

Abstract
The catalytic asymmetric sulfenylation of double bonds has been achieved using a BINAM-based phosphoramide catalyst and electrophilic sulfur source. Simple alkenes as well as styrenes afforded sulfenylated tetrahydrofurans and tetrahydropyrans by closure with pendant hydroxyl or carboxyl groups. Intermolecular thiofunctionalization was also achieved with simple alcohols or carboxylic acids as the nucleophiles.
Olefins are among the most versatile building blocks in organic synthesis. Their utility largely derives from the myriad functionalization reactions that simultaneously form carbon-carbon or carbon-heteroatom bonds along with the creation of two new stereo-centers.1 This family of reactions includes the functionalization of alkenes with boron, nitrogen and oxygen moieties stereo-, regio-, and enantioselectively.2 However, the stereocontrolled transfer of sulfur3 and selenium,4 to alkenes still remains underdeveloped; in fact, no example of a catalytic, asymmetric thiofunctionalization of an unactivated olefin is extant.5,6 Given the prevalence of sulfur in certain classes of natural products,7 as well as the rich chemistry of sulfur that allows for further manipulations,8 a reliable and highly enantioselective method for vicinal thiofunctionalization is of interest.
The successful development of a catalytic, enantioselective sulfenylation reaction requires that certain critical mechanistic features be established. First, the AdE thiofunctionalization of alkenes with electrophilic sulfur(II) sources is now universally accepted to involve thiiranium ions.9 Because of the distribution of positive charge in these highly-strained, electrophilic species, attack of a nucleophile can occur at both the carbon and the sulfur centers. Reaction at the carbon centers forms stereodefined, vicinally functionalized products by an invertive ring-opening10 whereas reaction at sulfur regenerates the alkene along with a sulfenylated nucleophile. Experimental data shows that nucleophilic attack at the carbon atoms of thiiranium ions is preferred.11 Second, the configurational stability of thiiranium ions must be sufficient that, if generated in enantiomerically enriched form under catalytic conditions, they could be intercepted by nucleophiles without erosion in enantiopurity. A recent report from these laboratories confirmed that enantiomerically enriched thiiranium ions, generated stoichiometrically in situ, are configurationally stable at −20 °C and can be captured by a variety of nucleophiles with complete enantiospecificity.12 Furthermore, the configurational stability of thiiranium ions persists in the presence of an alkene, implying that racemization via olefin-to-olefin transfer is slow at −20 °C.11,13 Hence, if enantiomerically enriched thiiranium ions could be formed catalytically, their rate of racemization would be sufficiently slow such that capture by nucleophiles could form enantioenriched sulfides.12
The engineering of a catalytic, enantioselective thiofunctionalization first requires a functioning catalytic process. Previous studies from these laboratories demonstrated that seleniranium ions could be generated catalytically from weakly electrophilic selenium(II) sources by Lewis bases to form reactive ionic selenylating species in accord with the principle of Lewis base activation of Lewis acids.4b,14 Subsequent studies showed that chiral Lewis bases were capable of delivering an arylselenium moiety to simple olefins with modest enantioselectivity.4c We envisioned designing an analogous system wherein judicious choice of a sulfur(II) electrophile, along with a chiral Lewis base, would allow for the formation of enantio-enriched thiiranium ions, which would then be captured by a variety of nucleophiles to effect a catalytic asymmetric thiofunctionalization (Scheme 1).
Scheme 1.

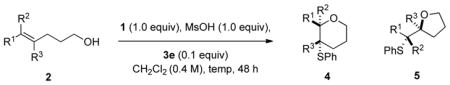
Orienting experiments employed stable, crystalline, commercially available sulfenylating agent N-phenylsulfenyl-phthalimide and (1) alcohol 2a. 4c,15 Because previous studies established the need for Lewis bases and Brønsted acids to effect chalcogenoetherifications,4c a survey of both components was undertaken. A suitable catalyst must balance sufficient Lewis basicity (to function as a sulfenyl transfer agent) with low Brønsted basicity (to remain active in the presence of a Brønsted acid). Tetrahydrothiophene (THT) was selected to initiate the survey in view of its established competence as a group transfer agent16 along with its weak Brønsted basicity (Table 1).17 Thus, 1, 2a, THT and TFA were combined at room temperature and thioether 4a was detected in significant amounts (entry 1).15b The use of a stronger Brønsted acid, MsOH (pKa (TFA) = 3.75, pKa (MsOH) = 1.6, in DMSO)18 allowed for complete conversion of 2a in less than 3 h (entry 2). Importantly, barely any reaction was detected after 24 h with either TFA or MsOH alone, excluding the possibility of simple Brønsted acid catalysis (entries 3 and 4). Next, a variety of Lewis bases containing different donor atoms were surveyed to determine their catalytic efficacy. HMPA proved ineffective, whereas DMPU(S), a thiourea, displayed moderate reactivity (entry 5). Considering the importance of phosphorus(V) compounds for selenofunctionalization,4b,c a number of phosphine chalcogenides were tested. Both Ph3P(S) and Cy3P(S) showed high catalytic activity, leading to complete conversion of 2a within 3 h (entries 6 and 7). Phosphoramides HMPA(S) and HMPA(Se) also showed potential; the HMPA(Se) catalyzed reaction was complete within 24 h (entries 8 and 9).
Table 1.
Survey of Achiral Lewis Bases
 | ||||
|---|---|---|---|---|
| entry | acid | Lewis base | conv., % | |
| 3 ha | 24 ha | |||
| 1 | TFA | THT | 33 | 70 |
| 2 | MsOH | THT | 100 | 100 |
| 3 | TFA | None | 0 | 0 |
| 4 | MsOH | None | trace | 10 |
| 5 | MsOH | DMPU(S) | 7 | 55 |
| 6 | MsOH | Ph3P(S) | 100 | 100 |
| 7 | MsOH | Cy3P(S) | 100 | 100 |
| 8 | MsOH | HMPA(S) | trace | 35 |
| 9 | MsOH | HMPA(Se) | 31 | 100 |
Conversion was determined by assuming 2a was converted only to 4a as no other significant product was detected by 1H NMR spectroscopy.
The next stage of reaction development was to identify structural elements that lead to high enantioselectivity in the thioetherification process. Because the softer sulfur and selenium donor atoms displayed good catalytic activity, chiral catalysts incorporating simple sulfides and phosphine sulfides (selenides) were investigated. Surprisingly, THT analogue 3a (Chart 1) and phosphine sulfide 3b both gave poor enantioselectivity (Table 2, entries 1, 2). Thio-phosphoramide 3c displayed good selectivity but poor reactivity (entry 3). Replacing sulfur with selenium (3d) increased the reactivity of the catalyst (entry 4) as expected. Further optimization identified 3e which afforded 4a with useful enantioselectivity (84:16) in 3 h.19 The enantioselectivity could be further improved by lowering the reaction temperature to −20 °C (with an attendant increase in reaction time, entries 6 and 7). Finally, the catalyst loading could be lowered to 0.1 equiv without adversely affecting the enantioselectivity (entries 8 and 9). Although sulfenylating agents other than 1 were tried, no difference in enantioselectivity was observed.20
Chart 1.

Table 2.
Survey of Chiral Lewis Bases
 | |||||
|---|---|---|---|---|---|
| entry | Lewis base | equiv | temp, °C | time,a h | e.r.b |
| 1 | 3a | 0.2 | 23 | 24 | 40:60 |
| 2c | 3b | 0.2 | 23 | 1 | 46:54 |
| 3 | 3c | 0.2 | 23 | 27 | 82:18 |
| 4 | 3d | 0.2 | 23 | 3 | 79:21 |
| 5 | 3e | 0.2 | 23 | 3 | 84:16 |
| 6 | 3e | 0.2 | −20 | 24 | 91:9 |
| 7 | 3e | 0.2 | −30 | N/A | 91:9 |
| 8 | 3e | 0.05 | −20 | 24 | 89:11 |
| 9 | 3e | 0.1 | −20 | 24 | 91:9 |
Time to full conversion.
Ratio of (2R, 3S)/(2S, 3R), absolute configuration established by independent synthesis of 4a, see the Supporting Information.
N-Phenylthiobenzotriazole and TFA were used.
With the feasibility of the catalytic asymmetric thioetherification demonstrated, the next stage was to establish the sensitivity of the reaction to the steric and electronic properties of the substrate. Under the optimal reaction conditions, substrate 2a provided pyran 4a in 80% yield with excellent constitutional and enantioselectivity (Table 3, entry 1).21 Electron-deficient alkene 2b reacted more sluggishly (41% conversion in 48 h), whereas electron-rich alkene 2c behaved comparably to 2a. On the other hand, the electronic character of the double bonds had only a minimal effect on enantioselectivity (entries 2 and 3). Geminal dimethyl substituents in the tether did not significantly affect the rate or enantiomeric composition of the products as both 2d and 2e afforded thioethers 4d and 4e in good yields and nearly identical enantioselectivities compared to 4a (entries 4 and 5). The constitutional selectivity was somewhat eroded for 4e, most likely because of the increased sensitivity of the approaching alcohol to the steric difference between the alkyl and phenyl groups. Non-conjugated alkenes were also useful substrates. For trans non-conjugated alkenes, the constitutional selectivity was dependent on the difference in size of the alkene substituents (entries 6 and 7). The increased compression in the endo-transition state disfavors 4 for alkenes with bulkier C(5) substituents.22 Excellent enantioselectivities were obtained with these substrates. For cis nonconjugated alkene 2h, the reaction proceeded in 72% yield, 20:1 site selectivity, but poor enantioselectivity (entry 8).23
Table 3.
Scope of the Intramolecular Asymmetric Sulfenylation Reaction.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | substrate | R1 | R2 | R3 | temp,°C | product | yield,a % | 4/5b | e.r.c,d,e |
| 1 | 2a | Ph | H | H | −20 | 4a | 80 | 49:1 | 91:9 |
| 2 | 2b | 4-CF3C6H4 | H | H | −10 | 4b | 36f | 25:1 | 88:12 |
| 3 | 2c | 4-MeO-C6H4 | H | H | −20 | 4c | 84 | >10:1 | 91:9 |
| 4 | 2d |
|
−20 |
 4d |
94 | 19:1 | 92:8 | ||
| 5 | 2e |
|
−20 |
 4e |
84 | 13:1 | 92:8 | ||
| 6 | 2f | CH2CH2Ph | H | H | −20 | 4f | 88 | 5:1 | 96:4 (4f) 96:4 (5f) |
| 7 | 2g | i-Pr | H | H | −20 | 4g | 71 | 17:1 | 96:4 |
| 8 | 2h | H | CH2CH2Ph | H | −10 | 5h | 81 | 1:20 | 54:46 |
| 9 | 2i | H | H | Ph | −20 | 5i | 85 | <1:99 | 62:38 |
| 10 | 2j | H | H | H | −10 | 5j | 72 | 1:50 | 83:17 |
| 11 | 2k | Ph | H | Me | 23 | 5k | 24 | 18:1 | 60:40 |
| 12 | 2l | Ph | Me | H | −20 | 4l | 82 | 17:1 | 70:30 |
| 13 | 6 |

|
−10 |
 7 |
83 | >99:1 | 91:9 | ||
Yield of isolated products.
Pyran/furan selectivity was determined by 1H NMR spectroscopy on the product mixture prior to chromatographic separation.
The enantiomeric ratio for the major constitutional isomer 4 or 5.
The enantiomeric ratio was determined by CSP-SFC analysis, see Supporting Information.
The absolute configuration of 4a was assigned by X-ray crystallographic analysis, see Supporting Information, all other compounds are assigned by analogy.
55% of unreacted 2b was recovered.
The generality of the thiofunctionalization reaction for different alkene types was also investigated. The substitution pattern of the double bond was found to have a major effect on the enantiomeric composition of the products. Geminally disubstituted alkene 2i produced furan 5i in 85% yield but only 62:38 e.r., whereas terminal olefin 2j cyclized in 72% yield and with 83:17 e.r. Trisubstituted olefin 2k was the least reactive, and 60% of the starting material could be recovered. On the other hand, isomer 2l was more reactive, affording 5l in 82% yield and 70:30 e.r. These results indicate that the presence of a substituent cis to the phenyl group is detrimental to the enantioselectivity and rate of the reaction. Notably, catalytic, asymmetric thiolactonization was possible as carboxylic acid 6 cyclized to form lactone 7 in high yield, and with high endo selectivity and good enantioselectivity.
The intermolecular capture of thiiranium ions by nucleophiles can also be achieved with this catalytic system. Exposure of 4-octene to the reaction conditions in the presence of 1.0 equiv of MeOH afforded phenylthio methyl ether 8 in 93% yield and 92:8 e.r. (Table 4 entry 1). Whereas 1-octene was unreactive at −20 °C, at room temperature the reaction was complete within 48 h, producing 9 in 77% yield and with 82:18 e.r. (entry 2). In the reaction of (E)-2-methyl-3-heptene, a 4:1 mixture of constitutional isomers 10a and 10b was obtained with 58% overall yield. Acetic acid was an effective nucleophile for the intermolecular thiofunctionalization as well (entry 4).
Table 4.
Intermolecular Sulfenylation of Simple Alkenes
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | R1 | R2 | NuH | temp, °C | product | yield,a % | a/b ratiob | e.r.c |
| 1 | n-Pr | n-Pr | MeOH | −20 | 8 | 93 | - | 92:8 |
| 2 | H | n-Hex | MeOH | 23 | 9 | 77 | 10:1 | 82:18 |
| 3 | i-Pr | n-Pr | MeOH | −20 | 10 | 58 | 4:1 | 84:16 (a) 84:16 (b) |
| 4 | n-Pr | n-Pr | AcOH | −20 | 11 | 77 | - | 91:9 |
Yield of combined isomers.
Isomer ratio determined by 1H NMR spectroscopy on samples prior to chromatographic separation.
Enantiomeric ratio of major isomer determined by CSP-SFC analysis, see Supporting Information.
Preliminary mechanistic experiments were performed to determine whether the composition of the constitutionally isomeric products (4 vs. 5) was established under kinetic control. A wide spectrum of behavior was noted ranging from no isomerization at room temperature (5j) to rapid isomerization at room temperature, but no isomerization at −10 °C (5h) to slow isomerization at −10 °C (5f). Thus, the ratios of constitutional isomers listed in Table 3 may not represent products of kinetic control.24 Nonetheless, the enantiomeric composition of the products in the isomerization experiments was maintained.
A catalytic cycle for the thiofunctionalization is proposed in Figure 1. Sulfenylation of the Lewis base 3e by 1 mediated by MsOH generates the active catalytic species 12. Evidence for this species is provided by 31P NMR spectroscopy in which the diagnostic signal for 3e disappears (91.6 ppm) and a new signal at 60.4 ppm is observed. This value is in good agreement with related [(R2N)3PX-YAr]+ compounds that have been prepared.4c,25 Transfer of the sulfenium ion from 12 to an alkene forms intermediate thiiranium ion 13, which, under control of the catalyst architecture, represents the enantiodetermining step.4c The enantioenriched thiiranium ion then undergoes intra- or intermolecular nucleophilic capture to deliver the corresponding enantioenriched thioether.
Figure 1.

Proposed catalytic cycle for asymmetric sulfenofunctionalization.
In conclusion, the first catalytic, asymmetric sulfenylation of simple alkenes has been achieved using a chiral selenophosphoramide catalyst. Both inter- and intramolecular thiofunctionalizations are possible for a variety of olefin types. Efforts are underway to expand the substrate scope, improve the selectivity of the reaction, and elucidate the origins of enantioselectivity.
Supplementary Material
Acknowledgments
Financial support was provided by the National Institutes of Health (R01 GM08525). D.J.P.K. was partially supported by the John M. Witt Jr. Fellowship. T.V. thanks the Alexander von Humboldt Foundation for a Feodor Lynen Research Fellowship. We thank M. D. Cullen and D. Kalyani for pioneering experimental contributions and M. T. Burk and for helpful discussions.
Footnotes
Supporting Information Available: Full experimental procedures, and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Patai S, editor. The Chemistry of Double Bonded Functional Groups. Wiley; Chichester: 1997. [Google Scholar]
- 2.(a) Crudden CM, Edwards D. Eur J Org Chem. 2003:4695–4712. [Google Scholar]; (b) Muniz K. Chem Soc Rev. 2004;33:166–174. doi: 10.1039/b307102m. [DOI] [PubMed] [Google Scholar]; (c) Jacobsen EN, Wu M. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 2 Springer; Heidelberg: 1999. [Google Scholar]; (d) Marko IE, Svendsen JS. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 2 Springer; Heidelberg: 1999. [Google Scholar]
- 3.(a) Gais HJ, Böhme A. J Org Chem. 2002;67:1153–1161. doi: 10.1021/jo010668b. [DOI] [PubMed] [Google Scholar]; (b) Zheng S, Gao N, Liu W, Liu D, Zhao X, Cohen T. Org Lett. 2010;12:4454–4457. doi: 10.1021/ol101915b. [DOI] [PubMed] [Google Scholar]
- 4.(a) Wirth T. Angew Chem, Int Ed. 2000;39:3740–3749. doi: 10.1002/1521-3773(20001103)39:21<3740::aid-anie3740>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Collins WR. Org Lett. 2007;9:3801–3804. doi: 10.1021/ol701617d. [DOI] [PubMed] [Google Scholar]; (c) Denmark SE, Kalyani D, Collins WR. J Am Chem Soc. 2010;132:15752–15765. doi: 10.1021/ja106837b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For a stoichiometric, asymmetric thioetherification see: Lucchini V, Modena G, Pasquato L. J Chem Soc Chem Comm. 1994:1565–1566.
- 6.Organocatalytic methods employing proline derivatives have been developed for the catalytic, asymmetric α-sulfenylation of aldehydes and enals. See: Marigo M, Wabnitz TC, Fielenbach D, Jorgensen KA. Angew Chem, Int Ed. 2005;44:794–797. doi: 10.1002/anie.200462101.Zhao GL, Rios R, Vesely J, Eriksson L, Córdova A. Angew Chem, Int Ed. 2008;47:8468–8472. doi: 10.1002/anie.200802335.
- 7.(a) Corey EJ, Gin DY, Kania RS. J Am Chem Soc. 1996;118:9202–9203. [Google Scholar]; (b) Bernardo PH, Brasch N, Chai CLL, Waring P. J Biol Chem. 2003;278:46549–46555. doi: 10.1074/jbc.M304825200. [DOI] [PubMed] [Google Scholar]; (c) Joyner PM, Liu J, Zhang Z, Merritt J, Qi F, Cichewicz RH. Org Biomol Chem. 2010;8:5486–5489. doi: 10.1039/c0ob00579g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sasaki E, Ogasawara Y, Liu H. J Am Chem Soc. 2010;132:7405–7417. doi: 10.1021/ja1014037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Metzner P, Thuillier A. Sulfur Reagents in Organic Synthesis. Academic Press; 1994. [Google Scholar]; (b) Page P, editor. Organosulfur Chemistry: Synthetic Aspects. Academic Press; London: 1995. [Google Scholar]
- 9.(a) Smit VA, Zefirov NS, Bodrikov IV, Krimer MZ. Acc Chem Res. 1979;12:282–288. [Google Scholar]; (b) Rayner CM. In: Organosulfur Chemistry: Synthetic Aspects. Page P, editor. Chapter 3 Academic Press; London: 1995. [Google Scholar]
- 10.Destro R, Lucchini V, Modena G, Pasquato L. J Org Chem. 2000;65:3367–3370. doi: 10.1021/jo991731o. [DOI] [PubMed] [Google Scholar]
- 11.Lucchini V, Modena G, Pasquato L. J Am Chem Soc. 1988;110:6900–6901. [Google Scholar]
- 12.Denmark SE, Vogler T. Chem Eur J. 2009;15:11737–11745. doi: 10.1002/chem.200901377. [DOI] [PubMed] [Google Scholar]
- 13.Denmark SE, Collins WR, Cullen MD. J Am Chem Soc. 2009;131:3490–3492. doi: 10.1021/ja900187y. [DOI] [PubMed] [Google Scholar]
- 14.Denmark SE, Beutner GL. Angew Chem Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 15.(a) No background reaction was observed between these two components at room temperature. (b) No reaction was observed in the absence of TFA at room temperature.
- 16.Piccinini A, Kavanagh SA, Connon PB, Connon SJ. Org Lett. 2010;12:608–611. doi: 10.1021/ol902816w. [DOI] [PubMed] [Google Scholar]
- 17.Laurence C, Gal J-F. Lewis Basicity and Affinity Scales: Data and Measurement. Chapter 3 John Wiley & Sons; Chichester: 2009. [Google Scholar]
- 18.Bordwell FG. Acc Chem Res. 1988;21:456–463. [Google Scholar]
- 19.Other modifications of the BINAM structure, including substitution at the 3,3′ positions and saturation of the rings did not increase enantioselectivity.
- 20.N-Thiophenylbenzatriazole and N-thiophenylsaccharin gave 78:22 and 79:21 er, respectively with 0.2 equiv of catalyst 3c at 23 °C compared to 79:21 for 1.
- 21.Reactions proceeded to full conversion unless otherwise noted. Reactions that were not complete within 48 h at −20 °C, were instead executed at −10 °C.
- 22.For transition state calculations and cyclization rates of the analogous seleniranium ions, see: Gruttadauria M, Meo PL, Noto R. Tetrahedron. 2001;57:1819–1826.
- 23.With cis-styrenes extremely long reaction times (20% conv. at 4 d) were observed even at room temperature.
- 24.For a recent demonstration of the Brønsted acid dependence of constitutional site selectivity see Wang H, Huang D, Cheng D, Li L, Shi Y. Org Lett. 2011;13:1650–1653. doi: 10.1021/ol200127n.See also: Fox DJ, House D, Warren S. Angew Chem, Int Ed. 2002;41:2462–2482. doi: 10.1002/1521-3773(20020715)41:14<2462::AID-ANIE2462>3.0.CO;2-3.
- 25.A 31-P chemical shift of a species containing a cationic (R3N)3P-Se-S-R subunit could not be found. The closest analogy is the cationic (R3N)3P-S-Se-Aryl subunit as in ref. 4c. See also: Godfrey SM, Ollerenshaw RTA, Pritchard RG, Richards CL. J Chem Soc, Dalton Trans. 2001;508–509
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.