

Abstract
Retinoid X receptors (RXRs) are involved in a number of signaling pathways as heterodimeric partners of numerous nuclear receptors. Hepatocytes express high levels of the RXRα isotype, as well as several of its putative heterodimeric partners. Germ-line disruption (knockout) of RXRα has been shown to be lethal in utero, thus precluding analysis of its function at later life stages. Hepatocyte-specific disruption of RXRα during liver organogenesis has recently revealed that the presence of hepatocytes is not mandatory for the mouse, at least under normal mouse facility conditions, even though a number of metabolic events are impaired [Wan, Y.-J., et al. (2000) Mol. Cell. Biol. 20, 4436–4444]. However, it is unknown whether RXRα plays a role in the control of hepatocyte proliferation and lifespan. Here, we report a detailed analysis of the liver of mice in which RXRα was selectively ablated in adult hepatocytes by using the tamoxifen-inducible chimeric Cre recombinase system. Our results show that the lifespan of adult hepatocytes lacking RXRα is shorter than that of their wild-type counterparts, whereas proliferative hepatocytes of regenerating liver exhibit an even shorter lifespan. These lifespan shortenings are accompanied by increased polyploidy and multinuclearity. We conclude that RXRα plays important cell-autonomous function(s) in the mechanism(s) involved in the lifespan of hepatocytes and liver regeneration.
Keywords: conditional somatic mutagenesis, Cre-Lox, liver, partial hepatectomy, polyploidy
Nuclear receptors are signal transducers that play crucial roles in vertebrate embryogenesis, organogenesis, cellular proliferation and differentiation, and homeostasis (1, 2). They belong to a superfamily of transcriptional regulators that includes ligand-dependent receptors for steroid and thyroid hormones, retinoic acids (the active forms of vitamin A), vitamin D3, and a variety of other lipophilic ligands, as well as orphan receptors for which no ligands have yet been found. Within this superfamily, retinoid X receptor (RXR) α, β, and γ play a central role as heterodimeric partners for several nuclear receptors, e.g., retinoic acid receptor α, β, and γ; thyroid hormone receptor α and β; peroxisome proliferator-activated receptor α, β, and γ; liver X receptor α and β; farnesoid X-activated receptor; and 1,25-dihydroxyvitamin D3 receptor as well as several orphan nuclear receptors (3, 4). These heterodimers represent the functional units that bind to cognate response elements located in the promoter regions of target genes to regulate their expression (1).
The liver, a key player in mammalian homeostasis, has the unique capacity to restore its mass in response to both toxin and inflammatory injuries, transplantation, and partial hepatectomy (PH) (5, 6). Many members of the nuclear receptor superfamily including RXRs are expressed in the liver, as well as a wealth of genes that contain response elements for RXR–nuclear receptor heterodimers (7, 8). Thus, as RXRα is the most abundant of the three RXR isotypes in the liver, it may play a prominent role in hepatic growth, regeneration, and homeostatic functions. However, the possible postnatal role of RXRα could not be analyzed in knockout mice lacking RXRα (RXRαKO), as RXRαKO fetuses die at embryonic days 13.5–16.5 from abnormal development of the ventricular myocardium (9, 10). The liver of RXRαKO fetuses appeared morphologically and histologically normal, but its development was delayed by about 24 h, compared with wild-type (wt) fetuses (9, 10). Interestingly, it was recently reported that mice, in which RXRα was selectively ablated in hepatocytes by using the Cre-Lox technology, were viable even though a number of metabolic events regulated by nuclear receptors that heterodimerize with RXR were impaired. No morphological defect could be evidenced in the liver of these conditional mutants (7, 8). This was surprising as there was some indication that RXRs and some of their heterodimeric partners might control hepatocyte proliferation and lifespan (refs. 11–16; see Discussion).
Our present study reports a detailed analysis of the liver of mice in which RXRα was selectively ablated in hepatocytes. It reveals that this ablation reduces the lifespan of adult hepatocytes, induces the formation of spots of focal necrosis in the liver of adult mice, but does not reduce the liver mass. Furthermore, upon partial hepatectomy, the lifespan of RXRα-ablated hepatocytes was further reduced, and additional cellular abnormalities could be detected.
Materials and Methods
Animals.
Alb-Cre (17), αAT-Cre-ERT (18), and RXRαL2/+ (19) mice and mouse genotyping were as described. Animals were maintained in a 12-h light-dark cycle, with food and water provided ad libitum.
Liver resection of the left and median lobes (PH) was performed after midventral laparotomy between 8 a.m. and 12 p.m.
DNA and RNA Analysis.
Southern blot analysis of Cre-mediated RXRα ablation was performed on genomic DNA, which was isolated from various tissues, digested with BamHI, and probed as described (20).
Total liver RNA was extracted by the guanidium-thiocyanate-phenol-chloroform method (21). Ten micrograms total RNA was electrophoresed on a 0.8% agarose gel, transferred to a nylon membrane, and hybridized with RXRα, RXRβ, RXRγ, ApoCIII, and 36B4-labeled probes, as described (20, 22).
Histology.
Liver, rinsed in PBS, was fixed with Bouin's solution and embedded in paraffin. Six-micrometer sections were stained with hematoxylin and eosin.
RXRα and Ki67 immunohistochemistry was performed on 10-μm cryosections, by using the biotinylated anti-RXRα 4RX3A2 mAb (1/1,000 dilution in 0.1% BSA/PBS) (19, 23) and the NCL-Ki67p polyclonal antibody (NovoCastra, Newcastle, U.K.), respectively, according to the manufacturer's instructions. The number of Ki67-labeled nuclei was determined by counting the Ki67-positive hepatocyte nuclei in at least three low magnification (×100) microscope fields for each sample. More than 2,000 hepatocytes were screened per field.
BrdUrd Incorporation.
Animals were injected i.p. with 50 mg/kg BrdUrd 2 h before dissection. Liver was rinsed in PBS and embedded in tissue-Tek OCT compound (Sakura, Zoeterwoude, The Netherlands). Ten-micrometer cryosections, fixed with 4% paraformaldehyde, were incubated with an antibody against BrdUrd (Boehringer Mannheim), which was 50-fold diluted in 0.1% BSA/PBS, revealed with CY3-conjugated donkey anti-rabbit IgG antibody, and mounted with Vectorshield medium (Vector Laboratories) containing 4′,6-diamino-2-phenylindole dihydrochloride. The number of BrdUrd-labeled hepatocytes was determined by counting the BrdUrd-positive hepatocyte nuclei in at least five low-magnification (×100) microscope fields for each sample. More than 1,800 hepatocytes were screened per field.
Liver Functions.
Alkaline phosphatase (ALP), alanine aminotransferase (ALT; also called glutamate/pyruvate transaminase, GPT), and aspartate aminotransferase (AST; also called glutamate/oxalacetate transaminase, GOT) enzymatic activities were determined on mouse serum with Sigma diagnostic kits, according to the manufacturer's protocols.
Mosaic Mice Study.
RXRα−/− embryonic stem (ES) cells were obtained after electroporation of RXRα+/− ES cells with a targeting vector similar to pHR(RXRα)0.2 (9), in which the neomycin resistance cassette was replaced by a hygromycin resistance cassette (24), as described (9). RXRα−/−, RXRα+/−, and RXRα+/+ 129/SV ES D4 cells were injected into C57BL/6 blastocysts, and proteins were extracted from organs of 10-month-old RXRα−/−, RXRα−/+, and RXRα+/+ chimeric mice derived from these blastocysts after implantation into C57BL/6 recipient females. Isoenzymes GPI-1a (from the 129/SV ES D4 cells) and GPI-1b (from the C57BL/6 host blastocysts) were resolved on cellugel (Helena Laboratories), and their enzymatic activities were revealed as described (25). The fraction of ES cell-derived cells was evaluated by comparing the intensity of the two isoenzyme activities.
Stastistical Analyses.
Values are reported as mean ± SEM. Statistical significance (P < 0.05) was determined by unpaired Student's t test (statview, Abacus Concepts, Berkeley, CA).
Results
Ablation of RXRα in Adult Hepatocytes Compromises Their Lifespan and Regenerative Capacity.
To investigate the role of RXRα in liver, we first produced chimeric animals by injecting either RXRα−/− or control (CT; RXRα+/+ or RXRα+/−) 129/SV ES cells into wt C57BL/6 blastocysts, followed by implantation into recipient females. The contribution of ES cells to various organs of the resulting chimeras was determined by glucose phosphate isomerase (GPI) analysis, as distinct GPI isoenzymes are expressed in cells derived from 129/SV ES cells and C57BL/6 cells. In 10-month-old animals, the CT and RXRα−/− ES cell contributions were similar in the various organs analyzed, with the exception of the liver, where it was 10-fold lower (Fig. 1A). This finding indicates that RXRα could play an important role in hepatocyte proliferative capacity and/or lifespan, even though it was not found to be essential for liver organogenesis (9, 10).
Figure 1.
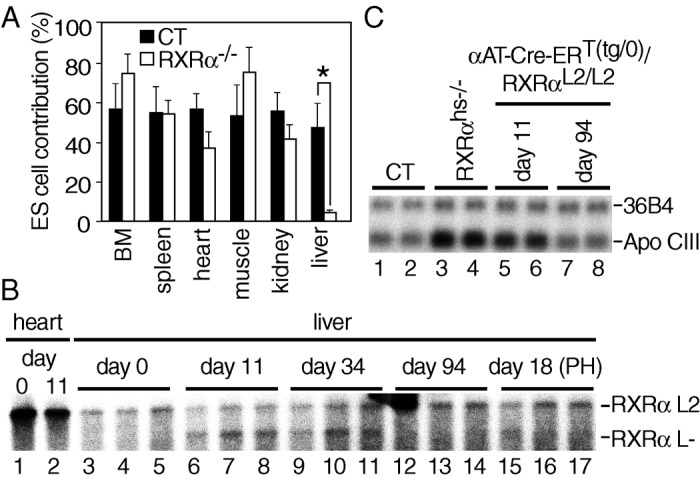
Impaired lifespan of RXRα null hepatocytes. (A) Low contribution of RXRα−/− cells in liver of chimeric mice. The fraction of RXRα−/− cells was evaluated in various organs of 10-month-old chimeric mice by the ratio of the distinct GPI isozymes characteristic of 129/SV and C57BL/6 mice. Six and nine chimeras obtained by injection of CT (RXRα+/+ or RXRα+/− 129/SV ES cells) and RXRα−/− 129/SV ES cells into C57BL/6 blastocysts, respectively, were analyzed. BM, bone marrow. Values are expressed as the mean ± SEM. *, P < 0.001. (B) Time-dependent loss of RXRα L− alleles in liver of Tam-treated αAT-Cre-ERT(tg/0)/RXRαL2/L2 mice. Cre-ERT-mediated RXRα ablation was determined by Southern blot analysis, performed on genomic DNA isolated from heart and liver of αAT-Cre-ERT(tg/0)/RXRαL2/L2 double-transgenic mice, taken before (day 0; lanes 1 and 3–5), and at the indicated days after the first Tam injection (3-month-old animals) (lanes 2 and 6–17). PH was performed 12 days after the first Tam injection (lanes 15–17). Liver DNA was extracted from three animals for each time point. The position of the fragments corresponding to the RXRα L2 and L− alleles is indicated. (C) Relief of down-regulation of ApoCIII expression in liver of RXRαhs−/− and Tam-treated αAT-Cre- ERT(tg/0)/RXRαL2/L2 mice. ApoCIII RNA levels were analyzed by Northern blot, performed on RNA extracted from livers of CT (Tam-treated αAT- Cre-ERT(0/0)/RXRαL2/L2 double-transgenic mouse or vehicle-treated αAT- Cre-ERT(tg/0)/RXRαL2/L2 double-transgenic mouse, lanes 1 and 2, respectively), RXRαhs−/− (lanes 3 and 4), and Tam-treated αAT-Cre-ERT(tg/0)/RXRαL2/L2 double-transgenic mice taken 11 (lanes 5 and 6) and 94 days (lanes 7 and 8) after the first Tam injection. The position of ApoCIII and 36B4 transcripts is indicated.
To investigate the possible role of RXRα on hepatocyte lifespan, RXRαL2/+ mice, in which the exon encoding the RXRα DNA binding domain is “floxed” on one allele (19), were crossed with αAT-Cre-ERT(tg/0) hemizygous transgenic mice, which express selectively the tamoxifen-inducible Cre-ERT recombinase into more than 50% of the hepatocytes (18). Three-month-old αAT-Cre-ERT(tg/0)/RXRαL2/L2 mice generated from these crosses were treated with 1 mg of tamoxifen (Tam) for 5 consecutive days to induce the Cre activity of Cre-ERT, and the fraction of cells in which RXRα was ablated was estimated by Southern blotting on DNA extracted at various times after Tam treatment. As expected, ≈50% of the RXRα L2 alleles were converted into L− alleles by day 11 after the first Tam injection (Fig. 1B, compare lanes 6–8 with lanes 3–5). However, the proportion of L− alleles decreased with time, and almost no RXRα L− alleles could be detected by day 94 (Fig. 1B, lanes 12–14). The progressive disappearance of hepatocytes in which RXRα was ablated was further confirmed by analysis of the expression of the ApoCIII gene, an RXRα target gene whose down-regulation in hepatocytes has been shown to be RXRα mediated (7). By day 11 after the first Tam injection, the amount of ApoCIII transcripts was 2.2-fold higher in livers of αAT-Cre-ERT(tg/0)/RXRαL2/L2 mice (Fig. 1C, lanes 5 and 6) than in those of Tam-treated αAT-Cre-ERT(0/0)/RXRαL2/L2 (lane 1) and vehicle-treated αAT-Cre-ERT(tg/0)/RXRαL2/L2 (lane 2) CT mice. Note that, as expected, this increase was about 2-fold lower than that observed in RXRαhs−/− mouse livers in which RXRα was ablated in all hepatocytes (see below; see also Fig. 1C, compare lanes 5 and 6 with lanes 3 and 4). In contrast, by day 94 after the first Tam injection, ApoCIII expression in αAT-Cre-ERT(tg/0)/RXRαL2/L2 mice was similar to that observed in CT mice (Fig. 1C, compare lanes 7 and 8 with lanes 1 and 2), in agreement with the disappearance of hepatocytes in which RXRα was ablated.
PH is well known to trigger liver regeneration (5, 6). Interestingly, 4 and 8 h after PH of 3-month-old wt mice, liver RXRα transcripts increased, whereas those of RXRβ and RXRγ were unchanged (Fig. 2A and data not shown). Furthermore, an increase in hepatocyte nuclear RXRα protein was observed 12–24 h after PH (Fig. 2B and data not shown), thus suggesting that RXRα could play a critical role during liver regeneration. PH was therefore performed 12 days after the first Tam treatment of αAT-Cre- ERT(tg/0)/RXRαL2/L2 mice. Most interestingly, almost all hepatocytes in which RXRα had been ablated disappeared within 6 days after PH [Fig. 1B; compare RXRα L− allele at day 11 (lanes 6–8), day 18 (PH) (lanes 15–17), and day 34 (lanes 9–11)]. Thus, RXRα may be required in hepatocytes of adult mice for a vital function(s), notably under proliferative conditions.
Figure 2.
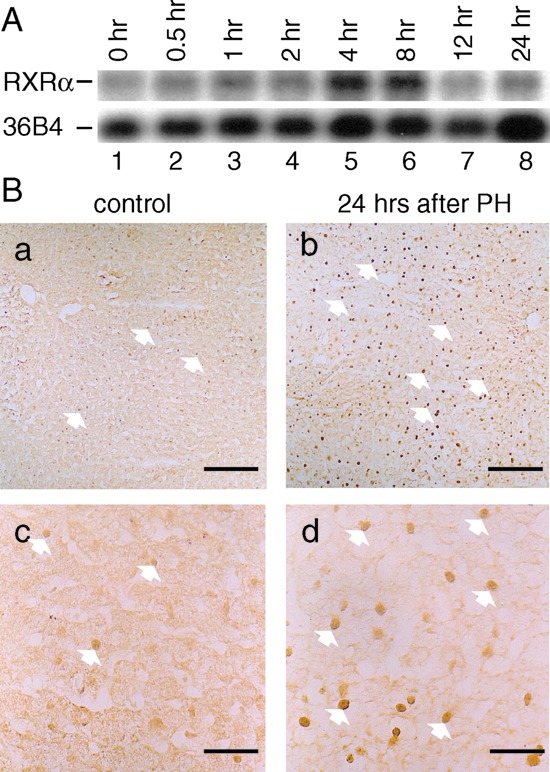
Analysis of RXRα expression during liver regeneration. (A) Increased RXRα RNA levels. RXRα RNA levels were analyzed by Northern blotting of liver RNA extracted from 129/SV mice before (0 h) or at the indicated times after PH. The positions of RXRα and 36B4 transcripts are indicated. (B) Increased RXRα protein levels. RXRα protein was revealed with the 4RX3A2 mAb on 129/SV mouse liver cryosections, 24 h after sham operation (control a and c) or after PH (b and d). [Scale bar: 160 μm (a and b) and 40 μm (c and d).] White arrows point to some of the hepatocyte RXRα-positive nuclei.
Cellular Abnormalities in Liver of Mice Lacking RXRα in Hepatocytes.
Taken together, the above observations indicate that the lifespan and regenerative capacity of adult hepatocytes are compromised in the absence of RXRα. In contrast, it was recently reported that mice in which RXRα was efficiently and selectively disrupted in hepatocytes during liver development did not exhibit any apparent morphological abnormality, even though several metabolic pathways that involve RXRα were impaired in the liver of these animals (7, 8). We therefore generated similar mice that do not express RXRα in hepatocytes to further investigate its role at the cellular level.
RXRαL2/+ mice were crossed with Alb-Cre(tg/0) hemizygous transgenic mice that express Cre under the control of the albumin promoter (17) to generate Alb-Cre(tg/0)/RXRαL2/L2 mice. Efficient excision of floxed DNA was previously shown to occur selectively in hepatocytes of these transgenic mice during liver ontogenesis (7, 17). Alb-Cre(tg/0)/RXRαL2/L2 mice (hereafter called RXRαhs−/− for hepatocyte-specific RXRαL−/L− genotype) were born at a Mendelian ratio, and no difference in viability and fertility was observed when compared with Alb-Cre(tg/0)/RXRα+/+, Alb- Cre(0/0)/RXRα+/+, Alb-Cre(0/0)/RXRαL2/L2, and Alb-Cre(tg/0)/ RXRαL2/+ littermates, which were all used as CT mice. As expected ≈80% of the RXRα L2 alleles were converted into RXRα-disrupted L− alleles in the liver of 8-week-old RXRαhs−/− animals, whereas no L− allele could be detected in other tested organs, nor in any organ from control animals (Fig. 3A and data not shown; see also ref. 7). Furthermore, no RXRα protein could be revealed by immunohistochemistry in hepatocyte nuclei of 8-week-old RXRαhs−/− mice, under conditions where it was readily detected in CT animals (Fig. 3B). Finally, in agreement with previous results (7), the expression of ApoCIII, one of the genes whose down-regulation is mediated by RXRα in the liver, was enhanced in RXRαhs−/− mice but not in CT Alb-Cre(tg/0)/RXRαL2/+ mice (Fig. 3C and data not shown). Thus, RXRα was efficiently and selectively disrupted in hepatocytes of RXRαhs−/− mice. Note that, in agreement with previous data (7), the expression levels of RXRβ and RXRγ were not grossly affected in the liver of RXRαhs−/− mice (Fig. 3C and data not shown).
Figure 3.

Hepatocyte-selective RXRα ablation in Alb-Cre(tg/0)/RXRαL2/L2 (RXRαhs−/−) mice. (A) Efficiency and selectivity of RXRα gene disruption in liver. Genomic DNA extracted from the indicated organs of 8-week-old Alb-Cre(0/0)/RXRαL2/L2 CT and Alb-Cre(tg/0)/RXRαL2/L2 (RXRαhs−/−) mice was analyzed by Southern blotting. The position of fragments corresponding to the RXRα L2 and L− alleles is indicated. (B) Hepatocytes from Alb- Cre(tg/0)/RXRαL2/L2 (RXRαhs−/−) mice do not express the RXRα protein. Liver cryosection of 8-week-old Alb-Cre(0/0)/RXRαL2/L2 CT and RXRαhs−/− mice were reacted with the 4RX3A2 anti-RXRα mAb. Arrows in CT section point to some of the RXRα-positive nuclei. (Scale bar, 80 μm.) (C) Expression of ApoCIII, RXRβ, and RXRγ in the liver of RXRαhs−/− mice. ApoCIII, RXRβ, and RXRγ RNA levels were analyzed by Northern blot on RNA extracted from liver of 2 CT (Alb-Cre(0/0)/RXRαL2/L2) and 2 RXRαhs−/− 8-week-old mice, as indicated. 36B4 RNA was used as an internal control. (D) Focal necrosis in RXRαhs−/− liver. Histological analysis of a liver section from a 3-month-old RXRαhs−/− mouse, stained with hematoxylin and eosin. The area of focal necrosis is surrounded by a line. (Scale bar, 160 μm.) (E) Increased ALP, ALT, and AST levels in RXRαhs−/− mice. ALP, ALT/GPT, and AST/GOT levels were determined on serum taken from 3-month-old CT (Alb-Cre(tg/0)/RXRα+/+, Alb-Cre(0/0)/RXRα+/+, Alb-Cre(0/0)/RXRαL2/L2) and RXRαhs−/− animals (n = 10 and 11, respectively). Average values (± SEM) are presented. *, P < 0.05. (F) Higher hepatocyte proliferative index in RXRαhs−/− liver. The percentage of BrdUrd-positive and Ki67-positive hepatocytes was determined on liver sections from CT (Alb-Cre(tg/0)/RXRα+/+, Alb-Cre(0/0)/RXRα+/+, Alb- Cre(0/0)/RXRαL2/L2), and RXRαhs−/− mice at the age of 3 months. Values are expressed as the mean ± SEM. *, P < 0.02. BrdUrd labeling was analyzed in 10 CT and 11 RXRαhs−/− mice. Ki67 labeling was analyzed in three CT and four RXRαhs−/− mice.
However, in contrast to the previous reports of Wan et al. (7, 8), restricted areas of focal necrosis were occasionally observed on liver sections of six of eleven 3-month-old RXRαhs−/− mice, whereas no such necrosis was observed on liver sections from control mice (Fig. 3D and data not shown). Note that by using the terminal deoxynucleotidyltransferase-mediated UTP end-labeling assay, we could not detect significant hepatocyte apoptosis in either CT or RXRαhs−/− mice (data not shown). The existence of cytolysis and necrosis in 3-month-old RXRαhs−/− mice was further supported by assays of ALP, alanine aminotransferase (ALT/GPT), and aspartate aminotransferase (AST/GOT) enzymatic activities (Fig. 3E). ALP and ALT/GPT levels were 1.4- and 1.3-fold higher in RXRαhs−/− than in CT mice, respectively (P < 0.05). AST/GOT levels were also higher in RXRαhs−/− than in CT mice, but the difference may not be statistically significant.
BrdUrd incorporation was used to estimate the proliferative index (PI) of hepatocytes in 3-month-old CT and RXRαhs−/− mice. A 1.7-fold increase in the fraction of BrdUrd-positive hepatocytes was observed in RXRαhs−/− mice (Fig. 3F), under conditions where the weight and lipid content of RXRαhs−/− and control livers were similar (data not shown; see also ref. 7). This hepatocyte PI increase in RXRαhs−/− mice was confirmed by immunostaining for Ki-67, a nuclear protein known to be expressed by proliferating cells (26) (Fig. 3F). Thus, hepatocytes in which RXRα is ablated exhibit both a shorter lifespan and an increased PI.
Liver Regeneration Is Impaired in Mice Lacking RXRα in Hepatocytes.
We next examined liver regeneration in adult RXRαhs−/− mice. Interestingly, although PH postoperative morbidity and mortality were similar in 3-month-old CT and RXRαhs−/− mice (less than 2%), ALP, ALT/GPT, and AST/GOT enzymatic activities were markedly increased in sera of RXRαhs−/− mice when compared with control mice (Fig. 4A), thus indicating increased liver necrosis in RXRαhs−/− mice. In contrast to control mice, histological analysis of liver sections 36–60 h after PH revealed the presence of cytolysis and neutrophil infiltration in RXRαhs−/− mice, as well as large areas of focal necrosis (more than 10% of the area of liver sections of RXRαhs−/− mice and less than 0.5% in control littermates; Fig. 4B, compare a and e and b and f; Fig. 4C; data not shown). Interestingly, 48–72 h after PH, highly polyploid (≥8C) hepatocytes were 5-fold more numerous in RXRαhs−/− mice than in CT mice (Fig. 4 B, c and g and D; data not shown). In addition, periodic acid-Schiff positive giant multinucleated hepatocytes were also observed 7 days after PH on sections of RXRαhs−/− livers but not on sections of control livers (Fig. 4B, compare d and h; data not shown). BrdUrd incorporation indices were similar in control and RXRα-ablated hepatocytes 36 h post-PH, which corresponds to maximal BrdUrd incorporation in hepatocytes of control mice (Fig. 4E; data not shown). However, 48 and 60 h after PH, the fraction of hepatocytes that incorporated BrdUrd was 1.3- and 1.5-fold higher in RXRαhs−/− mice than in control mice (Fig. 4E). One week after PH, RXRαhs−/− and control mice had a similar liver weight (liver/body weight ratio: 3.9 ± 0.1% and 4.1 ± 0.1%, respectively).
Figure 4.
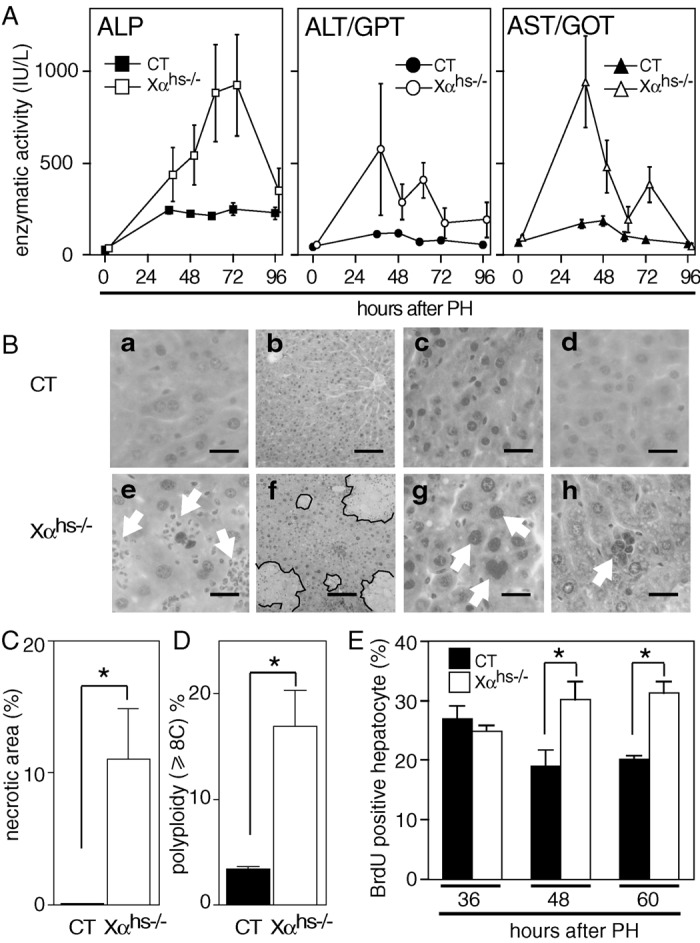
Cell defects in RXRαhs−/− mice during liver regeneration. (A) Increased ALP, ALT, and AST activities in RXRαhs−/− mice after PH. ALP, ALT/GPT, and AST/GOT enzymatic activities were determined on sera from 3-month-old CT (Alb-Cre(tg/0)/RXRα+/+, Alb-Cre(0/0)/RXRα+/+, Alb-Cre(0/0)/RXRαL2/L2), and RXRαhs−/− (Xαhs−/−) mice, at various times after PH. Values are expressed as the mean ± SEM (n = 3–5). (B) Highly polyploid hepatocytes, giant multinucleated cells, neutrophil infiltration, and focal necrosis in RXRαhs−/− mice after PH. Histological analysis of hematoxylin and eosin-stained sections of liver taken 36 h (a and e), 48 h (b and f), 72 h (c and g), and 1 week (d and h) after PH of CT (a–d) and RXRαhs−/− (e–h) mice. Arrows indicate neutrophil infiltration in a, polyploid (≥8C) hepatocyte in g, and giant multinucleated cell in h. Lines surround areas of focal necrosis in f. [Scale bar, 20 μm (a, c–e, g, and h) and 80 μm (b and f).] (C) Quantification of necrotic areas of focal necrosis in RXRαhs−/− mice. Necrotic areas were determined on liver sections from five CT (Alb- Cre(tg/0)/RXRα+/+, Alb-Cre(0/0)/RXRα+/+, Alb-Cre(0/0)/RXRαL2/L2) and five RXRαhs−/− (Xαhs−/−) animals. Samples were taken 36 and 48 h after PH, from three and two animals from each group, respectively, and three fields containing more than 1,800 hepatocytes each were analyzed for each animal. (D) Quantification of polyploid hepatocytes in RXRαhs−/− mice. Polyploid hepatocytes (≥8C) were determined on liver sections from five CT (Alb- Cre(tg/0)/RXRα+/+, Alb-Cre(0/0)/RXRα+/+, Alb-Cre(0/0)/RXRαL2/L2) and five RXRαhs−/− (Xαhs−/−) animals. Samples were taken 60 and 72 h after PH, from two and three animals from each group, respectively, and three fields containing more than 1,800 hepatocytes each were analyzed for each animal. Note that the number of polyploid hepatocytes is underevaluated as 6-μm sections were analyzed. *, P < 0.001. (E) Increased hepatocyte proliferative index in RXRαhs−/− mice after PH. Hepatocyte proliferation was analyzed by BrdUrd incorporation 36, 48, and 60 h after PH. The percentage of BrdUrd-positive hepatocytes was determined on liver sections from five 3-month-old CT (Alb-Cre(tg/0)/RXRα+/+, Alb-Cre(0/0)/RXRα+/+, Alb-Cre(0/0)/RXRαL2/L2) and five RXRαhs−/− (Xαhs−/−) animals. Values are expressed as mean ± SEM. *, P < 0.05.
Discussion
RXRα null (RXRαKO) mutant fetuses die in utero at embryonic days 13.5–16.5, most probably from heart failure caused by abnormalities of ventricular myocardium (9, 10). However, the liver of RXRαKO mutant fetuses appeared essentially normal both morphologically and histologically, although its development may be delayed by about 24 h when compared with wt littermates (9, 10). Thus, RXRα did not appear to be critically required for the formation of hepatocytes during liver organogenesis, but it remained to be determined whether RXRα could be required for further fetal and postnatal liver development. It was also unknown whether RXRα could be required for the maintenance of adult hepatocytes that have a low rate of cell turnover, with a lifespan of at least 300–400 days (27–29), and/or for their remarkable proliferative capacity during the liver regeneration that can be induced by PH or a variety of injuries (5, 6, 30).
Our present analysis of adult mouse chimeras, obtained through injection of RXRα−/− ES cells into wt blastocytes, clearly demonstrates that RXRα−/− cells are selectively under-represented in livers of these chimeras when compared with other tissues. This may originate from impaired proliferation of RXRα−/− hepatocytes during liver postnatal growth and/or their premature death. This latter possibility was supported by studies in which the Tam-inducible Cre-ERT recombinase system was used to selectively ablate RXRα in ≈50% of the hepatocytes of adult mice. Indeed, these RXRα-ablated hepatocytes disappear in less than 90 days, which is much shorter than the lifespan of wt hepatocytes. Moreover, we also show that RXRα levels are increased in hepatocytes shortly after PH and, interestingly, that the selective disappearance of RXRα-ablated hepatocytes is even faster during liver regeneration, thus suggesting that proliferating hepatocytes lacking RXRα could be particularly prone to premature death.
The conclusion that RXRα plays an important cell-autonomous function(s) in the mechanism(s) involved in the lifespan of hepatocytes was unexpected, as it was recently reported that selective ablation of RXRα in all hepatocytes during liver development has no effect on adult liver morphological characteristics (7, 8). However, a detailed histological analysis of similar adult mutants generated in our laboratory (RXRαhs−/− mice) reveals the existence of occasional focal necrotic areas, whose presence is correlated with increased serum levels of enzymes known to be released during liver necrosis. Concomitantly, the PI of RXRαhs−/− hepatocytes is significantly increased. Moreover, PH-induced liver regeneration in adult RXRαhs−/− generates much more pronounced enzymatic and histological stigmas of liver necrosis accompanied by neutrophil infiltration. Twenty-four hours post-PH, the PI of hepatocytes is similarly increased in CT and RXRαhs−/− livers, indicating that their replicative capacity is identical at an early stage of liver regeneration. However, this PI becomes higher in mutant hepatocytes at later stages of regeneration, most probably reflecting a compensatory growth subsequent to necrosis of hepatocytes lacking RXRα, to restore the optimal liver mass.
Polyploidization is a characteristic feature of the growth of liver in rats, as well as in other mammals, including humans (see refs. 12 and 30–34). In newborn rats, actively dividing diploid hepatocytes dominate. Binucleate hepatocytes, resulting from acytokinetic mitosis, markedly increase over the next few weeks. Mononucleate tetraploid cells resulting from disturbance in karyokinesis then appear (60–70% in adults), followed by binucleated cells with two tetraploid nuclei (≈20% in adults) and finally by mononucleated octoploid cells (1–3% in adults), whereas a small fraction of diploid cells (≈10%) is still present. In contrast to developmental growth, PH-induced regeneration growth is characterized by a marked decrease of binuclearity as a consequence of a reduced rate of binucleation (31, 34). The biological significance of hepatic polyploidy is unclear, but it is generally considered that advanced polyploidy in mammalian cells is an indication of terminal differentiation and senescence of cells (33, 34). In this respect, it is remarkable that the PH-induced proliferative hepatocytes that lack RXRα exhibit much higher degrees of polyploidy than their wt counterparts and that this increased polyploidy is correlated with an increase in their death rate. In any event, our present data show that the lifespan of hepatocytes depends on cell-autonomous events mediated by RXRα. The lifespan of adult hepatocytes lacking RXRα is shorter than that of their wt counterparts, whereas PH-induced proliferative hepatocytes lacking RXRα have an even shorter lifespan. Whether two distinct RXRα-mediated mechanisms are involved in the death of “quiescent” and “replicative” RXRα-ablated hepatocytes, or whether the same RXRα-mediated mechanism would operate in the two cases and be magnified just under replicative conditions, remains to be determined.
Which RXRα heterodimer(s) could mediate the events that prevent early death of quiescent and proliferative hepatocytes is unknown. The liver contains a wealth of members of the nuclear receptor superfamily that can heterodimerize with RXRα (2, 7–10). Among these RXR partners, retinoic acid receptors and thyroid hormone receptors are potential candidates. First, it has been shown that PH of vitamin A-deficient rats leads to focal necrosis of the liver in the course of its regeneration (15) and that this necrosis could be prevented by vitamin A administration just before PH (14). Second, Torres et al. (12) have shown that, in the rat, normal hepatocyte polyploidization is regulated by the T3 thyroid hormone. Whether retinoic acid receptors (α, β, or γ) and/or thyroid hormone receptors (α or β) are RXR partners in the regulation of hepatocyte lifespan and replication will require further studies in which hepatocyte-specific somatic ablation of these receptor genes will be performed, remembering that double mutants might be required to circumvent problems linked to functional redundancy between receptor isotypes.
Acknowledgments
We thank M. A. Magnuson for Albumin-Cre mice, J. Auwerx for ApoCIII probe, and R. Lorentz, C. Gérard, and S. Bronner for technical assistance; we also thank M. LeMeur and the animal facility staff for animal care and the secretarial staff for preparation of the manuscript. This work was supported by funds from Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale, the Collège de France, the Hôpital Universitaire de Strasbourg, the Association pour la Recherche sur le Cancer, the Fondation pour la Recherche Médicale, the Human Frontier Science Program, the European Economic Community, and the Ministère de l'Éducation Nationale de la Recherche et de la Technologie. T.I. was supported by postdoctoral fellowships from the Centre National de la Recherche Scientifique, the Fondation de la Recherche Médicale, and the Toyobo Science Foundation. M.J. was supported by fellowships from the Association pour la Recherche sur le Cancer and the Institut National de la Santé et de la Recherche Médicale.
Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- ApoCIII
apolipoprotein CIII
- AST
aspartate aminotransferase
- Cre-ERT
fusion protein between the Cre recombinase and a mutated ligand binding domain of the human estrogen receptor
- CT
control
- ES
embryonic stem
- GOT
glutamate/oxalacetate transaminase
- GPI
glucose phosphate isomerase
- GPT
glutamate/pyruvate transaminase
- PH
partial hepatectomy
- PI
proliferative index
- RXR
retinoid X receptor
- RXRαKO
RXRα knockout
- Tam
tamoxifen
- wt
wild type
References
- 1.Mangelsdorf D J, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans R M. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kastner P, Mark M, Chambon P. Cell. 1995;83:859–869. doi: 10.1016/0092-8674(95)90202-3. [DOI] [PubMed] [Google Scholar]
- 3.Chambon P. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- 4.Giguère V. Endocr Rev. 1999;20:689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- 5.Steer C J. FASEB J. 1995;9:1396–1400. doi: 10.1096/fasebj.9.14.7589980. [DOI] [PubMed] [Google Scholar]
- 6.Michalopoulos G K, DeFrances M C. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 7.Wan Y-J Y, An D, Cai Y, Repa J J, Chen T H P, Flores M, Postic C, Magnuson M A, Chen J, Chien K R, et al. Mol Cell Biol. 2000;20:4436–4444. doi: 10.1128/mcb.20.12.4436-4444.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wan Y-J, Cai Y, Lungo W, Fu P, Locker J, French S, Sucov H M. J Biol Chem. 2000;275:28285–28290. doi: 10.1074/jbc.M000934200. [DOI] [PubMed] [Google Scholar]
- 9.Kastner P, Grondona J, Mark M, Gansmuller A, LeMeur M, Décimo D, Vonesch J L, Dollé P, Chambon P. Cell. 1994;78:987–1003. doi: 10.1016/0092-8674(94)90274-7. [DOI] [PubMed] [Google Scholar]
- 10.Sucov H M, Dyson E, Gumeringer C L, Price J, Chien K R, Evans R M. Genes Dev. 1994;8:1007–1018. doi: 10.1101/gad.8.9.1007. [DOI] [PubMed] [Google Scholar]
- 11.Ohmura T, Columbano G L, Columbano A, Katyal S L, Locker J, Shinozuka H. Pharmacol Lett Life Sci. 1996;58:211–216. doi: 10.1016/0024-3205(96)00039-2. [DOI] [PubMed] [Google Scholar]
- 12.Torres S, Diaz B P, Cabrera J J, Diaz-Chico J C, Diaz-Chico B N, Lopez-Guerra A. Am J Physiol. 1999;276:G155–G163. doi: 10.1152/ajpgi.1999.276.1.G155. [DOI] [PubMed] [Google Scholar]
- 13.Francavilla A, Carr B I, Azzarone A, Polimeno L, Wang Z, Van Thiel D H, Subbotin V, Prelich J G, Starzl T E. Hepatology. 1994;20:1237–1241. [PubMed] [Google Scholar]
- 14.Evarts R P, Hu Z, Omori N, Omori M, Marsden E R, Thorgeirsson S S. Am J Pathol. 1995;147:699–706. [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Z, Fujio K, Marsden E R, Thorgeirsson S S, Evarts R P. Cell Growth Differ. 1994;5:503–508. [PubMed] [Google Scholar]
- 16.Desvergnes B, Wahli W. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 17.Postic C, Magnuson M A. Genesis. 2000;26:149–150. doi: 10.1002/(sici)1526-968x(200002)26:2<149::aid-gene16>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 18.Imai T, Chambon P, Metzger D. Genesis. 2000;26:147–148. doi: 10.1002/(sici)1526-968x(200002)26:2<147::aid-gene15>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 19.Li M, Chiba H, Warot X, Messaddeq N, Gérard C, Chambon P, Metzger D. Development (Cambridge, UK) 2001;128:675–688. doi: 10.1242/dev.128.5.675. [DOI] [PubMed] [Google Scholar]
- 20.Imai T, Jiang M, Chambon P, Metzger D. Proc Natl Acad Sci USA. 2001;98:224–228. doi: 10.1073/pnas.011528898. . (First Published December 26, 2000; 10.1073/ pnas.011528898) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 22.Peters J M, Hennuyer N, Staels B, Fruchart J C, Fievet C, Gonzalez F J, Auwerx J. J Biol Chem. 1997;272:27307–27312. doi: 10.1074/jbc.272.43.27307. [DOI] [PubMed] [Google Scholar]
- 23.Rochette-Egly C, Lutz Y, Pfister V, Heyberger S, Scheuer I, Chambon P, Gaub M P. Biochem Biophys Res Commun. 1994;204:525–536. doi: 10.1006/bbrc.1994.2491. [DOI] [PubMed] [Google Scholar]
- 24.te Riele H, Maandag E R, Clarke A, Hooper M, Berns A. Nature (London) 1990;348:649–651. doi: 10.1038/348649a0. [DOI] [PubMed] [Google Scholar]
- 25.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder J C. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlüter C, Duchrow M, Wohlenberg C, Becker M H G, Key G, Flad H-D, Gerdes F. J Cell Biol. 1993;123:513–522. doi: 10.1083/jcb.123.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bralet M-P, Branchereau S, Brechot C, Ferry N. Am J Pathol. 1994;144:896–905. [PMC free article] [PubMed] [Google Scholar]
- 28.MacDonald R A. Arch Intern Med. 1961;107:335–343. doi: 10.1001/archinte.1961.03620030023003. [DOI] [PubMed] [Google Scholar]
- 29.Ponder K P. FASEB J. 1996;10:673–684. doi: 10.1096/fasebj.10.7.8635684. [DOI] [PubMed] [Google Scholar]
- 30.Columbano A, Shinozuka H. FASEB J. 1996;10:1118–1128. doi: 10.1096/fasebj.10.10.8751714. [DOI] [PubMed] [Google Scholar]
- 31.Saeter G, Schwarze P E, Seglen P O. J Natl Cancer Inst. 1998;80:950–957. doi: 10.1093/jnci/80.12.950. [DOI] [PubMed] [Google Scholar]
- 32.Saeter G, Schwarze P E, Nesland J M, Juul N, Pettersen E O, Seglen P O. Carcinogenesis. 1988;9:939–945. doi: 10.1093/carcin/9.6.939. [DOI] [PubMed] [Google Scholar]
- 33.Gupta S. Semin Cancer Biol. 2000;10:161–171. doi: 10.1006/scbi.2000.0317. [DOI] [PubMed] [Google Scholar]
- 34.Gerlyng P, Grotmol A T, Erikstein B, Huitfeldt H S, Stokke T, Seglen P O. Cell Prolif. 1993;26:557–565. doi: 10.1111/j.1365-2184.1993.tb00033.x. [DOI] [PubMed] [Google Scholar]