to achieve this long sought-after goal, as cellular biosynthetic pathways produce glycoproteins as a heterogeneous mixture of glycoforms. Glycoprotein structures with optimal bioactivity are difficult to identify from such complex mixtures and impossible to generate exclusively using conventional cell hosts. The tools of synthetic biology have been harnessed to construct heterologous glycan biosynthetic pathways in simple host cells.5,6 While these systems have produced glycoproteins that are globally modified with a single glycan type, no recombinant expression system can yet produce proteins with discrete glycans at individual sites and with complete homogeneity.
Chemical synthesis is currently the only plausible approach to achieve homogeneously glycosylated proteins bearing tailored glycans at specific sites.7−10 Several groups have successfully combined frontier protein ligation techniques with chemical or enzymatic glycan and glycopeptide synthesis methods to construct glycoproteins that duplicate natural structures.11−13 The valor of these efforts should not be underestimated since the difficulties inherent to glycan and protein syntheses amplify each other when both biopolymers coexist in the same synthetic target.14,15 A more practical, though less biologically authentic, approach involves conjugating glycans to recombinant proteins via unnatural but synthetically tractable linkages.16−18 Site-specificity in the modification can be ensured by outfitting the protein with a noncanonical amino acid side chain capable of chemoselective ligation with an appropriately functionalized glycan.19−27
We and others have explored the oxime-forming reaction of aminooxy glycans with peptide-associated aldehydes or ketones as a means of site-specific glycosylation.25,28,29 This chemistry proceeds in mildly acidic aqueous buffers that are compatible with most proteins and poses little risk of side reactivity with endogenous protein or glycan functionalities.30,31 Despite promising initial work, however, the generality of the approach remains limited by two challenges: (1) the need for a practical means of introducing aldehydes or ketones into recombinant proteins using standard expression systems, and (2) a reliable route to synthesize higher-order aminooxy glycans.
The recently developed aldehyde tag methodology answers the first challenge, as it enables site-specific introduction of aldehyde-bearing formylglycine (fGly) residues into any recombinant protein of interest using conventional Escherichia coli(32) or mammalian(33) expression systems. The aldehyde tag is a five-residue consensus sequence (CxPxR, where x is variable) that is recognized by the formylglycine generating enzyme (FGE).34−36 Cotranslationally, FGE oxidizes the genetically encoded cysteine residue to fGly, thereby providing a uniquely reactive site for chemical modification. Although FGE is native to most cell types, coexpression of additional FGE alongside a recombinant protein substrate ensures high Cys-to-fGly conversion yields. Use of the aldehyde tag method simply requires cloning the consensus sequence into the protein’s gene at the desired site of modification, followed by protein production in FGE-expressing cells.
In addition to its practicality, the aldehyde tag method benefits from the resemblance of the fGly side chain to naturally glycosylated amino acid side chains. Most secreted and cell-surface vertebrate glycoproteins are modified at Asn residues with N-glycans or at Ser/Thr residues with O-glycans (Figure 1A). We envisioned oxime-linked glycoproteins deriving from conjugation of the fGly aldehyde with synthetic aminooxy glycans as illustrated in Figure 1B. Glycan–protein linkages comprising fGly-derived oximes are close structural mimics of N-glycan linkages and just one atom longer than O-glycan linkages.
Figure 1.

Natural glycan linkages resemble the linkage produced by protein glycoengineering via the reaction of aminooxy sugars with the aldehyde tag. (A) Linkages naturally found in the most abundant mammalian N- and O-linked glycan structures. (B) The linkage formed upon the reaction of an aminooxy glycan with the aldehyde of an fGly-containing protein. The resulting oxime side chain bears high structural similarity to the natural glycoprotein attachments.
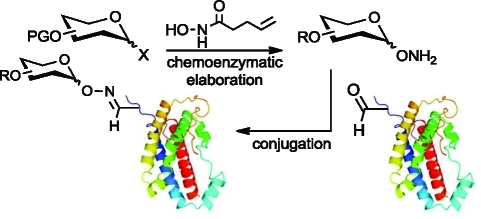
To capitalize on the aldehyde tag as a tool for site-specific protein glycosylation, we sought to answer the second challenge—a practical and general route to synthesize complex aminooxy glycans. Here we report on the use of glycosyl N-pentenoyl hydroxamates as intermediates in the synthesis of higher-order aminooxy glycans. These analogues function well in both chemical and enzymatic glycosylation reactions, enabling the efficient and high-yielding production of both simple and elaborated aminooxy glycans as substrates for protein conjugation. The new synthetic approach, together with the aldehyde tag technology, enabled the generation of recombinant proteins modified site-specifically with homogeneous glycans.
Results and Discussion
Evaluation of N-Hydroxypentenamide as a Masked Aminooxy Group
In previous work, aminooxy glycosides were synthesized by displacement of a glycosyl halide with N-hydroxysuccinimide (NHS) or N-hydroxyphthalimide (NHPht).29,37,38 Reaction yields were acceptable using monosaccharide substrates but, in our hands, fell precipitously with increasing glycan complexity. Additionally, glycosylations of NHS and NHPht with other donors, such as glycosyl acetimidates and thioglycosides, were low yielding, even with monosaccharide substrates. In addition to falling short as glycosyl acceptors, NHS and NHPht were inadequate aminooxy protecting groups. They were labile to basic and reductive conditions, which undermined their use during complex glycan synthesis. Moreover, we envisioned using one-pot multienzyme methods(39) to convert simple aminooxy glycans into more complex structures. However, such reactions would require protection of the aminooxy group to avoid oxime formation with the latent aldehydes or ketones associated with free sugars. The lability of the NHS and NHPht groups undermined our efforts to deprotect late-stage glycan intermediates without loss of the aminooxy protecting group.
Therefore, we sought to identify an alternative aminooxy protecting group that possessed better chemical stability overall but was readily removable under mild conditions that are compatible with free sugars. Ideally, the nucleophilicity of the masked aminooxy group would be superior to that of the N-hydroxyimides as well, thus enhancing reactivity as a glycosyl acceptor. After screening a variety of options, we found the N-pentenoyl protecting group to be an excellent candidate. Originally introduced by Fraser-Reid and co-workers,40−42 the pentenoyl amide is stable to many reaction conditions and is removed by gentle treatment with aqueous iodine. Despite its potential as a synthetic substrate, N-pentenoyl hydroxamic acid (1, Table 1, also termed N-hydroxypentenamide, which we abbreviate NHPent) has seldom been utilized in synthesis(43) and, to our knowledge, never as a masked aminooxy group.
Table 1. Reaction of Various Glycosyl Donors with N-Pentenoyl Hydroxamic Acid 1.

Reactions corresponding to entries 1–7 were conducted in CH2Cl2 (unless specified) at 0 °C, and those corresponding to entries 8–13 were performed at −20 °C.
Isolated yields after column chromatography.
Ratio determined by isolated yields or 1H NMR spectroscopy.
To determine the scope of compound 1's reactivity as a glycosyl acceptor, we subjected it to glycosylation reactions using donor substrates that included glycosyl bromides and fluorides, thioglycosides, and glycosyl acetimidates (Table 1). Most substrates gave acceptable yields of NHPent glycoside product. The highest yields were obtained using glycosyl N-phenyl trifluoroacetimidates,44−46 as previously observed in the syntheses of glycosyl amides(44) and hydroxamates.47,48 By contrast, the less stable glycosyl trichloroacetimidates gave low yields in glycosylation reactions with 1 under similar conditions (data not shown).
We anticipated that the mild conditions reported for cleavage of N-pentenoyl amides would be compatible with unprotected glycans. To confirm that such conditions can cleave N-alkoxypentenamides without harm to free sugars, we performed model reactions on the test substrate N-pentenoyl aminooxy lactose (15, prepared from the known disaccharide aminooxy lactose(28) as shown in Scheme S1). Treatment of N-pentenoyl aminooxy lactose with 3 equiv of I2 in 1:1 tetrahydrofuran (THF)/H2O as reported(41) resulted in iodocyclization of the NHPent group, but hydrolysis of the iodoimidate intermediate to afford the free aminooxy disaccharide was sluggish. Fortunately, the use of nonaqueous solvent mixtures and acid catalysis, as shown previously,49,50 alleviated this problem. We identified the optimal deprotection conditions to be 3 equiv of I2 in wet MeCN/MeOH with 0.1% formic acid (FA) at room temperature, which afforded quantitative deprotection of N-pentenoyl aminooxy lactose in under 1 h.
Chemical Synthesis of Aminooxy Lewis x
The utility of NHPent glycosides as substrates for complex aminooxy glycan synthesis was demonstrated with the construction of the trisaccharide Lewis x (Galβ1,4(Fucα1,3)GlcNAc), a leukocyte and stem cell marker (Scheme 1).51,52 We began the synthesis with the coupling of known trichloroacetimidate 16 to thioglycoside 17 to generate the lactosamine precursor 18 following related, published routes.53,54 As seen with similarly protected glucosamine derivatives,53,54 only glycosylation at the more reactive 4-hydroxyl of 17 was observed. The fucose residue was introduced in the form of 19, which was outfitted with a p-methoxybenzyl (PMB) ether at the 2-position to enhance cis-glycosylation. Similar to a previous report,(55) TfOH-catalyzed coupling of 19 with the free C-3 hydroxyl of 18 proceeded smoothly to afford 20. The PMB group was removed with 10% trifluoroacetic acid, thereby avoiding reductive or oxidative conditions that are incompatible with the pentenoyl and thioglycoside groups, and the 2-hydroxyl group of the fucose residue was acetylated to afford 21.
Scheme 1. Chemical Synthesis of Aminooxy Lewis x.

Motivated by the results in Table 1, we converted thioglycoside 21 to the corresponding glycosyl N-phenyl trifluoroacetimidate, 22, for coupling with compound 1.(46) The glycosylation reaction was conducted with 1 equiv of TMSOTf in dichloromethane at −20 °C to afford N-hydroxypentenoyl trisaccharide 23. The Troc protecting group was removed with Zn in acidic MeCN, and the amino group was acetylated. Notably, we observed no reduction of the N–O bond during the Zn reduction step, in sharp contrast to our previous experience with NHPth glycosides. Removal of the TBDPS and acetyl groups of 24 proceeded in high yield and without damage to the N-hydroxypentenamide as well. Finally, aminooxy Lewis x was obtained by treatment of 25 with I2 in acidic MeCN/MeOH.
Chemoenzymatic Synthesis of Complex Aminooxy Glycans
Enzymatic elaboration of synthetic core structures is an increasingly popular strategy for complex glycan assembly.39,56 Enzymes provide tight stereo- and regiochemical control and obviate the need for intermediate protecting group manipulations and harsh chemical conditions. The impact of enzymatic glycosylation methods is most evident in cases where the chemical counterpart is notoriously difficult, such as in the addition of terminal sialic acid residues to a complex glycan.(57) Given the biological importance of sialosides,(58) we sought to construct sialylated aminooxy glycans as substrates for glycoprotein engineering.
Trisaccharides 29 and 30 (Scheme 3), termed aminooxy 2,6- and 2,3-sialyllactose, respectively, were chosen as initial targets with which to evaluate the performance of NHPent glycosides as substrates for enzymatic sialylation. We used the one-pot, three-enzyme system introduced by Chen and co-workers59,60 to append sialic acid to N-pentenoyl aminooxy lactose (15, Scheme 2). This methodology allows for the in situ generation of costly cytidine 5′-monophosphate (CMP)-sialic acid from ManNAc, pyruvate, and cytidine 5′-triphosphate (CTP) by the combined use of E. coli aldolase and a CMP-sialic acid synthase, NmCSS.(61) In the same pot, bacterial α-2,6-sialyltransferase (Pd2,6ST) or α-2,3-sialyltransferase (PmST1) transferred sialic acid to 15, affording NHPent glycosides 27 and 28, respectively, in excellent yields. Notably, free aminooxy glycans would not be viable substrates for this procedure, as the aminooxy group would undergo side reactions with free reducing sugars as well as pyruvate. Finally, we employed our previously optimized deprotection conditions to obtain trisaccharides 29 and 30 (Scheme 3).
Scheme 3. Deprotection of NHPent on Sialosides.

Scheme 2. Enzymatic Sialylation of N-Pentenoyl Aminooxy Lactose.

A higher level of complexity was embodied in the tetrasaccharide sialyl Lewis x (Siaα2,3Galβ1,4(Fucα1,3)GlcNAc, abbreviated SLex), a glycan known for its involvement in cancer progression and leukocyte homing.62,63 We envisioned generating aminooxy-functionalized SLex by chemical synthesis of an NHPent-modified disaccharide core, followed by enzymatic installation of the terminal sialic acid and fucose residues. Toward this end, compound 18 was transformed into NHPent lactosamine derivative 33 by acetylation of its one free hydroxyl group, conversion of the thioglycoside to the N-phenyl trifluoroacetimidate, and glycosylation with compound 1 (Scheme 4). Deprotection and N-acetylation gave N-acetyllactosamine NHPent glycoside 35 as a substrate for enzymatic glycosylation. The α-2,3-sialoside 36 was generated enzymatically as described above for compound 28. After purification by size exclusion chromatography, the fucose residue was installed using the one-pot, two-enzyme system described by Wu and co-workers.(64) Briefly, guanosine 5′-diphosphate (GDP)-fucose was generated in situ from adenosine triphosphate (ATP), guanosine 5′-triphosphate (GTP), and fucose by the Bacteroides fragilis fucose kinase phosphorylase (FKP). Subsequent glycosylation using Helicobacter pylori α-1,3-fucosyltransferase (Hp1,3FT) generated tetrasaccharide 37. Finally, deprotection of 37 with I2 in acidic MeCN/MeOH gave aminooxy SLex38 in 65% isolated yield.
Scheme 4. Chemoenzymatic Synthesis of Aminooxy SLex.

Conjugation of Aminooxy Glycans to Aldehyde-Tagged Human Growth Hormone
With a route to complex aminooxy glycans established, we sought to combine the synthetic sugars with recombinant aldehyde-tagged proteins as shown schematically in Figure 2A. As a protein substrate, we used a previously reported construct of the human growth hormone (hGH) that contains a C-terminal aldehyde tag as well as an N-terminal His tag. The native protein is a well-established biotherapeutic used in the treatment of human developmental diseases among others.65,66 Site-specific chemical modification of hGH is a potential route to improving its pharmacokinetic properties.67,68 Indeed, an artificially glycosylated hGH variant was recently shown to have a prolonged serum half-life in mice.(69)
Figure 2.

Generation of site-specifically glycosylated hGH. (A) Aldehyde-tagged hGH was reacted with synthetic aminooxy glycans to yield the oxime-glycosylated conjugates. (B) Western blot of aldehyde-tagged hGH (lane 1) or the C→A mutant (lane 2) after reaction with aminooxy lactose S1 (5% MeCN, 0.02% FA, 0.26 mg/mL aldehyde-tagged hGH, 1 mM S1, 16 h, 37 °C). The blot was probed with SBA-FITC (top), and total protein was detected by Ponceau stain (bottom). (C) ESI-MS spectrum of the crude reaction mixture showing ions in the 20+ and 21+ charge states. Ions corresponding to aldehyde-tagged hGH (hGH) and glycosylated hGH (hGH+Lac) are shown with arrows. (D) Western blot of aldehyde-tagged hGH (lane 1) or the C→A mutant (lane 2) after reaction with aminooxy SLex38 (50 mM NaCit, pH 3.5, 0.26 mg/mL aldehyde-tagged hGH, 1 mM 38, 16 h, 37 °C). The blot was probed with an anti-SLex antibody KM93 followed by a horseradish peroxidase (HRP)-conjugated secondary antibody (top), and then stripped and reprobed with AAL-biotin followed by a FITC-conjugated antibiotin antibody (middle). Total protein loading was confirmed by Ponceau staining (bottom). (E) ESI-MS spectrum of the SLex crude reaction mixture showing ions in the 20+ and 21+ charge states. Ions corresponding to aldehyde-tagged hGH (hGH) and glycosylated hGH (hGH+SLex) are shown with arrows.
We produced aldehyde-tagged hGH with >95% Cys-to-fGly conversion in E. coli with coexpression of the FGE from Mycobacterium tuberculosis.(32) The protein was purified by chromatography on Ni-NTA agarose and then reacted with aminooxy lactose (S1) in 5% aqueous MeCN with 0.02% FA (pH ∼3.8). The formation of the lactose glycoconjugate was confirmed by Western blot probing with the galactose-binding lectin, soybean agglutinin conjugated to fluorescein isothiocyanate (SBA-FITC) (Figure 2B). Significant SBA reactivity was observed only for products of aldehyde-tagged protein conjugation. A control conjugation reaction using an hGH construct in which the Cys residue required for the aldehyde installation was mutated to Ala (C→A) produced no SBA-reactive product. This result confirmed the necessity of the Cys-derived fGly residue for site-specific chemical glycosylation with aminooxy lactose. Further, we analyzed the oxime conjugate by electrospray ionization mass spectrometry (ESI-MS) (Figure 2C). Ions corresponding to glycosylated (hGH+Lac) and unreacted (hGH) aldehyde-tagged hGH were observed, the relative intensities of which indicated a reaction yield of ∼81%. The protein glycoconjugate was found to be highly stable when stored at −20 °C, and <10% hydrolysis of the conjugate was observed after 8 days at 4 °C (Table S1).
We performed a similar reaction using the more complex glycan aminooxy SLex (38). The formation of the glycosylated hGH product was confirmed by Western blot probing with a SLex-specific antibody as well as with the fucose-binding lectin, Aleuria aurantia lectin (AAL), conjugated to biotin (Figure 2D). Using the above conjugation conditions (5% MeCN, 0.02% FA), the yield of glycosylated hGH as determined by ESI-MS was a modest 20–25%. However, use of a sodium citrate (NaCit) buffer at pH 3.5 improved the conjugation efficiency to ∼64%. Despite the acidity of the reaction buffer, we did not detect any loss of the potentially labile sialic acid and fucose moieties or any damage to the aldehyde-tagged protein.
Conclusion
In summary, the facile generation of homogeneous glycoproteins was made possible by an improved route to aminooxy glycans featuring glycosyl N-pentenoyl hydroxamate intermediates, combined with the genetically encoded aldehyde tag. The versatility of the aldehyde tag with respect to expression system, protein target, and site of aldehyde placement makes this approach broadly useful for protein glycoengineering. In principle, multiple aldehyde tags can be introduced into a single protein, allowing for the production of multivalent glycoforms, a work currently in progress.
The method joins a growing toolkit for site-specific modification of proteins with synthetic glycans, which combined could produce myriad protein glycoforms with discrete glycans at different positions. Other chemoselective ligation reactions, such as azide–alkyne cycloadditions and disulfide exchange reactions, have proven useful for modifying a single protein with multiple moieties.(19) Oxime formation is orthogonal to these and other chemoselective ligation reactions (thiol–ene additions, olefin metathesis, Suzuki couplings, etc.). Exploring the tandem and parallel use of these reactions for multiplexed protein modification is a subject of future interest.
Experimental Section
Phenyl (2,3,4,6-Tetra-O-benzoyl-β-d-galactopyranosyl)-(1→4)-6-O-(tert-butyldiphenylsilyl)-3-O-acetyl-2-deoxy-2-(2,2,2-trichloroethoxy)carbonylamino-1-thio-β-d-glucopyranoside (31)
A solution of disaccharide 18 (400 mg, 0.32 mmol) in pyridine/Ac2O (2:1, v:v; 4.5 mL) was stirred for 3 h at room temperature under N2. Upon completion, the reaction was concentrated and coevaporated with toluene to remove excess Ac2O and AcOH. The crude residue was purified by silica flash chromatography (Hex/EtOAc gradient) to obtain fully protected disaccharide 31 as a white powder (400 mg, 97%): 1H NMR (500 MHz, CDCl3) δ 8.18 (d, J = 7.4 Hz, 2H), 8.07 (d, J = 7.4 Hz, 2H), 7.93–7.79 (m, 6H), 7.68–7.49 (m, 12H), 7.47–7.34 (m, 5H), 7.33–7.19 (m, 5H), 7.09 (t, J = 7.5 Hz, 2H), 7.03 (t, J = 7.3 Hz, 1H), 6.85 (t, J = 7.7 Hz, 2H), 6.69 (d, J = 9.0 Hz, 1H), 6.02 (d, J = 2.9 Hz, 1H), 5.81 (dd, J = 10.1, 8.2 Hz, 1H), 5.58 (dd, J = 10.4, 3.1 Hz, 1H), 5.50 (t, J = 9.8 Hz, 1H), 5.27 (d, J = 8.0 Hz, 1H), 5.11 (d, J = 10.4 Hz, 1H), 4.90 (q, J = 12.1 Hz, 2H), 4.74 (dd, J = 11.2, 4.6 Hz, 1H), 4.55–4.39 (m, 2H), 4.30–4.23 (m, 1H), 4.09 (q, J = 10.2 Hz, 1H), 3.95 (d, J = 11.2 Hz, 1H), 3.77 (d, J = 11.0 Hz, 1H), 3.40 (d, J = 9.7 Hz, 1H), 2.24 (s, 3H), 1.09 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 171.54, 166.00, 165.56, 165.32, 164.45, 154.59, 136.05, 135.67, 135.29, 133.70, 133.52, 133.33, 133.17, 132.91, 131.69, 130.86, 130.25, 129.84, 129.77, 129.74, 129.51, 129.13, 128.77, 128.69, 128.41, 128.29, 128.22, 128.16, 127.78, 126.74, 100.10, 95.78, 85.76, 78.61, 77.41, 74.52, 74.18, 73.61, 72.02, 71.43, 69.90, 68.22, 61.95, 60.96, 54.87, 26.76, 21.24, 19.35; HRMS (ESI) calcd for C67H65NO16Cl3SSi [M+H]+m/z = 1304.2853, found 1304.2883.
N-Hydroxypent-4-enamide (2,3,4,6-Tetra-O-benzoyl-β-d-galactopyranosyl)-(1→4)-6-O-(tert-butyldiphenylsilyl)-3-O-acetyl-2-deoxy-2-(2,2,2-trichloroethoxy)carbonylamino-β-d-glucopyranoside (33)
Thioglycoside 31 (290 mg, 0.22 mmol) was dissolved in acetone/CH2Cl2/H2O (15:1:1, v:v; 6 mL), and N-iodosuccinimide (NIS) (75 mg, 0.33 mmol) was added while stirring at room temperature. The reaction was monitored by thin-layer chromatography (TLC) (2:1, Hex/EtOAc), and N-bromosuccinimide was added in 2 equiv portions until the reaction was complete. Upon full hydrolysis, the resulting hemiacetal was diluted in CH2Cl2 and washed sequentially with Na2S2O3 solution, NaHCO3 solution, and brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The resulting residue was dissolved in CH2Cl2 (3 mL) and Cs2CO3 (150 mg, 0.44 mmol) was added, followed by N-phenyl trifluoroacetimidoyl chloride (72 μL, 0.44 mmol) dropwise. The reaction mixture was stirred for 12 h at room temperature under N2, after which it was diluted with CH2Cl2, filtered, and concentrated under vacuum. The crude residue was purified by silica chromatography to give the trifluoroacetimidate 32 as a yellow oil (267 mg, 87%): 1H NMR (500 MHz, CDCl3) δ 8.16–8.03 (m, 4H), 7.87–7.68 (m, 7H), 7.68–7.31 (m, 14H), 7.19 (dddd, J = 28.0, 16.4, 10.1, 3.9 Hz, 7H), 6.98 (dt, J = 30.6, 15.6 Hz, 1H), 6.71 (dd, J = 23.5, 7.7 Hz, 1H), 6.00–5.92 (m, 1H), 5.83–5.73 (m, 1H), 5.60–5.40 (m, 1H), 5.33 (t, J = 9.9 Hz, 1H), 5.25 (dd, J = 22.4, 14.4 Hz, 1H), 5.18 (dt, J = 19.0, 9.4 Hz, 1H), 4.76 (dt, J = 28.6, 14.3 Hz, 1H), 4.63–4.51 (m, 2H), 4.47–4.35 (m, 1H), 4.29–4.17 (m, 1H), 4.13 (dt, J = 13.7, 7.1 Hz, 1H), 4.05–3.69 (m, 2H), 3.58–3.26 (m, 1H), 2.20–1.92 (m, 3H), 1.16–1.02 (m, 9H); 13C NMR (126 MHz, CDCl3) δ 173.12, 166.12, 165.51, 165.50, 164.72, 154.46, 136.03, 135.47, 133.88, 133.84, 133.62, 133.45, 133.35, 130.54, 130.02, 129.92, 129.88, 129.70, 129.58, 129.17, 129.03, 128.87, 128.73, 128.55, 128.44, 128.36, 127.96, 100.52, 95.48, 74.75, 73.37, 73.24, 72.20, 71.66, 69.90, 68.23, 62.14, 60.53, 54.23, 26.90, 20.97, 19.50. A mixture of trifluoroacetimidate 32 (257 mg, 0.186 mmol) and N-pentenoyl hydroxamic acid 1 (32 mg, 0.28 mmol) was dried by coevaporation with anhydrous toluene and left under high vacuum. To the dried mixture was added 3 Å MS with stirring in CH2Cl2 (4 mL) for 1 h at room temperature under N2. The solution was cooled to −20 °C, and TMSOTf (34 μL, 0.19 mmol) was added dropwise. The reaction was allowed to warm to room temperature over 1.5 h and was quenched with triethylamine, filtered, and concentrated under vacuum. The resulting residue was purified by silica flash chromatography (toluene/acetone gradient) to obtain N-hydroxypentenoyl disaccharide 33 as a white solid (160 mg, 66%): 1H NMR (500 MHz, CDCl3) δ 8.45 (s, 1H), 8.15–8.02 (m, 4H), 7.83–7.74 (m, 4H), 7.74–7.65 (m, 4H), 7.60 (dt, J = 21.6, 6.4 Hz, 2H), 7.54–7.36 (m, 12H), 7.25–7.20 (m, 2H), 7.20–7.16 (m, 2H), 5.95 (d, J = 3.2 Hz, 1H), 5.82–5.69 (m, 2H), 5.69–5.61 (m, 1H), 5.48 (dd, J = 10.3, 3.2 Hz, 1H), 5.20 (t, J = 9.3 Hz, 1H), 5.12 (d, J = 8.0 Hz, 1H), 5.01 (d, J = 16.5 Hz, 1H), 4.96 (d, J = 10.3 Hz, 1H), 4.86 (d, J = 12.0 Hz, 1H), 4.68 (t, J = 9.1 Hz, 2H), 4.53 (d, J = 6.4 Hz, 2H), 4.34 (t, J = 9.3 Hz, 1H), 4.17 (t, J = 6.0 Hz, 1H), 3.94–3.77 (m, 3H), 3.27 (d, J = 9.4 Hz, 1H), 2.35–2.15 (m, 4H), 2.12 (s, 3H), 1.08 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 171.32, 170.85, 166.00, 165.51, 165.43, 165.21, 164.72, 136.58, 136.08, 136.06, 136.02, 135.95, 135.48, 135.44, 133.81, 133.51, 133.46, 133.43, 133.29, 130.54, 130.15, 130.09, 130.04, 129.98, 129.93, 129.85, 129.65, 129.49, 128.91, 128.83, 128.68, 128.64, 128.60, 128.45, 128.41, 128.32, 128.11, 127.85, 127.77, 115.81, 100.22, 95.41, 75.63, 74.87, 74.71, 73.14, 71.91, 71.50, 69.83, 68.08, 62.01, 60.64, 54.00, 53.53, 28.98, 26.86, 20.95, 19.42; HRMS (ESI) calcd for C66H67N2O18Cl3Si [M+H]+m/z = 1309.3296, found 1309.3315.
N-Hydroxypent-4-enamide (2,3,4,6-Tetra-O-benzoyl-β-d-galactopyranosyl)-(1→4)-6-O-(tert-butyldiphenylsilyl)-3-O-acetyl-2-deoxy-2-acetamido-β-d-glucopyranoside (34)
To a solution of N-hydroxypentenoyl disaccharide 33 (140 mg, 0.11 mmol) in 10% FA/MeCN (3 mL) was added activated Zn (500 mg), and the mixture was stirred for 3 h. The suspension was filtered to remove catalyst, concentrated under vacuum, and coevaporated with toluene to remove excess FA. The crude free amine was dissolved in anhydrous MeOH (3 mL), followed by dropwise addition of Ac2O (55 μL, 0.54 mmol) and N,N-diisopropylethylamine (DIPEA) (22 μL, 0.13 mmol) at 0 °C under N2. After 1.5 h, the reaction was allowed to warm to room temperature, diluted with MeOH, and concentrated under vacuum. The resulting residue was purified by silica flash chromatography (toluene/acetone gradient) to give the N-acetylated disaccharide 34 as a white solid (97 mg, 77%): 1H NMR (500 MHz, CDCl3) δ 9.32 (s, 1H), 8.13–8.03 (m, 4H), 7.82–7.73 (m, 4H), 7.72–7.67 (m, 2H), 7.66–7.54 (m, 6H), 7.52–7.45 (m, 8H), 7.41–7.37 (m, 2H), 7.23 (t, J = 7.8 Hz, 3H), 7.17 (t, J = 7.8 Hz, 2H), 6.19 (d, J = 8.4 Hz, 1H), 5.96 (d, J = 3.1 Hz, 1H), 5.82–5.69 (m, 2H), 5.48 (dd, J = 10.4, 3.4 Hz, 1H), 5.11 (dd, J = 17.2, 7.9 Hz, 2H), 5.00 (dd, J = 17.2, 1.3 Hz, 1H), 4.94 (d, J = 10.2 Hz, 1H), 4.60 (d, J = 8.5 Hz, 1H), 4.58–4.47 (m, 2H), 4.36 (t, J = 9.1 Hz, 1H), 4.20–4.10 (m, 2H), 3.88 (q, J = 11.2 Hz, 2H), 3.26 (d, J = 9.4 Hz, 1H), 2.38–2.15 (m, 4H), 2.14 (s, 3H), 2.02 (s, 3H), 1.06 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 172.53, 171.81, 170.88, 166.04, 165.46, 164.88, 137.93, 136.75, 136.06, 135.74, 135.43, 133.81, 133.56, 133.43, 133.40, 131.23, 130.58, 130.15, 129.94, 129.89, 129.85, 129.54, 129.11, 128.98, 128.82, 128.73, 128.65, 128.52, 128.41, 128.30, 128.26, 127.78, 125.38, 115.62, 102.03, 100.27, 75.89, 72.99, 72.35, 71.86, 71.59, 69.83, 68.17, 62.07, 60.56, 52.23, 32.87, 29.04, 26.82, 23.35, 21.53, 21.03, 19.41; HRMS (ESI) calcd for C65H68N2O17Si [M+H]+m/z = 1177.4360, found 1177.4396.
N-Hydroxypent-4-enamide (β-d-Galactopyranosyl)-(1→4)-2-deoxy-2-acetamido-β-d-glucopyranoside (35)
A solution of N-hydroxypentenoyl disaccharide 34 (94 mg, 0.080 mmol) in THF (4 mL) was cooled to 0 °C, and tetrabutylammonium fluoride (1 M in THF, 160 μL) was added dropwise under N2. The mixture was allowed to warm to room temperature and stirred for 18 h, after which it was diluted with EtOAc and concentrated under reduced pressure. The resulting residue was dissolved in MeOH (4 mL), and NaOMe (25% in MeOH, 40 μL) was added dropwise with stirring at room temperature. After 8 h, the reaction was neutralized with Dowex 50W-X8 (H+) and filtered to remove resin. The crude product was concentrated under vacuum and purified by silica column chromatography (10–25% MeOH/CH2Cl2). The purified residue was filtered to remove trace silica and lyophilized to yield the deprotected N-hydroxypentenoyl LacNAc 35 as a white powder (32 mg, 84%): 1H NMR (500 MHz, D2O) δ 5.80 (ddt, J = 12.8, 10.3, 6.5 Hz, 1H), 5.10–4.98 (m, 2H), 4.75 (d, J = 8.8 Hz, 1H), 4.44 (d, J = 7.8 Hz, 1H), 3.98–3.92 (m, 1H), 3.92–3.80 (m, 3H), 3.77–3.66 (m, 5H), 3.63 (dd, J = 10.0, 3.4 Hz, 1H), 3.61–3.54 (m, 1H), 3.50 (dd, J = 9.8, 7.9 Hz, 1H), 2.32 (dd, J = 13.1, 6.5 Hz, 2H), 2.28–2.21 (m, 2H), 2.02 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 174.72, 172.85, 136.46, 115.89, 103.79, 102.82, 77.78, 75.33, 75.06, 72.45, 72.22, 70.91, 68.51, 61.00, 59.77, 52.90, 31.72, 28.81, 22.18; HRMS (ESI) calcd for C19H32N2O12 [M+H]+m/z = 481.2028, found 481.2036.
N-Hydroxypent-4-enamide (5-Acetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-β-d-galactopyranosyl-(1→4)-2-deoxy-2-acetamido-β-d-glucopyranoside (36)
To N-pentenoyl aminooxy LacNAc 35 (21 mg, 0.044 mmol) were added N-acetylmannosamine (15 mg, 0.066 mmol), sodium pyruvate (24 mg, 0.22 mmol), and CTP·Na (37 mg, 0.066 mmol), and they were dissolved in H2O (3 mL). A concentrated stock of Tris-HCl buffer pH 8.5 with MgCl2 was added to a final concentration of 100 mM Tris, 20 mM MgCl2. Recombinant E. coli K12 sialic acid aldolase (4 U), Neusserua meningitidis CMP-sialic acid synthetase (4 U), and Pasteurella multocida α-2,3-sialyltransferase (2 U) were added, followed by H2O, to bring the volume to 4 mL. The reaction mixture was incubated at 37 °C for 2 h, followed by shaking at room temperature for 14 h. The reaction was monitored by TLC (4:2:1 EtOAc/MeOH/H2O), and upon completion, calf alkaline phosphatase was added to remove remaining nucleotide phosphate. After further incubation at 37 °C for 2 h, the reaction mixture was quenched with cold MeOH (4 mL) and incubated on ice for 20 min. The mixture was centrifuged, precipitates were removed, and the solution concentrated under vacuum. The resulting residue was passed through a BioGel P-2 size exclusion column and eluted with ddH2O to obtain 36 (28.9 mg, 86%) as a white, fluffy powder after lyophilization: 1H NMR (600 MHz, D2O) δ 5.80 (ddt, J = 16.9, 10.3, 6.6 Hz, 1H), 5.09–4.99 (m, 2H), 4.74 (d, J = 8.8 Hz, 1H), 4.52 (d, J = 7.9 Hz, 1H), 4.08 (dd, J = 9.9, 3.1 Hz, 1H), 3.96 (app d, J = 10.7 Hz, 1H), 3.92 (d, J = 3.0 Hz, 1H), 3.88–3.79 (m, 5H), 3.76–3.70 (m, 3H), 3.69–3.64 (m, 3H), 3.62–3.52 (m, 5H), 2.73 (dd, J = 12.4, 4.6 Hz, 1H), 2.31 (dd, J = 13.5, 6.7 Hz, 2H), 2.24 (app t, J = 7.0 Hz, 2H), 2.02 (s, 3H), 2.00 (s, 3H), 1.77 (t, J = 12.1 Hz, 1H); 13C NMR (151 MHz, D2O) δ 175.00, 174.73, 173.83, 172.51, 136.54, 115.87, 103.81, 102.54, 99.79, 77.73, 75.46, 75.16, 75.08, 72.87, 72.20, 71.74, 69.34, 68.31, 68.08, 67.45, 62.57, 61.01, 59.81, 59.42, 52.91, 51.67, 39.62, 31.73, 28.81, 22.19, 22.01; HRMS (ESI) calcd for C30H49N3O20 [M–H]−m/z = 770.2837, found 770.2812.
N-Hydroxypent-4-enamide (5-Acetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-β-d-galactopyranosyl-(1→4)[(1→3)-α-l-fucopyranosyl]-2-deoxy-2-acetamido-β-d-glucopyranoside (37)
To a 5 mL solution of 100 mM Tris buffer (pH 7.5) containing 5 mM l-fucose (0.025 mmol), 5 mM MgSO4 (0.025 mmol), 10 mM GTP·Na (0.05 mmol), and 10 mM ATP·Na (0.05 mmol) were added Saccharomyces cerevisiae inorganic pyrophosphatase (40 U) and B. fragilis FKP (4 U). The mixture was incubated at 37 °C for 2 h to preform GDP-fucose and monitored by TLC (2:1:1 iPrOH/AcOH/H2O). To this was added N-pentenoyl aminooxy sialoside 36 (5 mg, 0.07 mmol) and H. pylori α-1,3 fucosyltransferase (1 U), followed by incubation at room temperature for 16 h. The reaction was monitored by TLC (4:2:1 EtOAc/MeOH/H2O), and upon completion, calf alkaline phosphatase was added to remove remaining nucleotide phosphate. After further incubation at 37 °C for 2 h, the reaction mixture was quenched with cold MeOH (5 mL) and incubated on ice for 20 min. The mixture was centrifuged, precipitates were removed, and the solution was concentrated under vacuum. The resulting residue was passed through a BioGel P-2 size exclusion column and eluted with ddH2O to obtain 37 (4.6 mg, 78%) as a white, fluffy powder after lyophilization: 1H NMR (600 MHz, D2O) δ 5.88–5.77 (m, 1H), 5.06 (dd, J = 26.6, 14.1 Hz, 3H), 4.81 (app s, 1H), 4.51 (d, J = 7.7 Hz, 1H), 4.12–4.01 (m, 2H), 3.97 (dd, J = 20.8, 10.7 Hz, 2H), 3.92–3.82 (m, 7H), 3.75 (d, J = 12.5 Hz, 1H), 3.70–3.55 (m, 10H), 3.51 (t, J = 8.7 Hz, 1H), 2.79–2.70 (m, 1H), 2.37–2.30 (m, 2H), 2.30–2.22 (m, 2H), 2.02 (d, J = 4.1 Hz, 6H), 1.78 (t, J = 12.2 Hz, 1H), 1.15 (d, J = 6.1 Hz, 3H); 13C NMR (151 MHz, D2O) δ 175.04, 174.51, 173.79, 172.53, 136.55, 115.88, 103.70, 101.62, 99.64, 98.61, 75.65, 75.57, 74.91, 74.58, 72.92, 71.89, 71.83, 69.24, 69.19, 68.26, 68.12, 67.70, 67.30, 66.70, 62.62, 61.44, 59.52, 59.33, 53.56, 51.70, 39.78, 31.73, 28.80, 22.29, 22.03, 15.25; HRMS (ESI) calcd for C36H59N3O24 [M–H]−m/z = 916.3416, found 916.3394.
Aminooxy (5-Acetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-β-d-galactopyranosyl-(1→4)[(1→3)-α-l-fucopyranosyl]-2-deoxy-2-acetamido-β-d-glucopyranoside (38)
A solution of N-hydroxypentenoyl SLex37 (4.2 mg, 0.0046 mmol) in MeCN/MeOH/FA (3:1:0.001, v:v; 1.6 mL) was stirred at room temperature with dropwise addition of I2 (0.014 mmol, 0.5 M solution in THF). The mixture was heated to 35 °C and stirred for 2 h, after which the reaction was quenched with the addition of aqueous NH4HCO3 (500 mM) and Na2S2O3 (50 mM) until the disappearance of color. The solvent was removed under reduced pressure, and the remaining residue was purified by silica flash chromatography (4:2:1 EtOAc/MeOH/H2O). The desired fractions were pooled and concentrated under vacuum. The purified product was dissolved in ddH2O and lyophilized to give the free aminooxy SLex38 (2.5 mg, 65%) as a white powder: 1H NMR (600 MHz, D2O) δ 5.11 (d, J = 4.0 Hz, 1H), 4.72 (d, J = 8.7 Hz, 1H), 4.52 (d, J = 7.8 Hz, 1H), 4.09 (dd, J = 9.9, 3.0 Hz, 1H), 4.04 (dd, J = 14.8, 4.7 Hz, 1H), 3.98 (dd, J = 18.7, 9.6 Hz, 2H), 3.94–3.92 (m, 1H), 3.92–3.87 (m, 5H), 3.86 (dd, J = 13.6, 6.3 Hz, 2H), 3.78 (app d, J = 2.8 Hz, 1H), 3.70–3.63 (m, 8H), 3.61–3.56 (m, 3H), 3.55–3.51 (m, 1H), 2.76 (dd, J = 12.4, 4.6 Hz, 1H), 2.03 (s, 6H), 1.80 (t, J = 12.2 Hz, 1H), 1.17 (d, J = 6.6 Hz, 3H); 13C NMR (151 MHz, D2O) δ 175.06, 174.51, 173.78, 103.04, 101.65, 99.95, 99.66, 98.61, 75.68, 75.37, 74.92, 74.71, 73.18, 72.95, 71.90, 71.84, 69.27, 69.20, 68.27, 68.15, 67.72, 67.32, 66.72, 62.65, 61.46, 59.69, 53.94, 51.72, 43.35, 39.79, 22.18, 22.05, 15.27; HRMS (ESI) calcd for C31H52N3O23 [M–H]−m/z = 834.2994, found 834.2997.
General Procedure for Coupling Aminooxy Sugars to Aldehyde-Tagged Protein
To a solution of 5% MeCN, 0.02% FA, or 50 mM NaCit, pH 3.5, was added 0.26 mg/mL aldehyde-tagged protein and 1 mM aminooxy sugar. The reaction was incubated at 37 °C for 18 h, at which point the reaction was quenched with 4× protein loading dye and resolved by SDS-PAGE. The protein was transferred to nitrocellulose and probed with anti-sialyl Lewis x IgM (KM93, Chemicon, 1:500 dilution) followed by anti-mouse IgM-HRP (Southern Biotech, 1:10000) in 5% dry milk in phosphate-buffered saline/0.1% Tween (PBST). The presence of galactose was probed with SBA-FITC (EY, 1:1000 dilution) in 3% bovine serum albumin (BSA)/PBST. For fucose analysis, the nitrocellulose was probed with AAL-biotin (EY Labs, 1:1000 dilution) followed by rabbit anti-biotin FITC (Abcam, 1:1000) in 3% BSA/PBST. Membranes were developed by chemiluminescence using the SuperSignal West Pico kit (Thermo) or scanned for fluorescence by a Typhoon 9410 imaging system (Amersham). For mass spectrometry analysis, the protein was reacted in 5% MeCN, 0.02% FA, or NaCit at 1 mg/mL with 2 mM aminooxy sugar for 18 h at 37 °C. The reaction was quenched with 1 M Tris buffer pH 8, and the crude protein was purified by reversed-phase high-performance liquid chromatography (PLRP-S, Polymer Laboratories) with elution in aqueous MeCN. The multicharged peaks of the full length protein were obtained by ESI-MS (Bruker/Agilent Esquire) and identified by mass spectrometry analysis software. For stability analysis, the purified glycoconjugate was diluted to 10 μM in PBS at pH 7.2 and stored at −20 or 4 °C. Aliquots were taken at various time points and analyzed by matrix-assisted laser desorption/ionization mass spectrometry to determine the remaining glycoconjugate.
Acknowledgments
We thank Dr. Xi Chen and Dr. Hai Yu for the gift of plasmids encoding the NmCSS, Pd26ST, and PmST1 enzymes; Dr. Peng Wu for supplying enzyme and plasmids for the FKP, Hp1,3FT, and pyrophosphatase; Dr. David King for MS analysis and expertise; and Brian Belardi, Karen Dehnert, David Rabuka, and Brian Carlson for materials, helpful discussion, and manuscript critique. This work was funded by NIH/NIGMS (R01GM059907). J.E.H. was supported by a predoctoral fellowship from the U.S. National Science Foundation.
Supporting Information Available
Experimental procedures for 1–30 and spectroscopic data for all new compounds; complete ref (68). This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Spiro R. G. Glycobiology 2002, 12, 43R–56. [DOI] [PubMed] [Google Scholar]
- van Kooyk Y.; Rabinovich G. A. Nat. Immunol. 2008, 9, 593–601. [DOI] [PubMed] [Google Scholar]
- Wu C. Y.; Wong C. H. Chem. Commun. 2011, 47, 6201–6207. [DOI] [PubMed] [Google Scholar]
- Li H.; d’Anjou M. Curr. Opin. Biotechnol. 2009, 20, 678–684. [DOI] [PubMed] [Google Scholar]
- Wildt S.; Gerngross T. U. Nat. Rev. Microbiol. 2005, 3, 119–128. [DOI] [PubMed] [Google Scholar]
- Rich J. R.; Withers S. G. Nat. Chem. Biol. 2009, 5, 206–215. [DOI] [PubMed] [Google Scholar]
- Kiessling L. L.; Splain R. A. Annu. Rev. Biochem. 2010, 79, 619–653. [DOI] [PubMed] [Google Scholar]
- Gamblin D. P.; Scanlan E. M.; Davis B. G. Chem. Rev. 2009, 109, 131–163. [DOI] [PubMed] [Google Scholar]
- Buskas T.; Ingale S.; Boons G. J. Glycobiology 2006, 16, 113R–136. [DOI] [PubMed] [Google Scholar]
- Grogan M. J.; Pratt M. R.; Marcaurelle L. A.; Bertozzi C. R. Annu. Rev. Biochem. 2002, 71, 593–634. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Chen J.; Wan Q.; Wilson R. M.; Danishefsky S. J. Biopolymers 2010, 94, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne R. J.; Wong C. H. Chem. Commun. 2010, 46, 21. [DOI] [PubMed] [Google Scholar]
- Hirano K.; Macmillan D.; Tezuka K.; Tsuji T.; Kajihara Y. Angew. Chem., Int. Ed. 2009, 48, 9557–9560. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Schmidt R. R. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Mitchell H. J. Angew. Chem., Int. Ed. 2001, 40, 1576–1624. [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen A.; Dawson P. E. Curr. Opin. Chem. Biol. 2008, 12, 760–766. [DOI] [PubMed] [Google Scholar]
- Chalker J. M.; Bernardes G. J. L.; Davis B. G.. Acc. Chem. Res. 2011, in press. [DOI] [PubMed]
- van Kasteren S. I.; Kramer H. B.; Jensen H. H.; Campbell S. J.; Kirkpatrick J.; Oldham N. J.; Anthony D. C.; Davis B. G. Nature 2007, 446, 1105–1109. [DOI] [PubMed] [Google Scholar]
- Watt G. M.; Lund J.; Levens M.; Kolli V. S. K.; Jefferis R.; Boons G. J. Chem. Biol. 2003, 10, 807–814. [DOI] [PubMed] [Google Scholar]
- Pozsgay V.; Vieira N. E.; Yergey A. Org. Lett. 2002, 4, 3191–3194. [DOI] [PubMed] [Google Scholar]
- Ichikawa Y.; Matsukawa Y.; Isobe M. J. Am. Chem. Soc. 2006, 128, 3934–3938. [DOI] [PubMed] [Google Scholar]
- Lee D. J.; Mandal K.; Harris P. W. R.; Brimble M. A.; Kent S. B. H. Org. Lett. 2009, 11, 5270–5273. [DOI] [PubMed] [Google Scholar]
- Grayson E. J.; Bernardes G. J. L.; Chalker J. M.; Boutureira O.; Koeppe J. R.; Davis B. G. Angew. Chem., Int. Ed. 2011, 50, 4127–4132. [DOI] [PubMed] [Google Scholar]
- Liu H.; Wang L.; Brock A.; Wong C. H.; Schultz P. G. J. Am. Chem. Soc. 2003, 125, 1702–1703. [DOI] [PubMed] [Google Scholar]
- Kaya E.; Gutsmiedl K.; Vrabel M.; Müller M.; Thumbs P.; Carell T. ChemBioChem 2009, 10, 2858–2861. [DOI] [PubMed] [Google Scholar]
- Lin Y. A.; Chalker J. M.; Floyd N.; Bernardes G. J. L.; Davis B. G. J. Am. Chem. Soc. 2008, 130, 9642–9643. [DOI] [PubMed] [Google Scholar]
- Rodriguez E. C.; Marcaurelle L. A.; Bertozzi C. R. J. Org. Chem. 1998, 63, 7134–7135. [DOI] [PubMed] [Google Scholar]
- Marcaurelle L. A.; Shin Y.; Goon S.; Bertozzi C. R. Org. Lett. 2001, 3, 3691–3694. [DOI] [PubMed] [Google Scholar]
- Dirksen A.; Dawson P. E. Bioconjugate Chem. 2008, 19, 2543–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia J.; Raines R. T. Angew. Chem., Int. Ed. 2008, 47, 7523–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrico I. S.; Carlson B. L.; Bertozzi C. R. Nat. Chem. Biol. 2007, 3, 321–322. [DOI] [PubMed] [Google Scholar]
- Wu P.; Shui W.; Carlson B. L.; Hu N.; Rabuka D.; Lee J.; Bertozzi C. R. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 3000–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabuka D. Curr. Opin. Chem. Biol. 2010, 14, 790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierks T.; Dickmanns A.; Preusser-Kunze A.; Schmidt B.; Mariappan M.; von Figura K.; Ficner R.; Rudolph M. G. Cell 2005, 121, 541–552. [DOI] [PubMed] [Google Scholar]
- Roeser D.; Preusser-Kunze A.; Schmidt B.; Gasow K.; Wittmann J. G.; Dierks T.; von Figura K.; Rudolph M. G Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S.; Tropper F. D.; Roy R. Tetrahedron 1995, 51, 6679–6686. [Google Scholar]
- Renaudet O.; Dumy P. Tetrahedron Lett. 2001, 42, 7575–7578. [Google Scholar]
- Muthana S.; Cao H.; Chen X. Curr. Opin. Chem. Biol. 2009, 13, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez J. C.; Fraser-Reid B. J. Chem. Soc., Chem. Commun. 1991, 159. [Google Scholar]
- Debenham J. S.; Madsen R.; Roberts C.; Fraser-Reid B. J. Am. Chem. Soc. 1995, 117, 3302–3303. [Google Scholar]
- Madsen R.; Roberts C.; Fraser-Reid B. J. Org. Chem. 1995, 60, 7920–7926. [Google Scholar]
- Donohoe T. J.; Callens C. K. A.; Thompson A. L. Org. Lett. 2009, 11, 2305–2307. [DOI] [PubMed] [Google Scholar]
- Yu B.; Sun J. Chem. Commun. 2010, 46, 4668–4679. [DOI] [PubMed] [Google Scholar]
- Yu B.; Tao H. J. Org. Chem. 2002, 67, 9099–9102. [DOI] [PubMed] [Google Scholar]
- Hanashima S.; Castagner B.; Esposito D.; Nokami T.; Seeberger P. H. Org. Lett. 2007, 9, 1777–1779. [DOI] [PubMed] [Google Scholar]
- Tanaka H.; Iwata Y.; Takahashi D.; Adachi M.; Takahashi T. J. Am. Chem. Soc. 2005, 127, 1630–1631. [DOI] [PubMed] [Google Scholar]
- Thomas M.; Gesson J. P.; Papot S. J. Org. Chem. 2007, 72, 4262–4264. [DOI] [PubMed] [Google Scholar]
- Limanto J.; Shafiee A.; Devine P. N.; Upadhyay V.; Desmond R. A.; Foster B. R.; Gauthier D. R.; Reamer R. A.; Volante R. P. J. Org. Chem. 2005, 70, 2372–2375. [DOI] [PubMed] [Google Scholar]
- Blot V.; Reboul V.; Metzner P. J. Org. Chem. 2004, 69, 1196–1201. [DOI] [PubMed] [Google Scholar]
- Lanctot P. M.; Gage F. H.; Varki A. P. Curr. Opin. Chem. Biol. 2007, 11, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel S. R.; Wiese G. K.; Cho J.; Opperman M. J.; Hays D. L.; Siddiqui J.; Pienta K. J.; Furie B.; Dimitroff C. J. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 19491–19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellervik U.; Magnusson G. J. Org. Chem. 1998, 63, 9314–9322. [Google Scholar]
- Clinch K.; Evans G. B.; Furneaux R. H.; Rendle P. M.; Rhodes P. L.; Roberton A. M.; Rosendale D. I.; Tyler P. C.; Wright D. P. Carbohydr. Res. 2002, 337, 1095–1111. [DOI] [PubMed] [Google Scholar]
- Demchenko A.; Stauch T.; Boons G. J. Synlett 1997, 1997, 818–820. [Google Scholar]
- Wang L. X.; Huang W. Curr. Opin. Chem. Biol. 2009, 13, 592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Varki A. ACS Chem. Biol. 2009, 5, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. Nature 2007, 446, 1023–1029. [DOI] [PubMed] [Google Scholar]
- Yu H.; Chokhawala H.; Karpel R.; Yu H.; Wu B.; Zhang J.; Zhang Y.; Jia Q.; Chen X. J. Am. Chem. Soc. 2005, 127, 17618–17619. [DOI] [PubMed] [Google Scholar]
- Yu H.; Huang S.; Chokhawala H.; Sun M.; Zheng H.; Chen X. Angew. Chem., Int. Ed. 2006, 45, 3938–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.; Chokhawala H. A.; Huang S.; Chen X. Nat. Protoc. 2006, 1, 2485–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pudelko M.; Bull J.; Kunz H. ChemBioChem 2010, 11, 904–930. [DOI] [PubMed] [Google Scholar]
- Sackstein R.; Merzaban J. S.; Cain D. W.; Dagia N. M.; Spencer J. A.; Lin C. P.; Wohlgemuth R. Nat. Med. 2008, 14, 181–187. [DOI] [PubMed] [Google Scholar]
- Wang W.; Hu T.; Frantom P. A.; Zheng T.; Gerwe B.; del Amo D. S.; Garret S.; Seidel R. D.; Wu P. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 16096–16101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson A. G.; Svensson J.; Johannsson G. Growth Horm. IGF Res. 2007, 17, 441–462. [DOI] [PubMed] [Google Scholar]
- Quigley C. A. Endocrinol. Metab. Clin. North Am. 2007, 36, 131–186. [DOI] [PubMed] [Google Scholar]
- Chang T. K.; Jackson D. Y.; Burnier J. P.; Wells J. A. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 12544–12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H.; et al. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 9060–9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Okada S.; Tan L.; Goodrum K. J.; Kopchick J. J.; Kieliszewski M. J. Transgenic Res. 2010, 19, 849–867. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.