

Abstract
The halophilic γ-proteobacterium Halomonas elongata DSM 2581T thrives at high salinity by synthesizing and accumulating the compatible solute ectoine. Ectoine levels are highly regulated according to external salt levels but the overall picture of its metabolism and control is not well understood. Apart from its critical role in cell adaptation to halophilic environments, ectoine can be used as a stabilizer for enzymes and as a cell protectant in skin and health care applications and is thus produced annually on a scale of tons in an industrial process using H. elongata as producer strain. This paper presents the complete genome sequence of H. elongata (4 061 296 bp) and includes experiments and analysis identifying and characterizing the entire ectoine metabolism, including a newly discovered pathway for ectoine degradation and its cyclic connection to ectoine synthesis. The degradation of ectoine (doe) proceeds via hydrolysis of ectoine (DoeA) to Nα-acetyl-l-2,4-diaminobutyric acid, followed by deacetylation to diaminobutyric acid (DoeB). In H. elongata, diaminobutyric acid can either flow off to aspartate or re-enter the ectoine synthesis pathway, forming a cycle of ectoine synthesis and degradation. Genome comparison revealed that the ectoine degradation pathway exists predominantly in non-halophilic bacteria unable to synthesize ectoine. Based on the resulting genetic and biochemical data, a metabolic flux model of ectoine metabolism was derived that can be used to understand the way H. elongata survives under varying salt stresses and that provides a basis for a model-driven improvement of industrial ectoine production.
Introduction
Concentrated salt solutions like salt or soda lakes, coastal lagoons or human-made salterns are extreme environments, inhabited by only a few forms of higher life, but usually maintain dense microbial populations comprising species from all three domains of life (Oren, 2002; 2008;Butinar et al., 2005). Global salt deposits show that evaporation of marine saltwater and the development of hypersaline habitats has been an ongoing process for millions of years, providing ample time for the evolution of halophilic Bacteria and Archaea that can flourish at high salt concentrations. Halophilic Bacteria and Archaea have developed two basically different osmoregulatory mechanisms to cope with ionic strength and the considerable water stress, namely the ‘salt-in-cytoplasm’ mechanism and the organic osmolyte mechanism (Oren, 2002). Organisms following the salt-in-cytoplasm mechanism adapt the interior protein chemistry of the cell to high salt concentration (Lanyi, 1974; Dennis and Shimmin, 1997; Kennedy et al., 2001; Tebbe et al., 2005). The osmotic adjustment of the cell can be achieved by raising the salt concentration (KCl) in the cytoplasm according to the environmental osmolarity. In contrast, microorganisms applying the organic osmolyte mechanism keep their cytoplasm, to a large extent, free of KCl and the design ofthe cell's interior remains basically unchanged. Instead, organisms of this group accumulate highly water-soluble organic compounds, in order to maintain an osmotic equilibrium with the surrounding medium (Roberts, 2006). These molecules do not disturb the cell's metabolism, even at high cytoplasmic concentrations, and are thus aptly named ‘compatible solutes’ (Brown, 1976).
The predominant compatible solutes in halophilic Bacteria are the amino acid derivatives glycine-betaine and ectoine (Roberts, 2006; Oren, 2008). Most chemoheterotrophic Bacteria can readily use glycine-betaine as a compatible solute if it is available in the environment. However, only a few are capable of de novo synthesis of glycine-betaine (Nyyssöläet al., 2000). Far more prokaryotes synthesize the aspartate derivative ectoine (1,4,5,6,tetra-2-methyl-4-pyrimidonecarboxylic acid) as their main compatible solute (Galinski et al., 1985; Severin et al., 1992; Roberts, 2006), which can also be utilized as an energy source by halophilic bacteria such as Halomonas elongata and Chromohalobacter salexigens (Vargas et al., 2006). Compatible solutes are beneficial for bacterial cells not only as osmoregulatory solutes, but also as protectants of proteins by mitigating detrimental effects of freezing, drying and high temperatures (Lippert and Galinski, 1992; Borges et al., 2002). The beneficial effect is explained by the unfavourable interaction of compatible solutes with the protein's peptide backbone. The lower affinity of compatible solutes, compared with water, for the protein surface is termed the osmophobic effect and results in a thermodynamic force that contributes to protein folding and increased protein stability (Bolen and Baskakov, 2001; Bolen, 2004). Ectoine possesses additional protective properties compared with other compatible solutes, and stabilizes even whole cells against stresses such as UV radiation or cytotoxins (Buommino et al., 2005; Furusho et al., 2005; Kanapathipillai et al., 2005; Kolp et al., 2006). It also protects against nanoparticle-induced inflammation in lung epithelia (Sydlik et al., 2009), and damage to the small bowel from ischaemia and reperfusion injury (Wei et al., 2009). Its protective properties make ectoine a valuable compound, which is marketed in health care and skin care products. Ectoine is therefore produced annually on a scale of tons by industry in a biotechnological process with the halophilic γ-proteobacterium H. elongata used as producer strain (Vreeland et al., 1980; Lentzen and Schwarz, 2006).
Halomonas elongata, which can tolerate salt concentrations well above 10% (1.7 M) NaCl, synthesizes ectoine via a pathway utilizing enzymes specified by three genes, ectABC (Göller et al., 1998). Cells do not rely only on de novo synthesis of ectoine for adaptation to high saline environments, but can also take up compatible solutes or precursors thereof from the medium. To enable solute uptake, H. elongata is equipped with a set of compatible solute transporters (Kunte, 2004) of which only one accepts ectoine as a substrate, namely, the ectoine-specific transporter TeaABC (Grammann et al., 2002; Tetsch and Kunte, 2002; Kuhlmann et al., 2008). TeaABC is not only required for the osmoregulatory accumulation of external ectoine, but also counterbalances an unknown system responsible for excreting endogenous ectoine to the medium (Grammann et al., 2002). Discovery of this system was based on the finding that strains of H. elongata with an inoperable TeaABC transporter constantly release ectoine to the surrounding medium. Despite this, cells of this mutant are able to keep the internal ectoine concentration at the same level as the wild-type strain. Apparently, the mutation of teaABC not only causes excretion of ectoine to the medium but also results in overproduction of ectoine. This observation led to the hypothesis that TeaABC might be linked to the regulation of ectoine synthesis (Kunte, 2006) and helped to design an ectoine production strain of H. elongata with higher productivity in ectoine synthesis than the wild-type strain (Kunte et al., 2002). However, the mechanism of ectoine excretion is still unknown. Because of the industrial importance of ectoine and its producer organism H. elongata, it is necessary to gain a deeper insight into ectoine transport across the cytoplasmic membrane, ectoine metabolism and the metabolic fluxes in H. elongata in order to increase ectoine production, e.g. by metabolic engineering (Marin-Sanguino et al., 2009). To attain deeper insight, a joint project of industry and research was established to sequence and analyse the genome of H. elongata DSM 2581T. The genome sequence helped to elucidate the degradation pathway of ectoine, which is presented in this paper for the first time. Furthermore, we provide a comparative analysis of the genes involved in ectoine synthesis and ectoine degradation from this genome with corresponding genes from microorganisms found in marine and saline environments and in soil. Finally, using a systems biology approach, a metabolic model on the genome data from H. elongata is introduced. This provides a basis for a model-driven improvement of ectoine production.
Results and discussion
Genome organization
The genome of H. elongata type strain DSM 2581T consists of a single chromosome of 4 061 296 bp (Table 1) with a high GC content (63.6%). For this chromosome, a total of 3473 protein-coding genes were predicted using the Reganor program (McHardy et al., 2004), which integrates data obtained from Critica (Badger and Olsen, 1999) and Glimmer (Delcher et al., 1999).
Table 1.
H. elongata DSM 2581T genome summary
| Chromosomes | 1 |
| DNA, total number bases | 4 061 296 |
| Coding density (%) | 87.1% |
| G + C content (%) | 63.6% |
| Genes, total number | 3 555 |
| Protein coding genes | 3 473 |
| Total RNA genes | 82 |
| 5S rRNA | 4 |
| 16S rRNA | 4 |
| 23S rRNA | 4 |
| tRNA genes | 68 |
| Other RNA genes | 2 |
| Genes with assigned EC number | 1 265 |
The most prominent duplications are the four rRNA operons. The 16S rRNA and 5S rRNA sequences are identical in all four operons, while the 23S rRNA sequences contain nine polymorphic sites. There are only seven other large repeats (1.4–3.0 kb with approximately 88–99% sequence identity). In all cases, these repeats represent duplications of genes, e.g. of enzymes that participate in central intermediary metabolism. In some cases, the duplicated genes are affected by a frameshift or diverge beyond the end of the duplicated region. The H. elongata chromosome shows a typical GC skew plot with two inflections (data not shown), indicating the origin of replication and the termination point (position 1835573). The ectoine biosynthesis genes ectABC are only 5 kb from the termination point.
Genome comparison
A comparison of the predicted protein sequence set of H. elongata with the NR database (downloaded from NCBI October 4, 2009) emphasizes a very close relationship of H. elongata with C. salexigens, a halophilic γ-proteobacterium of the family Halomonadaceae (Arahal et al., 2001). About half of the proteins could be reliably assigned at the species level (1672 out of 3473) using protein BLAST (Altschul et al., 1997) and MEGAN (Huson et al., 2007). The vast majority of the assigned proteins are related to C. salexigens (1544 assignments, corresponds to 92%). The few remaining MEGAN assignments are relatively equally distributed among a number of different species with Marinobacter sp. ELB17 (2.6%) dominating any other species (< 1%). For an overview, see Fig. S1. For 290 sequences, no assignment was reported to any taxon by MEGAN because no significant hit to the NR database was found.
Approximately two-thirds of the H. elongata proteins have an ortholog in C. salexigens as indicated by bidirectional best BLAST results (2367 proteins, 68.1%). They are very closely related, with an average of 69% sequence identity. Gene order is also well conserved. On closer examination of the set of H. elongata proteins that have an ortholog in C. salexigens, it was found that when two genes encoding such orthologous proteins are the nearest genetic neighbours in H. elongata then their corresponding partners in C. salexigens are also the nearest genetic neighbours in 70% of the cases. This high level of synteny is also evident from a MUMmer alignment of the two chromosomal sequences (Fig. S2). This results in a prominent X-alignment. Such X-alignments have been described for several interspecies comparisons (Eisen et al., 2000). The prominence of the X-alignment may indicate a close relationship between the two species, which is astonishing as both organisms are classified into distinct genera.
We used the Metanor tool of the GenDB genome annotation system (Meyer et al., 2003) for automatic function prediction. For enzymes, these were cross-checked with the data obtained by the PRIAM program (Claudel-Renard et al., 2003). A total of 1265 complete or partial EC numbers were assigned. Central metabolism and the biosynthetic pathways related to ectoine biosynthesis and degradation were manually curated in detail.
Halomonas elongata contains a complete set of ribosomal proteins. There are tRNA ligases for 19 of the 20 canonical amino acids. Asparagine (Asn) is not loaded as Asn but as aspartate with subsequent amidation to Asn by a gatABC-encoded enzyme.
To further analyse the protein set from H. elongata, we assigned cluster of orthologous groups of proteins (COGs) using the eggNOG 2.0 dataset as a reference (Muller et al., 2010). The assignment procedure, using an in-house script to analyse BlastP results, is described in Experimental procedures and in Supporting information.
We found a number of high-occupancy COGs that contain many different proteins from H. elongata (Table S1). Six COGs have more than 20 proteins of which COG0583 is most highly occupied with 57 members. The 20 most highly occupied COGs belong to four functional classes: (i) transcription regulators (six COGs), (ii) ‘general function’ enzymes (five COGs), (iii) transporters (seven COGs) and (iv) two-component systems (two COGs).
The most highly occupied COG0583 contains lysR family transcription regulators. The five high-occupancy COGs with ‘general function’ enzymes code for short-chain alcohol dehydrogenases, aldehyde dehydrogenases, acetyltransferases, methyltransferases and FAD-dependent oxidoreductases. One of the acetyltransferases in the frequent COG0454 is EctA, the first enzyme of the ectoine biosynthesis pathway. The seven high-occupancy COGs with transporters include the three subunits of TRAP transporters (COG1638, COG1593, COG3090, TRAP-type C4-dicaroxylate transport system, periplasmic component, large and small permease component). The teaABC ectoine transporter belongs to this set of COGs. The other frequently occurring transporters are MFS superfamily permeases and DMT superfamily permeases as well as two subunits of ABC-type transporters (ATPase, permease).
We searched for COGs, which are preferentially carried by halophilic/marine bacteria by comparing the proteins of 3 halophiles and 10 marines to 14 non-halophilic species (Supporting information, Table S2). Among the 97 COGs identified by this approach were many secondary transport systems that are thought to be dependent on Na+ symport, and sodium-proton exchangers. The transport systems that can be mainly found in halophiles belong to the NSS family and the TRAP family of transporters. The distribution of TRAP transporters was analysed by Mulligan and colleagues (2007) and they found that these uptake systems are extensively used in marine bacteria. There is evidence that TRAP transporters are powered by sodium symport (Mulligan et al., 2007) and perhaps the utilization of sodium-dependent transporters could be advantageous for halophilic bacteria that have a sodium gradient across their membrane. Members of the NSS family of transporters can be found in Eukarya, Bacteria and Archaea and transport nitrogenous substances (Beuming et al., 2006; Quick et al., 2006). In bacteria, NSS transporters catalyse the high-affinity uptake of amino acids by a sodium-symport mechanism (Androutsellis-Theotokis et al., 2003). Again, the preference of marine bacteria for these transporters can be explained by their dependency on sodium for transport. Both the NSS and TRAP transporter, also have a high affinity for their substrates (Androutsellis-Theotokis et al., 2003; Chae and Zylstra, 2006; Kuhlmann et al., 2008), which might be required in the marine environment with sometimes low solute concentrations.
High-salt adaptation may result in protein adaptation, e.g. by adjusting protein pI values. We analysed whether H. elongata has an unusual average pI when compared with each of the 27 organisms selected for identification of halophile-specific COGs (Supporting information, Table S3). If there is any pI shift in H. elongata proteins, the shift is only very slight and towards the acidic direction. These results indicate that an overall acidic proteome is not required for salt adaptation of halophilic bacteria employing the organic osmolyte mechanism.
Ectoine synthesis
Halomonas elongata synthesizes ectoine (1,4,5,6,tetra-2-methyl-4-pyrimidonecarboxylic acid) as its main compatible solute (Severin et al., 1992). Ectoine is synthesized from aspartate-semialdehyde, the central intermediate in the synthesis of amino acids belonging to the aspartate family (Fig. 1). Ectoine formation comprises three enzymatic steps (Peters et al., 1990; Ono et al., 1999). First, aspartate-semialdehyde is transaminated to 2,4-diaminobutyric acid (DABA) with glutamate as amino-group donor. The transamination is catalysed by DABA transaminase (EctB). Then, an acetyl group is transferred to DABA from acetyl-CoA by DABA-Nγ-acetyltransferase (EctA) in order to synthesize Nγ-acetyl-l-2,4-diaminobutyric acid. Finally, ectoine synthase (EctC) catalyses the cyclic condensation of Nγ-acetyl-l-2,4-diaminobutyric acid, which leads to the formation of ectoine. Under certain stress conditions (e.g. elevated temperatures) H. elongata converts some of the ectoine to 5-hydroxyectoine by ectoine hydroxylase (EctD) (Inbar and Lapidot, 1988; Wohlfarth et al., 1990).
Fig. 1.
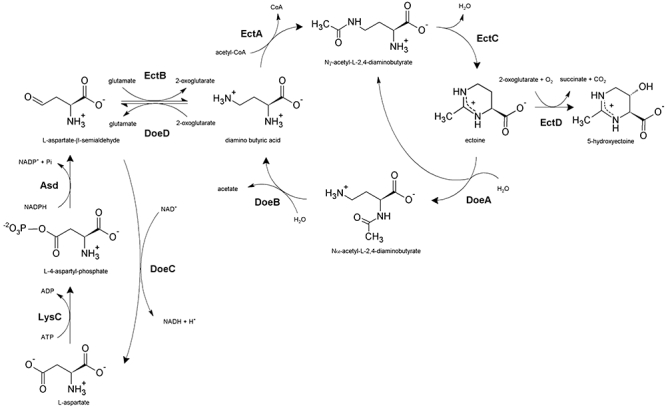
Metabolic pathway of the compatible solute ectoine in H. elongata. The degradation pathway is based on genetic and chromatographic analysis carried out in this study. Shown here is the hydrolysis of ectoine that leads directly to Nγ- and Nα-acetyl-l-2,4-diaminobutyric acid. For more details on DoeA activity see text. The depicted ectoine biosynthesis pathway is according to studies previously published (Peters et al., 1990; Göller et al., 1998; Ono et al., 1999). LysC: aspartate kinase; Asd: β-aspartate-semialdehyde-dehydrogenase; EctB: l-2,4-diaminobutyric acid transaminase; EctA: l-2,4-diaminobutyric acid Nγ-acetyltransferase; EctC: ectoine synthase; EctD: ectoine hydroxylase; DoeA: ectoine hydrolase; DoeB: Nα-acetyl-l-2,4-diaminobutyric acid deacetylase; DoeD: l-2,4-diaminobutyric acid transaminase; DoeC: aspartate-semialdehyde dehydrogenase.
The genes encoding the enzymes for ectoine de novo synthesis in H. elongata were identified by transposon mutagenesis, and the nucleotide sequence of ectAB as well as a partial sequence of ectC were published in 1998 (Göller et al., 1998). The genomic region containing these ectoine biosynthesis genes (ectA: Helo_2588, ectB: Helo_2589, ectC: Helo_2590) is shown in Fig. 2. The ectD gene (Helo_4008) encoding the hydroxylase for hydroxyectoine synthesis (Prabhu et al., 2004; Bursy et al., 2007) is located apart from the ectABC cluster. Immediately downstream of the 414 nt comprising ectC gene a further ORF is located (Helo_2591) that is predicted to encode a transcriptional regulator of the AraC family.
Fig. 2.

Ectoine synthesis gene organization in H. elongata and other prokaryotes. Shown are the ect genes from the subdivisions of the proteobacteria, the phyla Actinobacteria, Firmicutes, Planctomycetes and Thaumarchaeota. Only ectoine synthesis genes from prokaryotes with known genome sequences are depicted. l-2,4-diaminobutyric acid Nγ-acetyltransferase genes (ectA) are blue, diaminobutyric acid transaminase genes (ectB) are yellow, ectoine synthase (ectC) genes are green, ectoine hydroxylase genes (ectD, ectE) are red and aspartate kinase genes (ask) are pink. The accession numbers of all ect and ask genes are listed in Table S6.
The ectA gene encodes a 192-residue protein with a calculated molecular mass of 21.2 kDa. According to the studies of Ono and colleagues (Ono et al., 1999), DABA-acetyltransferase EctA displays a high specificity for its substrate DABA. EctA from H. elongata is a rather acidic protein with a calculated pI value of 4.8.
Similar acidic EctA proteins can be found in most of the marine and halophilic bacteria (e.g. C. salexigens, pI 5.3; Bacillus halodurans, pI 5.5; Halorhodospira halophila, pI 5.4; Blastospirellula marina pI 4.7) as well as in soil bacteria from the Actinomyces group (e.g. Nocardia farcinica, pI 5.0; Mycobacterium gilvum, pI 5.9; Streptomyces coelicolor, pI 5.0). In contrast, all but one of the remaining non-halophilic bacteria analysed in this study possess EctA proteins with a neutral or alkaline pI (e.g. Bacillus clausii, pI 7.5; Pseudomonas stutzeri, pI 8.0; Wolinella succinogenes, pI 9.0; Bordetella parapertussis, pI 8.4; Phenylobacterium zucineum, pI 8.9).
The ectB gene encodes a 421-residue protein with a molecular mass of 46.1 kDa, which requires K+ for its transaminase activity and for protein stability. Gel filtration experiments with purified protein from H. elongata indicate that the DABA aminotransferase EctB might form a homohexamer in the native state (Ono et al., 1999). The preferred amino group donor of EctB in the formation of DABA is glutamate, while in the reverse reaction DABA and γ-aminobutyrate are the preferred amino group donors to α-ketoglutarate.
The ectC gene encodes ectoine synthase, a 137-residue protein with a calculated molecular weight of 15.5 kDa and a pI value of 4.9. The EctC protein belongs to the enzyme family of carbon-oxygen lyases. In vitro experiments with purified EctC revealed that ectoine-synthase activity and affinity to its substrate are strongly affected by NaCl (Ono et al., 1999). N-acetylated amino acids having a carbon skeleton with one (ornithine derivatives) or two (lysine derivatives) atoms more than Nγ-acetyl-diaminobutyric acid are not suitable substrates for EctC. Galinski and co-workers, who described the ectoine biosynthetic pathway for the first time, demonstrated the reversibility of the ectoine synthase reaction when measured in crude cell extracts of Halorhodospira (formerly Ectothiorhodospira) halochloris (Peters et al., 1990). However, Ono et al. characterized purified EctC from H. elongata as an enzyme that is unable to carry out the reverse reaction from ectoine to Nγ-acetyl-diaminobutyric acid when ectoine was offered as substrate in the range of 10 mM to 1 M (Ono et al., 1999).
The ectD encoded ectoine hydroxylase consists of 332 amino acids and has a molecular weight of 37.4 kDa. The EctD protein is a member of an oxygenase subfamily within the non-heme-containing, iron (II)- and α-ketoglutarate-dependent dioxygenase superfamily. Ectoine hydroxylase was shown to catalyse the direct hydroxylation of ectoine to 5-hydroxyectoine (Bursy et al., 2007).
Transcriptional regulation of ectABC
Recent studies on the transcriptional regulation of ectABC in Bacillus pasteurii and Halobacillus halophilus revealed that ectABC is organized as one operon (Kuhlmann and Bremer, 2002; Saum and Müller, 2008), while in the halophilic γ-proteobacterium C. salexigens the transcriptional organization of the ect-cluster turned out to be rather complex (Calderón et al., 2004). Calderón et al. mapped a total of five promoters regulating ectABC transcription. Two σ70-controlled promoters, one σs-dependent promoter and a promoter of unknown specificity are located upstream of ectA, while a fifth promoter was found upstream of ectB.
To gain further information on the transcriptional regulation of the ectABC gene-cluster, we mapped the transcriptional initiation sites in H. elongata by RACE-PCR and found a different, but also complex promoter assembly. Two transcriptional initiation sites could be pinpointed in front of ectA, and one was mapped immediately upstream of ectC (Fig. 3). The two transcription initiation sites before ectA are located 25 and 92 bp, respectively, upstream from the ectA start codon. Inspection of the DNA sequence upstream of the first ectA initiation site (25 bp) revealed the presence of putative −10 and −35 sequences that resemble the binding site for the vegetative sigma factor σ70. The −10 and −35 sequences are separated by 17 bp, a typical spacing for promoters controlled by σ70. Upstream of the second initiation site (92 bp), a −10 DNA sequence was found that resembles σ38-controlled promoters. In addition to the −10 region, a so-called G-element exists at position −35. G-elements are characteristic for osmotically induced σ38 promoters (Lee and Gralla, 2004). RACE-PCR at the ectC gene mapped a transcription start point 47 bp upstream from the start codon (Fig. 3B). Upstream of the initiation site putative −12 and −24 sequences were found that are typical for σ54-controlled promoters. σ54-controlled promoters are often involved in transcription of nitrogen-regulated genes (Ausubel, 1984; Bordo et al., 1998). The two sequences of the putative σ54 promoter are appropriately spaced by four nucleotides. In addition, a sequence of 18 bp was found −111 to −128 bp upstream of the initiation site that resembles a consensus sequence required for transcription activation of some σ54 promoters controlled by FleQ (Hu et al., 2005). The transcriptional regulation of ectABC by an osmoregulated σ38 promoter and a σ54 promoter is in agreement with physiological observations made with other bacteria, such as Corynebacterium glutamicum and H. halochloris. In these organisms, it was shown that synthesis of the compatible solutes proline and glycine-betaine, respectively, is not only determined by salinity but also by nitrogen supply (Galinski and Herzog, 1990; Wolf et al., 2003).
Fig. 3.
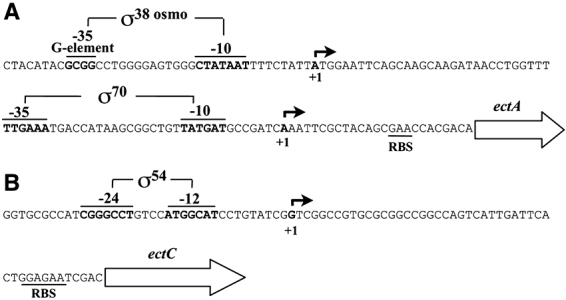
Transcription initiation sites and putative promoters of the ectoine synthesis genes ectABC. Nucleotide sequences of the ectA (A) and ectC (B) promoter regions. Arrows indicate the transcription initiation sites (+ 1), which were mapped by RACE-PCR. The −35 and −10 sequences of the σ70 and σ38 promoters upstream of ectA, and the −24 and −12 sequences of the σ54 promoter upstream of ectC are written in bold. (B) An 18 bp sequence GCCCGCTGACCATT is located at −111 to −128 bp upstream of the ectC transcription initiation site that resembles the consensus sequence of σ54 promoters that are controlled by FleQ (not displayed).
Organization of ectABC in different prokaryotes
The ectABC genes and the proteins for ectoine synthesis are very conserved among ectoine-producing bacteria. A study published recently by Lo and colleagues (2009) analysed the phylogenetic distribution of ect genes and showed that the prevalent organization of these genes is in a single cluster of at least three genes (ectABC), consistent with the analysis presented in this study. However, comparison of the H. elongata genome with other genomes revealed that the ectABC genes are not always organized in this way. A first analysis carried out by Vargas and co-workers with the genome of C. salexigens came to a similar result (Vargas et al., 2008). We extended this study and found that in Nitrosococcus oceani, ectC is located at a site different from ectAB. Marinobacter hydrocarbonoclasticus DSM 11845 [formerly aquaeolei VT8 (Márquez and Ventosa, 2005)] carries three ectC ORFs, but these are located at different sites within the genome and none are close to ectAB. In Alkalilimnicola ehrlichii, gene ectC is located downstream of ectAB but on the opposite strand. In all other genomes that were considered in this study, the ectABC components were clustered similar to ectABC from H. elongata. In summary, the way the ect-genes are organized in these organisms can be classified as follows (Fig. 2):
Bacillus halodurans, Ruegeria[formerly Silicibacter (Yi et al., 2007)] sp. TM1040, and all Vibrio species analysed in this study possess only one single ectABC cluster and are missing ectD that encodes the ectoine hydroxylase.
In H. elongata and two other members of the Oceanospirillales (C. salexigens, Alcanivorax borkumensis SK2), in γ-proteobacterium Saccharophagus degradans, and in B. clausii, the ectABC cluster and gene ectD can be found at separate sites within the genome. C. salexigens differs from H. elongata and the other members of this group in having a second ectD-like ORF named ectE. Similar to ectD, locus ectE is separated from ectABC.
The majority of genomes that were checked in this study, including the chromosome of the crenarchaeote Nitrosopumilus maritimus, contain a single ect-cluster comprising the genes ectABC and D.
In roughly half of the genomes that were compared with the H. elongata genome an additional ORF (ask) can be found encoding a putative aspartate kinase. The ORF ask is located downstream of ectB, ectC or ectD. All bacterial genomes in this study that contain ask are equipped with at least one further aspartate kinase (LysC). It is therefore tempting to speculate whether ask next to the ect components is coding for a specific kinase involved in ectoine synthesis. H. elongata and its halophilic relative C. salexigens do not possess such an ask. They rely on only one type of aspartate kinase, LysC (Helo_3742), responsible for the synthesis of ectoine and the amino acids lysine, threonine and methionine.
Homology analysis of aspartate kinases by Lo and colleagues (2009) revealed a separation of two subhomology divisions, which are denoted ASKα and ASKβ. Amino acid sequence analysis showed that LysC from H. elongata belongs to the ASKβ homology division and is most closely related to the ASKβ aspartate kinase from C. salexigens. According to the allosteric-specifity grouping of ASKβ enzymes, LysC of H. elongata is sensitive to the allosteric regulation of Thr and Lys. The gene encoding LysC in H. elongata is associated with the genes recA recX alaS lysC crsA tRNA, which is a conserved gene arrangement among γ-proteobacteria. This gene cluster is not associated with genes involved in the amino acid metabolism of the aspartate family (Lo et al., 2009) and our analysis of the lysC neighbourhood in H. elongata could not find any connections with the ectoine metabolism.
In our opinion, allosteric regulation alone is not a suitable mechanism in regulating LysC activity and thereby the internal ectoine concentration. Feedback or allosteric regulation is used in biological systems to achieve a certain optimal concentration of a metabolite. However, the ectoine content has to be adjusted constantly to match the external osmolarity and it is known that there is essentially a linear relationship between compatible solute content and the salt concentration of the medium (Kuhlmann and Bremer, 2002). Therefore, if an aspartate kinase were involved in controlling ectoine synthesis, then it should be also an osmoregulated enzyme and models explaining the osmoregulatory control of ectoine synthesis have been proposed (Kunte, 2006).
Ectoine degradation
Ectoine can be accumulated up to molar concentration by H. elongata depending on the salinity of the surrounding medium. Furthermore, ectoine can also be utilized as both a carbon and a nitrogen source by H. elongata and when ectoine is offered as a nutrient, it still serves as compatible solute (Göller, 1999). In order to find out how ectoine is degraded, the genome of H. elongata was compared with Sinorhizobium meliloti.
In S. meliloti, a cluster of five ORFs named eutABCDE was described with hypothetical functions in ectoine catabolism (Jebbar et al., 2005). No exact function was assigned to any of these five ORFs but their deduced amino acid sequences indicate they are similar to arylmalonate decarboxylases (eutA), threonine dehydratases (eutB), ornithine cyclodeaminases (eutC), aminopeptidases (eutD) and glutamate-desuccinylases/aspartoacylases (eutE). A homologue for each of the eutBCDE genes can be found within the chromosome of H. elongata (Fig. 4), but no eutA homologue could be identified. The homologues of eutBC (Helo_3660, 3659) and eutDE (Helo_3665, doeA; Helo_3664, doeB) are organized in two clusters that are separated by three ORFs (Fig. 4). These three ORFs are homologues of genes annotated as transcriptional regulator (Helo_3663, doeX), dehydrogenase (Helo_3662, doeC) and transaminase (Helo_3661, doeD) respectively.
Fig. 4.
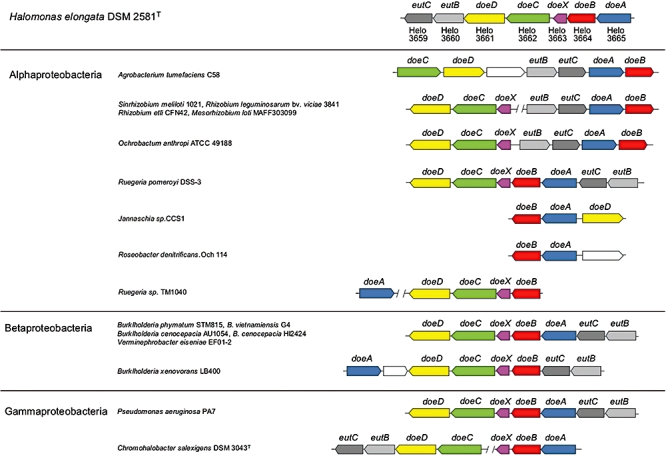
Ectoine-degradation gene organization in H. elongata and other bacteria. Ectoine hydrolase genes (doeA) are blue, Nα-acetyl-l-2,4-diaminobutyric acid deacetylase genes (doeB) are red, genes for AsnC/Lrp-like DNA-binding protein DoeX are pink (doeX), aspartate-semialdehyde dehydrogenase genes (doeC) are green, l-2,4-diaminobutyric acid transaminase genes (doeD) are yellow, eutB (putative threonine dehydratase) are light grey, eutC (putative ornithine cyclodeaminase) are dark grey. The gene doeA and doeB are homologues of eutD and eutE respectively. Only ectoine-degradation genes from bacteria with known genome sequences are depicted. Organisms carrying at least the two genes, doeA and doeB, could only be found in the proteobacteria domain. The accession numbers of all doe genes are listed in Table S6.
The ORFs Helo_3665, Helo_3664, Helo_3662 and Helo_3661 were chosen for mutation experiments, as the predicted enzymatic function of their gene products would make them candidates for the breakdown of ectoine into aspartate (Fig. 1). All four ORFs were deleted (in-frame null mutation) and the resulting mutants were either unable to utilize ectoine as a carbon source or they displayed reduced growth on ectoine. We therefore named the cluster doeABCD (degradation of ectoine ABCD, Fig. 4). Based on experiments (described below) and sequence homology, we named these ORFs doeA (Helo_3665, ectoine hydrolase), doeB (Helo_3664, Nα-acetyl-l-2,4-diaminobutyric acid deacetylase), doeC (Helo_3662, aspartate-semialdehyde dehydrogenase) and doeD (Helo_3661, diaminobutyric acid transaminase). A fifth ORF belonging to the doe cluster is located between doeB and doeC, and is named doeX (Helo_3663, Fig. 4). Deletion of Helo_3660 (eutB) and Helo_3659 (eutC) did not impair growth of the corresponding mutants on ectoine and we infer from these results that eutBC does not participate in ectoine degradation.
From the mutational and additional analytical experiments we concluded that the degradation of ectoine proceeds via hydrolysis of ectoine (DoeA) to the novel compound Nα-acetyl-l-2,4-diaminobutyric acid, deacetylation of Nα-acetyl-l-2,4-diaminobutyric acid (DoeB) to l-2,4-diaminobutyric acid, and a transaminase reaction (DoeD) leading to aspartate-semialdehyde. Finally, aspartate-semialdehyde is oxidized by DoeC to aspartate (Fig. 1). The proposed pathway is based on the following experimental and computational data:
(i)The doeA gene (Helo_3665) is a homologue to eutD and codes for a 399 aa protein (44.9 kDa, pI 5.0) that belongs to the peptidase-M24 family. Within that family, DoeA is similar to creatinase (creatine amidinohydrolase), which catalyses the hydrolysis of creatine to sarcosine and urea (Coll et al., 1990). Deletion of doeA created a mutant KB41 that was unable to grow on ectoine as carbon source. The doeA+ wild type could be restored in the ΔdoeA mutant by expressing doeA in trans from plasmid pKSB7 (pJB3Cm6::doeA), proving that no polar effect was causing the defect in ectoine catabolism.
To gain information on the enzymatic reaction catalysed by DoeA, the doeA gene was expressed in E. coli BL21 cells from plasmid pKSB11. After 16 h of expression, 1 mM ectoine was added to the salt medium (340 mM NaCl). E. coli is known to accumulate ectoine in the cytoplasm as compatible solute via osmoregulated transporters ProU and ProP but is unable to metabolize ectoine (Jebbar et al., 1992; Racher et al., 1999). The cytoplasmic fraction of E. coli was analysed by HPLC allowing for the detection of N-acetyl-l-2,4-diaminobutyric acid (N-Ac-DABA), the product of ectoine hydrolysis. Two forms of N-Ac-DABA were detected, which could be distinguished by comparison with a standard of Nγ-acetyl-l-2,4-diaminobutyric acid (Nγ-Ac-DABA) and Nα-acetyl-l-2,4-diaminobutyric acid (Nα-Ac-DABA). While neither Nα-Ac-DABA nor Nγ-Ac-DABA was formed in E. coli without doeA, both forms could be detected at a ratio of 2:1 in cells carrying doeA (Fig. 5). This unambiguously demonstrates that DoeA functions as ectoine hydrolase.
Fig. 5.
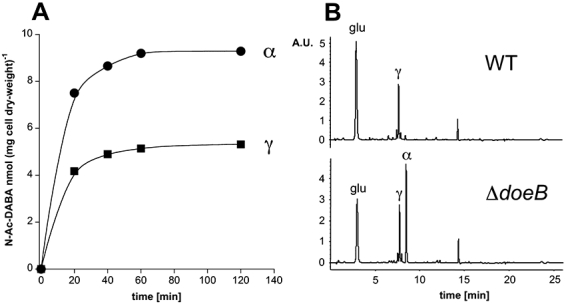
Chromatographic analysis of the cytoplasm from (A) E. coli expressing recombinant doeA and (B) H. elongata wild type and ΔdoeB-mutant strain KB42.A. Time course of Nα-Ac-DABA and Nγ-Ac-DABA formation from ectoine in E. coli expressing recombinant doeA. Cells were grown in mineral salt medium containing 340 mM NaCl. Ectoine was added (1 mM) and the cytoplasmic Nα-Ac-DABA (α) and Nγ-Ac-DABA (γ) content was determined by OPA-HPLC shortly before and 20, 40, 60 and 120 min after ectoine was added. Data presented are the mean from two independent experiments. No N-Ac-DABA was formed in E. coli cells without doeA.B. Chromatogram of the cytoplasm from H. elongata KB42 (ΔdoeB) and wild-type cells (WT), which were grown on mineral salt medium (1.03 M NaCl) in the presence of 500 µM glucose and 10 mM ectoine. After depletion of carbon-source glucose, the amino acid content of both strains was determined by OPA-HPLC. In wild-type cells glutamate (glu) and the ectoine precursor Nγ-Ac-DABA (γ) are detectable. In ΔdoeB mutant strain KB42 Nα-Ac-DABA (α) is accumulated as the predominant amino-reactive solute. Similar results were obtained when ectoine degradation was induced by hypoosmotic shock diluting the medium from 1.03 M down to 0.51 M NaCl. A.U., arbitrary units.
Similar results concerning ectoine hydolysis were obtained with H. elongataΔectA-mutant KB1 after growth with ectoine as carbon source. Although EctA-catalysed acetylation of DABA leading to Nγ-Ac-DABA is blocked, Nγ-Ac-DABA is accumulated in roughly the same concentration as in wild-type cells. In addition, Nα-Ac-DABA is also present although the pathway towards aspartate remained genetically unchanged. Apparently, ectoine hydrolase activity leads to the formation of Nα-Ac-DABA and Nγ-Ac-DABA also in the H. elongata background. Any significant contribution of ectoine synthase EctC to Nγ-Ac-DABA formation is rather unlikely, as purified EctC from H. elongata is described as an enzyme with no detectable reverse activity (Ono et al., 1999).
The specificity of ectoine hydrolase DoeA with respect to the isoforms of N-Ac-DABA remains somewhat ambiguous. While the results on DoeA expression in E. coli (Fig. 5) suggest the formation of both, Nα-Ac-DABA and Nγ-Ac-DABA, upon ectoine hydrolysis (as depicted in Fig. 1), cleavage of ectoine by DoeA could also produce only one isomer, which subsequently has to be converted into the corresponding isomer by an acetyltransferase. Whatever mechanism is employed by the cell, both are suitable and allow for the formation of Nα-Ac-DABA, which is the essential substrate for the subsequent catabolic enzyme DoeB (see below).
(ii)The 342 aa protein (36.6 kDa, pI 4.6) encoded by doeB (Helo_3664, homologue to eutE) is closely related to proteins of the succinyl-glutamate desuccinylase/aspartoacylase subfamily, which are part of the M14 family of metallocarboxypeptidases (Makarova and Grishin, 1999). The desuccinylase is involved in arginine catabolism while the aspartoacylase cleaves N-acetyl-aspartate into aspartate and acetate (Le Coq et al., 2008). Deletion of doeB resulted in a mutant KB42 that could not utilize ectoine as carbon and nitrogen source. The doeB+ wild type could be restored in mutant KB42 (ΔdoeB) by expressing doeB from plasmid pJSB3 (pJB3Cm6::doeB). Analysing the cytoplasmic fraction of wild type and ΔdoeB strain by HPLC revealed that Nα-acetyl-l-2,4-diaminobutyric acid is accumulated as the predominant amino-reactive solute in mutant KB42 while no Nα-Ac-DABA is detectable in the H. elongata wild type (Fig. 5). The Nγ-Ac-DABA concentration, however, remains still the same in KB42 and wild-type cells. Based on the results from the feeding experiments (no growth on ectoine as nitrogen-source) and the HPLC analysis (accumulation of Nα-Ac-DABA, unchanged Nγ-Ac-DABA level), we propose that the hydrolysis of ectoine is directly succeeded by DoeB-catalysed deacetylation of Nα-Ac-DABA.
While Nγ-Ac-DABA is not a substrate for DoeB, it serves again as a substrate for ectoine synthase EctC and can be converted back to ectoine. Nα-Ac-DABA, however, is removed from this cycle by deacetylation to DABA, which then can either flow off to aspartate or re-enter the ectoine synthesis pathway in wild-type cells. This closes the cycle of synthesis and degradation, which is powered by acetylation and deacetylation of DABA and Nα-Ac-DABA respectively (Fig. 1). As we will describe below, such a cycle provides a fast mechanism for the cell to regulate the cytoplasmic ectoine concentration.
(iii)The doeD gene (Helo_3661) encodes a putative aspartate aminotransferase (469 aa, 50.8 kDa, pI 5.6) with similarities to the PLP-dependent aspartate aminotransferase superfamily. Deletion of doeD resulted in mutant KB48 (ΔdoeD) that was impaired in growth on ectoine as a sole carbon source. In saline minimal medium (510 mM NaCl), the doeD deletion reduced the growth rate of strain KB48 threefold down to 0.077 h−1 compared with 0.248 h−1 observed with the wild type indicating a participation of DoeD in ectoine degradation. Deletion of the aminotransferase gene ectB, resulting in mutant SB1 (ΔectB), or in doeD-mutant KB48, resulting in the double knockout mutant SB1.1 (ΔectB, ΔdoeD), did not further reduce growth on ectoine. Because both mutant strains were still able to synthesize ectoine other transaminases (or amidases) are still active in H. elongata, converting DABA to L-aspartate-β-semialdehyde, and likewise, L-aspartate-β-semialdehyde to DABA, which can be readily explained by the rather broad substrate specificity of transaminases (Fotheringham, 2000).
(iv)The doeC gene (Helo_3662), located upstream of doeD, encodes a putative dehydrogenase (493 aa, 53.1 kDa, pI 4.8) and is most closely related to those dehydrogenases that act on aldehyde substrates. Knockout of doeC abolished growth of H. elongata strain KB47 (ΔdoeC) on medium containing ectoine as sole carbon source. Growth could be restored in doeC-mutant KB47 when ectoine was offered as nitrogen source in the presence of 10 mM glucose. This finding, together with the results obtained from the doeB mutant KB42, helped to determine the sequential order of the enzymatic reactions as depicted in Fig. 1, in which the dehydrogenase reaction and the transaminase reaction follow after the DoeB catalysed deacetylation of Nα-Ac-DABA.
However, according to the proposed pathway, acetate is split off from Nα-Ac-DABA in the second reaction. Acetate released in this reaction could serve as carbon source and should in principle allow dehydrogenase mutant KB47 to display at least some minimal growth on medium containing ectoine. We were able to prove through feeding experiments, that H. elongata can indeed utilize acetate as carbon source and that wild-type strain DSM 2581T, strain KB41 (ΔdoeA) and strain KB47 (ΔdoeC) are able to grow on medium containing 10 mM, 20 mM and 40 mM acetate respectively (Fig. S3). Adding 10 mM ectoine to the acetate medium suppressed growth of dehydrogenase-mutant KB47, while strain KB41 and KB42, as well as wild-type DSM 2581T, still managed to grow (Fig. S3). This finding explains why strain KB47 cannot grow on ectoine medium and fails to metabolize the internal acetate, which is split off from Nα-Ac-DABA during ectoine degradation. For now, we can only speculate about the underlying mechanism that causes the inability of strain KB47 to utilize acetate in the presence of ectoine. Internal accumulation of intermediates such as DABA or aspartate-semialdehyde to toxic levels cannot be ruled out as the reason for growth inhibition. However, because mutant KB47 can still feed on ectoine as nitrogen donor in the presence of 10 mM glucose, we favour the idea that some kind of catabolite repression is the reason for the failure of KB47 to grow on medium containing both acetate and ectoine.
Transcriptional organization of doeABX and DNA-binding protein DoeX
To characterize the genetic organization of the doe components, we carried out RT-PCR and RACE-PCR experiments. RT-PCR revealed that doeAB and the adjacent ORF, which was named doeX (Helo_3663), are organized as one operon (Fig. S4). The doeCD components are not part of the doeABX operon. The transcriptional initiation site of the doe-locus was mapped by RACE-PCR experiments and could be pinpointed 154 nt upstream of doeA. Inspection of the sequences upstream of the transcription start site revealed the presence of putative −10 and −35 sequences that resemble the consensus sequences of σ70-dependent promoters (Fig. S4).
The doeX gene was annotated as an ORF encoding a 158 aa transcriptional regulator (17.9 kDa, pI 5.8) belonging to the AsnC/Lrp family of DNA-binding proteins. Proteins of the AsnC/Lrp family are known to control the expression of a large number of operons, often in response to amino acid effectors, and can act as both transcriptional repressor and activator (Thaw et al., 2006). To test whether DoeX is a DNA-binding protein, recombinant DoeX protein was purified from E. coli and applied to electrophoretic mobility shift assays with labelled DNA (Fig. 6). DoeX was found to specifically bind to a 46 nt sequence located directly upstream to the doeA start codon.
Fig. 6.
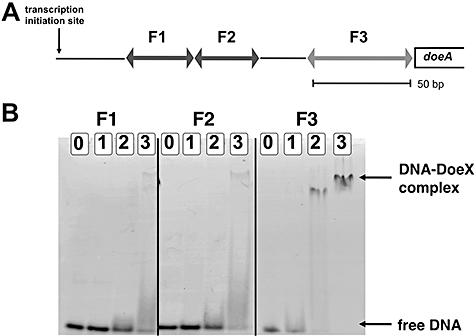
Electrophoretic mobility shift assays (EMSA) with DNA-binding protein DoeX.A. EMSA were performed to detect interactions of purified DoeX protein with labelled DNA fragments F1, F2 and F3 located upstream of the doeABX operon.B. In the presence of competitor DNA (500-fold to 1000-fold excess), DoeX specifically binds to fragment F3 resulting in a mobility shift on a non-denaturing polyacrylamide gel. Each assay (20 µl) contained 2 pmol labelled DNA. Lane 0, assay without DoeX protein (control); lane 1, 10 pmol DoeX; lane 2, 20 pmol DoeX; lane 3, 40 pmol DoeX.
Organization of doe genes in bacteria
From comparative genomic data, the ectoine degradation pathway described for H. elongata is mostly employed by non-halophilic organisms that, according to their genetic makeup, are unable to synthesize ectoine de novo. The doeA/eutD and doeB/eutE sites could be found within the genomes of 18 bacteria, all belonging to the proteobacteria (Fig. 4). Besides H. elongata, only two of them are ectoine-synthesizing organisms, namely C. salexigens and Ruegeria (Silicibacter) sp. TM1040. Homologues of both the doeA and doeB sites of H. elongata can be found in many other Bacteria, predominantly in the Rhizobiales of the α-proteobacterial domain and in the Burkholderiales of the β-proteobacteria. For further analysis, we selected only a few representatives from larger sets of highly similar sequences originating from species such as Burkholderia, which appear overrepresented in the list of completely sequenced genomes. The reduced set of sequences contains 18 members, which share a relatively high similarity (48% to > 80% sequence identity) with doeA of H. elongata (Helo_3665). With two exceptions (Ruegeria sp. TM1040 and Burkholderia xenovorans LB400), doeAB is clustered in all these genomes. A second set of nine doeA homologues with a lower similarity to Helo_3665 (30–45% sequence identity) was found within the Firmicutes, Euryarchaeota and the γ-proteobacteria groups. In these genomes, the doeA site is located in a different genetic neighbourhood with no other genes of the ectoine degradation pathway being present. This is illustrated in the computed doeA phylogeny (Fig. 7), where the two sequence sets form separate branches (drawn in black and red colour respectively). The two organisms in which doeA and doeB are not adjacent (Ruegeria sp. TM1040 and B. xenovorans) are highlighted (branches also drawn in red). For reference, organisms carrying the ectABC genes are marked in blue.
Fig. 7.
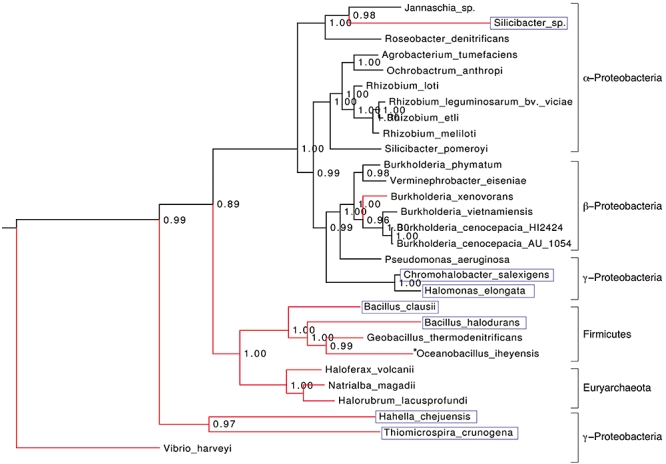
Phylogenetic tree of DoeA homologues and their position in the standard phylogeny (‘tree of life’). Branches marked in black correspond to genomes with the same ‘clustered’ organization of the doeAB genes. Red colour is used for those genomes where doeA is located separate from doeB <Ruegeria[previously Silicibacter (Yi et al., 2007)] sp. TM1040, B. xenovorans> or where doeB is not present at all. Organisms considered to be ectoine synthesizers based on the presence of the ectABC genes are boxed in blue. Numbers printed at the nodes are confidence values (varying from 0 to 1) derived from Bayesian statistics. *Oceanobacillus iheyensis carries an ectBC cluster but no ectA.
In most ectoine producers compared in our investigation (Fig. 2), either only doeA-like ORFs could be found, or none of the doe components were present. We therefore assume that either ectoine is not metabolized in these bacteria or that alternative pathways in ectoine degradation must exist with, and without, DoeA participation. In analogy to the glutamate-ornithine pathway, ectoine could be alternatively metabolized to aspartate via Nα-Ac-DABA, Nα-acetyl-aspartate-semialdehyde and Nα-acetyl-aspartate. Also, it cannot be ruled out that in organisms without Doe enzymes, a reverse synthesis pathway degrades ectoine, bypassing the irreversible EctA acetyltransferase reaction.
Ectoine metabolism: reconstruction, modelling and energetic considerations
The metabolic capabilities of an organism are one of the major aspects of cellular physiology. Genome annotation with an emphasis on metabolic reconstruction provides a basis to analyse metabolic capabilities and their impact on the specific adaptation of the organism to its natural environment. The immense amount of data generated, such as the 1265 complete or partial EC numbers assigned for H. elongata, calls for a computational strategy to drive a comprehensive analysis. As a first step, we have concentrated on the metabolism of ectoine, as this compatible solute is a key metabolite for survival and success of the halophilic bacterium H. elongata. A Flux Balance Analysis (Varma and Palsson, 1993a,b;) approach was selected to create a mathematical model of ectoine metabolism as detailed in Supporting information. Using this model, we have analysed two aspects of ectoine metabolism: (i) The energy balance of the glucose to ectoine conversion computed and compared with previous calculations reported in the literature (Oren, 1999). (ii) We attempted to predict the role of the proposed ectoine biosynthesis/degradation cycle.
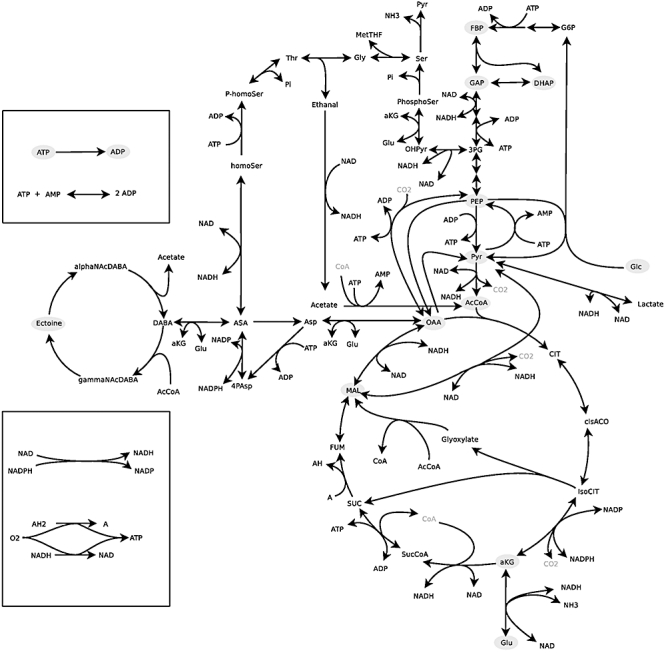
The metabolic network, reconstructed from the genome of H. elongata, contains a series of enzymes, which are involved in biosynthesis of ectoine from glucose (Fig. 8). Overall, glucose is converted to two molecules of PEP/pyruvate, one of which is converted to acetyl-CoA, the other to aspartate-semialdehyde via oxaloacetate. Ectoine is then produced from one molecule each of aspartate-semialdehyde and acetyl-CoA. The set of available enzymes allows for several distinct pathways, some of which result in an identical overall reaction while others differ with respect to the ATP balance. An exhaustive analysis is required to ensure that all alternatives have been considered. While this is difficult by manual inspection, this becomes a feasible task once a model is available so that mathematically sound techniques can be applied.
Fig. 8.
Depiction of the pathways considered in the metabolic model for ectoine.
In one of the possible pathways, PEP is converted to oxaloacetate through PEP carboxylase (EC 4.1.1.31). This particular case has been used for the previous calculations on the energetic costs of ectoine synthesis presented by Oren (1999). Oren's calculations assume nitrogen assimilation via glutamine synthetase (EC 1.4.1.13). With this assumption, the conversion of glucose to ectoine costs 2 ATP. However, an ATP-neutral pathway is possible using glutamate dehydrogenase (EC 1.4.1.2) as a more energy-efficient nitrogen assimilation reaction. This is based on the annotation of a NADH-dependent glutamate dehydrogenase (Helo_3049) in H. elongata. An ATP-neutral conversion of glucose to ectoine is consistent with a calorimetric analysis carried out by Maskow and Babel (2001), in which the authors conclude that an efficiency of approximately 100% was reached in the experiment.
The annotated genome allows for yet another solution, in which a total conversion of glucose into ectoine is possible with the simultaneous generation of ATP. The additional ATP can be obtained by using a different pathway, which proceeds via pyruvate to oxaloacetate and is catalysed by the malic enzyme (EC 1.1.1.38) and malate dehydrogenase (EC 1.1.1.37). An estimation of the reaction enthalpy for the conversion of glucose to ectoine shows that this pathway is not only stoichiometrically but is also thermodynamically feasible (see Supporting information). Currently, this hypothetical pathway is awaiting an experimental validation.
In summary, three alternative possibilities for conversion of glucose to ectoine differ in ATP production: (i) the pathway originally proposed by Oren (1999) requires two ATP for each molecule of ectoine, (ii) the Oren pathway with a modified nitrogen assimilation step is energy-neutral and (iii) the most energy-efficient pathway, which is thermodynamically feasible and has been revealed by modelling of ectoine biosynthesis, allows to generate one ATP per ectoine molecule. This has clear physiological implications. In case (ii), the energy-neutral pathway allows for a 100% ectoine yield only if no other ATP-utilizing process operates. This does not reflect the reality of a living cell, where maintenance processes require additional ATP generation. Only pathway (iii), which is the most energy-efficient, allows a 100% ectoine yield as long as other metabolic processes do not drain more than one ATP per glucose.
The novel degradation pathway of ectoine we introduced here may result in an apparent futile cycle, in which ectoine can be synthesized and degraded simultaneously resulting in a net conversion of acetyl-CoA into acetate. Acetate can be reconverted to acetyl CoA by acetate : CoA ligase (AMP-forming) (EC 6.2.1.1). Therefore, if such a cycle were active, it would result in an increased apparent cost of two ATPs per turn for ectoine synthesis.
The question remains why the cell would invest two high-energy phosphate bonds to run a cycle of ectoine synthesis and degradation. We believe that such a cycle is an elegant mechanism to control the speed of change in internal ectoine concentration as response to external changes in osmolarity. The turnover (or response) time of a metabolite, defined as the ratio between its concentration and the flux through it in the steady state, has been identified as a good indicator of the timescale of its transient responses (Nikerel et al., 2009). Metabolites with a high turnover tend to complete transitions faster than those with a low turnover. Thus, by keeping a flux through the synthesis/degradation cycle, the cell can achieve fast changes in ectoine levels to quickly respond to changes in external osmolality.
According to our model of a simultaneous activity of ectoine synthesis and degradation (ectoine cycle), disrupting the ectoine degradation pathway should lead to a lower ATP load for cells synthesizing ectoine and thereby result in higher ectoine productivity. In order to verify our proposed ectoine cycle and the mathematical model for ectoine synthesis, the doeA gene was deleted in the ectoine excreting mutant KB2.11 (ΔteaABC). Mutants with a dysfunctional or missing TeaABC ectoine transporter were shown to lose ectoine constantly to the medium (Grammann et al., 2002; Kunte et al., 2002). As the mathematical model was developed for non-growing cells, ectoine synthesis and export was analysed in minimal medium (510 mM NaCl) with non-growing cells (OD600 = 2.6; 100 µg ml−1 chloramphenicol) of mutant KB2.13 (ΔteaABC, ΔdoeA) and the parental strain KB2.11. While KB2.11 accumulated 40 mg ectoine l−1 h−1, the degradation mutant KB2.13 accumulated 50 mg ectoine l−1 h−1, which corresponds to a 20% higher productivity of strain KB2.13. This result supports our proposed regulatory ectoine cycle and demonstrates the usefulness of the mathematical model in predicting the effect of mutations on the bacterial metabolism.
Conclusion and outlook
Determining the complete genome sequence of H. elongata DSM 2581T has increased our understanding of the metabolism of ectoine and will be the first step in our work towards optimizing the industrial production of the compatible solute ectoine. We were able to completely reconstruct ectoine biosynthesis from glucose and ammonia, by annotating the genes encoding for the central metabolic enzymes and the pathways leading to the ectoine precursors aspartate and glutamate, including the enzymes responsible for the metabolite interconversions at the PEP-pyruvate-oxaloacetate node. In addition, we have identified a pathway for ectoine degradation and shown its cyclic connection to ectoine synthesis. On the basis of these data, a Flux Balance Analysis model of ectoine metabolism has been developed.
Initial steps in using this new data for strain optimization have been undertaken and are described briefly in this paragraph. First, the deletion of gene doeA, encoding the ectoine hydrolase protein, which catalyses the first step in the ectoine degradation pathway, has led to increased volumetric productivity of ectoine as predicted by our metabolic model. Furthermore, the genome sequence has enabled us to identify the gene for polyhydroxyalkanoate synthase phaC, which is important because Halomonas species are also known to synthesize polyhydroxyalkanoates in parallel to ectoine (Mothes et al., 2008). Finally, mutagenesis experiments are now underway to abolish polyhydroxyalkanoate biosynthesis by deleting phaC, which will hopefully enable new insights into metabolism of this energy storage compound and may also lead to a mutant strain that is able to convert glucose to ectoine more efficiently.
The metabolic model presented in this study only considers ectoine biosynthesis and the influence of different ATP-loads on its synthesis (Fig. S5). We are currently developing an extended model that will also include growth (increase of biomass). The extended model will help us to evaluate the effect of alternative C-sources on ectoine production, including biologically and environmentally relevant substances such as glutamate or glycerol. In addition, the use of extended models allows us to make specific changes in the metabolism of the organism, resulting in an increase of the carbon flux towards the formation of ectoine. Koffas and co-workers (Koffas et al., 2003) successfully used this strategy to improve lysine production in C. glutamicum. By coordinated overexpression of genes encoding two flux-controlling enzymes in central carbon metabolism and the lysine pathway, they were able to enhance the carbon flux towards its product lysine, resulting in an increase in lysine specific productivity by 250%.
The availability of genomic information paves the way for post-genome technologies such as DNA array and proteomics, which can now be employed with H. elongata. These technologies will accelerate the studies of osmoregulation in this halophilic bacterium and will enable us to manipulate its metabolism to create more efficient producer strains for ectoine.
Experimental procedures
Sequencing strategy
Whole genome sequencing was carried out in a two-step process. First, de novo sequencing was done using a Roche/454 GS 20 instrument (Margulies et al., 2005). Using the manufacturer's protocols random genomic DNA libraries were constructed and two sequencing runs were carried out with GS 20 chemistry (max. read length 100 bp). A total of 722 276 reads resulting in 70.6 Mb (average read length 98 bp) were assembled into 100 contigs of 4.0 Mb sequence using the Newbler assembler (Margulies et al., 2005). The resulting average coverage was 17.4 fold. As the applied assembly parameters were highly conservative, multicopy segments in the genome caused frequent termination of contig assembly in order to avoid misassemblies. Phred-like quality values were computed for the Newbler-generated contig sequences, similar to Sanger-based quality scores. The resulting contigs were then treated as if they were individual long sequencing reads in later phases of the assembly. In the second phase, a fosmid library was generated and end-sequenced. Phosmid end-sequences were co-assembled together with the 454-based contigs using the Phred-Phrap-Consed program package (Gordon et al., 1998). Fosmid end-sequences were used to order contigs. Remaining gaps were closed by either fosmid sequencing or by a PCR approach. The rRNA operons and other long repeats were solved by a mini-assembly strategy (Pfeiffer et al., 2008a). Each of the four rRNA operons was amplified as a long PCR product and sequenced on both strains by a nested set of primers. The individual rRNA operons were assembled and the resulting contigs were exported (including quality values) and added to the final assembly as if they were single reads.
Genome annotation
For gene prediction, the Reganor program (McHardy et al., 2004) from the annotation package GENDB (Meyer et al., 2003) was used, which integrates results from Critica (Badger and Olsen, 1999) and Glimmer (Delcher et al., 1999). Genes for transfer RNA (tRNAs) were predicted using tRNAscan (Lowe and Eddy, 1997). Genes encoding ribosomal RNA (rRNAs) were predicted using RNAmmer (Lagesen et al., 2007) except for the 16S rRNA 3′ end, which was set according to Lin and co-workers (Lin et al., 2008). Automated function prediction was performed using the Metanor tool of the GenDB genome annotation system (Meyer et al., 2003). For enzymes, these were compared with the data obtained by the PRIAM program (Claudel-Renard et al., 2003) in a manual curation effort. We used the HaloLex system (http://www.halolex.mpg.de) (Pfeiffer et al., 2008b) as a genome annotation platform.
Genome analysis
The protein set from H. elongata was compared with that of C. salexigens and other organisms using orthologous group analysis (COGs). Assignments for C. salexigens and other species were taken from eggNOG 2.0 (Muller et al., 2010). Assignments for H. elongata were made by a PERL script based on BlastP comparisons with the eggNOG 2.0 protein sequences. The same COG has been assigned for 98% of the 2367 proteins with a bidirectional best blast between H. elongata and C. salexigens. Assignment discrepancies are concentrated in distant bidirectional best blast pairs with reduced sequence identity values. For further details see the Supporting information.
We attempted to identify COGs, which are preferentially occupied in halophilic/marine organisms. We compared the occupancy between 3 halophilic/10 marine bacteria versus 14 standard bacteria (listed in Table S1). COGs were considered halophilic when the fraction of organisms having at least one COG member was twice that of the standard organisms. As an additional criterion, the COG had to be occupied in 2 of 3 halophiles or 5 of 13 halophile/marine organisms.
High-salt adaptation may result in protein adaptation, e.g. by adjusting protein pI values. We used the COG assignments to select protein pairs between H. elongata and those query organisms selected for identification of halophile-specific COGs. If there is only one COG member in the two organisms, the protein pair is used for subsequent computation of the difference in pI value. With this selection method, the pI difference could be averaged over c. 500 and 950 protein pairs for each of the 27 organisms included in the analysis (Table S2, Table S4).
Data analysis and comparative genomics
To find all genes encoding proteins involved in ectoine degradation and to compare gene clusters of ORFs encoding Doe-like proteins, BLAST queries (Altschul et al., 1997) of all available microbial genomes included at NCBI (http://ftp://ftp.ncbi.nih.gov/genomes/Bacteria) were conducted. Specifically, for the genomic doeA comparison, we identified homologues of the Helo_3665 amino acid sequence by performing a standard protein BLAST search (Altschul et al., 1997) in these databases. Out of all BLAST targets (with at least 30% sequence identity to Helo_3665) we compiled a selection of 29 sequences (including Helo_3665) by selecting only a few representative strains of species, which appear overrepresented in the list of completely sequenced genomes (e.g. Burkholderia). A peptidase sequence of Vibrio harveyi (20% sequence identity) was added and used as an out-group for phylogenetic analysis. For this set of 30 amino acid sequences a multiple alignment was computed using MUSCLE (Edgar, 2004). Poorly aligned and gapped positions were eliminated from the original MUSCLE alignment using the GBlocks algorithm (Castresana, 2000), which resulted in an alignment with 271 (54% of the original 497) positions. Finally, MrBayes (Huelsenbeck et al., 2001; Altekar et al., 2004) was employed for constructing the phylogenetic tree with the clade credibility values being based on Bayesian statistics. The final tree was drawn and rooted using the ATV/Archaeopteryx tool (Zmasek and Eddy, 2001). Using alternative strategies and software for alignment [e.g. employing T-Coffee (Notredame et al., 2000), ClustalW (Chenna et al., 2003), with and without GBlocks polishing] and phylogenetic analysis [e.g. employing PhyML (Guindon et al., 2005), PHYLIP (Felsenstein, 1989)] yielded qualitatively very similar results. The complete analysis pipelines were conducted with the help of the MIGenAS toolkit (Rampp et al., 2006).
Mathematical modelling
The mathematical modelling was carried out as outlined in ‘Network Reconstruction and Flux Balance Analysis’ and further detailed in Supporting information. All simulations were performed using Scilab 4.1.2 (INRIA ENPC). Degenerate solutions were explored using Flux Variability Analysis.
Bacterial strains, plasmids and growth conditions
Bacterial strains, vectors and recombinant plasmids used for this study are listed in Table S5. H. elongata strains were grown aerobically at 30°C on MM63 medium (Larsen et al., 1987) with glucose as carbon source. For certain experiments, ectoine was used as a carbon and nitrogen source, respectively, as specified. NaCl was added to concentrations as indicated. E. coli strains were grown aerobically at 37°C in Luria Bertani medium or MM63 minimal medium. Antibiotics with E. coli were used at the following concentrations: kanamycin 50 µg ml−1, ampicillin 100 µg ml−1, chloramphenicol 30 µg ml−1.
DNA isolation and manipulation
Total DNA from H. elongata was isolated according to a modified procedure of Marmur (Marmur, 1961). Routine manipulation of DNA, plasmid isolation, construction of recombinant plasmids, electrophoresis of DNA on agarose gels, and transformation were carried out according to standard procedures (Sambrook and Russell, 2001).
Purification of DoeX protein
Gene doeX was ligated into plasmid pET101 (Invitrogen), which allows the expression of recombinant protein with a C-terminal sequence of six consecutive histidine-residues (His-Tag) for subsequent purification by Ni-chelation chromatography. The resulting plasmid pKSB15 was transferred into E. coli BL21 (Invitrogen) and doeX expression was induced by the addition of IPTG (1 mM) to the medium of exponentially growing cells (OD540 1.0). After 5 h of induction, cells were harvested by centrifugation and disrupted using the BugBuster Reagent from Novagen (Darmstadt) according to the manufacturer's instruction. The soluble cell extract was loaded onto columns with Ni2+-resin (His-bind Resin, Novagen) and after washing, DoeX protein was released from the resin applying 1 M imidazole buffer. DoeX purification was monitored by SDS-PAGE and protein concentration was determined by applying the Bradford assay.
Generation of deletion mutants
DNA sequences upstream and downstream from the desired gene were joined together by applying the splicing by overlap extension PCR technique (Horton et al., 1989). The resulting PCR fragments were ligated into the shuttle vector pK18mobsacB (Schäfer et al., 1994) and transferred into H. elongata by E. coli S17-1 mediated conjugation (Simon et al., 1983; Kunte and Galinski, 1995). Deletion mutants, arising after double cross-over, were then selected for on LBG medium containing 22% (w/v) sucrose at 37°C. The deletion sites were verified by PCR and DNA-sequencing techniques.
Complementation experiments
To complement the mutations in strains KB41 (ΔdoeA) and KB42 (ΔdoeB), ORFs doeA and doeB, respectively, were expressed in trans with the help of shuttle vector pJB3Cm6 (Blatny et al., 1997). PCR-amplified doeA and doeB, respectively, were inserted into HindIII/XbaI-cut pJB3Cm6. The resulting recombinant plasmids pKSB7 (pJB3Cm6::doeA) and pJSB3 (pJB3Cm6::doeB) were transferred into the corresponding mutants via conjugation as described previously (Simon et al., 1983; Kunte and Galinski, 1995). H. elongata cells carrying the recombinant plasmid were selected on minimal medium containing chloramphenicol (30 µg ml−1) and tested for growth with ectoine as carbon source on mineral salt medium (680 mM NaCl).
RT-PCR and RACE-PCR
Total RNA was isolated from exponentially growing cells using a modified hot phenol method (Sambrook and Russell, 2001) and further purified using the NucleoSpin RNA-isolation kit (Macherey & Nagel) according to the manufacturer's instruction. cDNA for doeABX operon analysis was synthesized using the reverse primers (5′-TCGAACTTGACCAGGTAATCC-3′) binding in doeX and (5′-GGCACCGTACTCGACCTCAC-3′) binding to doeC. The transcriptional initiation sites were mapped by a modified RACE-PCR procedure based on a protocol by Gerhart and co-workers (Gerhart et al., 2005) using RNA from exponentially growing cells adapted to 680 mM NaCl. Transcription initiations site of doeA was mapped with reverse primer (5′-ACCAGTTGACCAGCGAGTTG-3′), ectA with reverse primer (5′-CGCTGATAGTGGTCTCG-3′), ectC with reverse primer (5′-TTCGCGGTGCACTTCGTT-3′), and a forward primer binding to the artificial RNA-adaptor attached to the 5′-end of the mRNAs. The transcription initiation site is defined by the boundary between the artificially attached RNA and the 5′-end of the transcript.
Electrophoretic mobility shift assay
Oligonucleotides used for this assay were fragment F1 (5′-TGTTAACAAATGTCATGACAATGAACAT-3′), F2 (5′-GGCCCTGACACGGTCGGCAAGTTAGCGC-3′), and F3 (5′-AGAGGCAGCCGGATATCGGTGACATGATCGTTTGGCGAGCGATTTC-3′). Double-stranded oligonucleotides of F1, F2 and F3 were generated by heating the complementary oligonucleotides in TA-buffer (330 mM TrisAc pH 7.8, 660 mM KAc, 100 mM MgAc, 5 mM DTT) for 10 min at 95°C and slowly cooling at room temperature. Double-stranded nucleotides were labelled by Fluorescein. Binding of DoeX to DNA was carried out in 20 µl SP1 buffer (4% glycerol, 1 mM MgCl2, 0,5 mM EDTA, 50 mM NaCl, 10 mM Tris, 0,5 mM DTT, pH 7.5) containing 10 pmol, 20 pmol or 40 pmol DoeX-protein and 2 pmol DNA (F1, F2, F3) in the presence of competitor DNA (poly dIdC, 500-fold to 1000-fold excess). After 30 min of incubation at 30°, protein–DNA complexes were resolved on a 6% polyacrylamide gel in Tris-borate EDTA buffer.
High-performance liquid chromatography (HPLC)
For identification and quantification of intracellular amino-reactive solutes, cells were harvested, freeze-dried and extracted with chloroform/water/methanol as described previously (Galinski and Herzog, 1990). Cellular extracts were analysed on an Agilent 1100 Series reversed-phase HPLC apparatus with o-phthalaldehyde (OPA) pre-column derivatization und UV detection according to the manufacture's instructions (Agilent; http://www.chem.agilent.com/Library/chromatograms/59801193.pdf). Standards of Nγ-Ac-DABA and Nα-Ac-DABA were obtained from alkaline hydrolysis of pure ectoine (50 mM) in 10 ml 0.1 KOH for 20 h at 50°C (Kunte et al., 1993).
Acknowledgments
We are grateful to T. Sommerfeld for his help with the HPLC measurements, to J. Straßner for support with the complementation experiments, to K. Grammann for her assistance in mutagenesis of teaABC, and to S. Taylor and M. Dyall-Smith for carefully reading the manuscript. We also would like to thank M. Dyall-Smith for providing the GC skew plot and the MUMmer alignment. This work was supported in part by a grant (KU 1112/3-1) from the Deutsche Forschungsgemeinschaft (DFG).
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Species-level assignment of H. elongata proteins by MEGAN analysis. The plot indicates the number of proteins assigned to the named species out of a set of 1672 proteins for which such an assignment was successful. The number of assigned proteins is given in parenthesis after the name of the species. Only 8% of the proteins are not assigned to C. salexigens.
Fig. S2. Alignment of the H. elongata and C. salexigens chromosomes. The chromosomes of H. elongata and C. salexigens were aligned using MUMmer software (Kurtz et al., 2004) and show a strong X-alignment. Such X-alignments have been described for several interspecies comparisons and attributed to genome inversions around the replication origin (Eisen et al., 2000). A prominent X-alignment probably indicates that the number of such genome rearrangement events was small, which is astonishing for species with such an evolutionary distance that they are classified into distinct genera.
Fig. S3. Utilization of acetate as carbon source by H. elongata strains in the absence and presence of ectoine. H. elongata wild type, mutant strains KB41 (ΔdoeA), KB42 (ΔdoeB) and KB47 (ΔdoeD) were incubated for 3 days at 30°C on mineral salt medium (0.51 M NaCl) containing 40 mM acetate (A) and 40 mM acetate plus 10 mM ectoine (B) respectively. All strains were able to grow on acetate as sole carbon source (A). In the presence of ectoine, mutant KB47 failed to grow with acetate, while KB41 and KB42 are still able to utilize acetate (B). The inability of doeD mutant KB47 to grow on acetate in the presence of ectoine explains why KB47 also fails to grow with ectoine alone although acetate should be still provided due to the deacetylase activity of DoeB (Fig. 3).
Fig. S4. RT-PCR and RACE-PCR analysis of the doeABXC region. (A) Genetic and physical organization of the doeABX locus. The position of the σ70-dependent promoter is indicated. Primer binding sites for RT-PCR are marked by triangles. Black triangles with dotted lines show successful generation of PCR products. For the pair of white triangles, no PCR product was obtained. Reverse primers were used for both, RT reaction and PCR. (B) RT-PCR analysis proving that doeABX is organized as one operon. A 1450 bp PCR product was amplified from cDNA and separated by agarose-gel electrophoresis (lane 1), which matched the size of the calculated doeABX PCR product (1473 bp). A corresponding doeX–doeC product could be amplified from genomic DNA with the same primer pair (positive control, lane 2). No PCR product could be amplified from cDNA using a primer pair to doeX and doeC, proving that doeC is not part of the doeABX operon (lane 3). A corresponding doeX–doeC product could be amplified from genomic DNA with the same primer pair (positive control, lane 4). A PCR product could be amplified with primers that both bind within the doeC ORF (control RT reaction, data not shown, product indicated in A). (C) Nucleotide sequence of the doeA promoter region. Arrows indicate the transcription initiation site (+ 1), which was mapped by RACE-PCR. The −35 and −10 sequences of the σ70-dependent promoter upstream of doeA are written in bold.
Fig. S5. Maximum ectoine yield as a function of the ATP load and turnover of ectoine in the synthesis degradation cycle (A) and flux distributions for maximum conversion into ectoine (B). (A) Points I to IV mark the flux distributions described in panel B. Horizontal axes represent total consumption of ATP by processes outside the model and the flux circulating through the ectoine synthesis/degradation cycle. The units are arbitrary, normalized for a glucose uptake of 100 (e.g. molecules). (B) I) One of the admissible flux distributions without ATP demand. II) Unique solution when total ATP demand is less than or equal to one ATP per glucose III) Example of solutions for higher ATP demands, superposition of II and maximum ATP production distribution. IV) Example of solutions for a turnover of ectoine, identical to III everywhere except the ATP load and cycle.
Table S1. The 20 COGs with the highest occupancy.
Table S2. Organisms used for the search for high salt COGs.
Table S3. Average pI difference between proteins from H. elongata and the indicated organisms.
Table S4. Enzymatic reactions used in the metabolic models.
Table S5. Bacterial strains and plasmids used in this study.
Table S6. Locus tag of genes similar to ect genes, ask, and doe genes depicted in Figs 2, 4 and 7, and accession data of genome sequences of corresponding organisms.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Altekar G, Dwarkadas S, Huelsenbeck JP, Ronquist F. Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics. 2004;20:407–415. doi: 10.1093/bioinformatics/btg427. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androutsellis-Theotokis A, Goldberg NR, Ueda K, Beppu T, Beckman ML, Das S, et al. Characterization of a functional bacterial homologue of sodium-dependent neurotransmitter transporters. J Biol Chem. 2003;278:12703–12709. doi: 10.1074/jbc.M206563200. [DOI] [PubMed] [Google Scholar]
- Arahal DR, García MT, Vargas C, Cánovas D, Nieto JJ, Ventosa A. Chromohalobacter salexigens sp. nov., a moderately halophilic species that includes Halomonas elongata DSM 3043 and ATCC 33174. Int J Syst Evol Microbiol. 2001;51:1457–1462. doi: 10.1099/00207713-51-4-1457. [DOI] [PubMed] [Google Scholar]
- Ausubel FM. Regulation of nitrogen fixation genes. Cell. 1984;37:5–6. doi: 10.1016/0092-8674(84)90294-0. [DOI] [PubMed] [Google Scholar]
- Badger JH, Olsen GJ. CRITICA: coding region identification tool invoking comparative analysis. Mol Biol Evol. 1999;16:512–524. doi: 10.1093/oxfordjournals.molbev.a026133. [DOI] [PubMed] [Google Scholar]
- Beuming T, Shi L, Javitch JA, Weinstein H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol Pharmacol. 2006;70:1630–1642. doi: 10.1124/mol.106.026120. [DOI] [PubMed] [Google Scholar]
- Blatny JM, Brautaset T, Winther-Larsen HC, Haugan K, Valla S. Construction and use of a versatile set of broad-host-range cloning and expression vectors based on the RK2 replicon. Appl Environ Microbiol. 1997;63:370–379. doi: 10.1128/aem.63.2.370-379.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolen DW. Effects of naturally occurring osmolytes on protein stability and solubility: issues important in protein crystallization. Methods. 2004;34:312–322. doi: 10.1016/j.ymeth.2004.03.022. [DOI] [PubMed] [Google Scholar]
- Bolen DW, Baskakov IV. The osmophobic effect: natural selection of a thermodynamic force in protein folding. J Mol Biol. 2001;310:955–963. doi: 10.1006/jmbi.2001.4819. [DOI] [PubMed] [Google Scholar]
- Bordo D, van Monfort RL, Pijning T, Kalk KH, Reizer J, Saier MH, Jr, Dijkstra BW. The three-dimensional structure of the nitrogen regulatory protein IIANtr from Escherichia coli. J Mol Biol. 1998;279:245–255. doi: 10.1006/jmbi.1998.1753. [DOI] [PubMed] [Google Scholar]
- Borges N, Ramos A, Raven ND, Sharp RJ, Santos H. Comparative study of the thermostabilizing properties of mannosylglycerate and other compatible solutes on model enzymes. Extremophiles. 2002;6:209–216. doi: 10.1007/s007920100236. [DOI] [PubMed] [Google Scholar]
- Brown AD. Microbial water stress. Bacteriol Rev. 1976;40:803–846. doi: 10.1128/br.40.4.803-846.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buommino E, Schiraldi C, Baroni A, Paoletti I, Lamberti M, De Rosa M, Tufano MA. Ectoine from halophilic microorganisms induces the expression of hsp70 and hsp70B' in human keratinocytes modulating the proinflammatory response. Cell Stress Chaperones. 2005;10:197–203. doi: 10.1379/CSC-101R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursy J, Pierik AJ, Pica N, Bremer E. Osmotically induced synthesis of the compatible solute hydroxyectoine is mediated by an evolutionarily conserved ectoine hydroxylase. J Biol Chem. 2007;282:31147–31155. doi: 10.1074/jbc.M704023200. [DOI] [PubMed] [Google Scholar]
- Butinar L, Santos S, Spencer-Martins I, Oren A, Gunde-Cimerman N. Yeast diversity in hypersaline habitats. FEMS Microbiol Lett. 2005;244:229–234. doi: 10.1016/j.femsle.2005.01.043. [DOI] [PubMed] [Google Scholar]
- Calderón MI, Vargas C, Rojo F, Iglesias-Guerra F, Csonka LN, Ventosa A, Nieto JJ. Complex regulation of the synthesis of the compatible solute ectoine in the halophilic bacterium Chromohalobacter salexigens DSM 3043T. Microbiology. 2004;150:3051–3063. doi: 10.1099/mic.0.27122-0. [DOI] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Chae JC, Zylstra GJ. 4-Chlorobenzoate uptake in Comamonas sp. strain DJ-12 is mediated by a tripartite ATP-independent periplasmic transporter. J Bacteriol. 2006;188:8407–8412. doi: 10.1128/JB.00880-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudel-Renard C, Chevalet C, Faraut T, Kahn D. Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res. 2003;31:6633–6639. doi: 10.1093/nar/gkg847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll M, Knof SH, Ohga Y, Messerschmidt A, Huber R, Moellering H, et al. Enzymatic mechanism of creatine amidinohydrolase as deduced from crystal structures. J Mol Biol. 1990;214:597–610. doi: 10.1016/0022-2836(90)90201-v. [DOI] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis PP, Shimmin LC. Evolutionary divergence and salinity-mediated selection in halophilic Archaea. Microbiol Mol Biol Rev. 1997;61:90–104. doi: 10.1128/mmbr.61.1.90-104.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA, Heidelberg JF, White O, Salzberg SL. Evidence for symmetric chromosomal inversions around the replication origin in bacteria. Genome Biol. 2000;1:RESEARCH0011.1–11.9. doi: 10.1186/gb-2000-1-6-research0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP – Phylogeny Inference Package. Cladistics. 1989;5:164–166. [Google Scholar]
- Fotheringham I. Engineering biosynthetic pathways: new routes to chiral amino acids. Curr Opin Chem Biol. 2000;4:120–124. doi: 10.1016/s1367-5931(99)00062-9. [DOI] [PubMed] [Google Scholar]
- Furusho K, Yoshizawa T, Shoji S. Ectoine alters subcellular localization of inclusions and reduces apoptotic cell death induced by the truncated Machado-Joseph disease gene product with an expanded polyglutamine stretch. Neurobiol Dis. 2005;20:170–178. doi: 10.1016/j.nbd.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Galinski EA, Herzog RM. The role of trehalose as a substitute for nitrogen-containing compatible solutes (Ectorhodospira halochloris) Arch Microbiol. 1990;153:607–613. [Google Scholar]
- Galinski EA, Pfeiffer HP, Trüper HG. 1,4,5,6-Tetrahydro-2-methyl-4-pyrimidinecarboxylic acid. A novel cyclic amino acid from halophilic phototrophic bacteria of the genus Ectothiorhodospira. Eur J Biochem. 1985;149:135–139. doi: 10.1111/j.1432-1033.1985.tb08903.x. [DOI] [PubMed] [Google Scholar]
- Gerhart E, Wagner H, Vogel J. Approaches to identify novel non-messenger RNAs in bacteria and to investigate their biological functions: functional analysis of identified non-mRNAs. In: Hartmann RK, Bindereif A, Westhof E, editors. Handbook of RNA Biochemistry. Weinheim, Germany: WILEY-VCH Verlag GmbH; 2005. pp. 614–642. [Google Scholar]
- Göller K. Institut Für Mikrobiolgie Und Biotechnologie. Bonn, Germany: Rheinische Friedrich-Wilhelms-Universität; 1999. Identifizierung und charaterisierung des ectoin-genclusters in Halomonas elongata. PhD Thesis. [Google Scholar]
- Göller K, Ofer A, Galinski EA. Construction and characterization of an NaCl-sensitive mutant of Halomonas elongata impaired in ectoine biosynthesis. FEMS Microbiol Lett. 1998;161:293–300. doi: 10.1111/j.1574-6968.1998.tb12960.x. [DOI] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Grammann K, Volke A, Kunte HJ. New type of osmoregulated solute transporter identified in halophilic members of the bacteria domain: TRAP transporter TeaABC mediates uptake of ectoine and hydroxyectoine in Halomonas elongata DSM 2581T. J Bacteriol. 2002;184:3078–3085. doi: 10.1128/JB.184.11.3078-3085.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Lethiec F, Duroux P, Gascuel O. PHYML Online – a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 2005;33:557–559. doi: 10.1093/nar/gki352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Hu RM, Yang TC, Yang SH, Tseng YH. Deduction of upstream sequences of Xanthomonas campestris flagellar genes responding to transcription activation by FleQ. Biochem Biophys Res Commun. 2005;335:1035–1043. doi: 10.1016/j.bbrc.2005.07.171. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F, Nielsen R, Bollback JP. Bayesian inference of phylogeny and its impact on evolutionary biology. Science. 2001;294:2310–2314. doi: 10.1126/science.1065889. [DOI] [PubMed] [Google Scholar]
- Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbar L, Lapidot A. The structure and biosynthesis of new tetrahydropyrimidine derivatives in actinomycin D producer Streptomyces parvulus. Use of 13C- and 15N-labeled L-glutamate and 13C and 15N NMR spectroscopy. J Biol Chem. 1988;263:16014–16022. [PubMed] [Google Scholar]
- Jebbar M, Talibart R, Gloux K, Bernard T, Blanco C. Osmoprotection of Escherichia coli by ectoine: uptake and accumulation characteristics. J Bacteriol. 1992;174:5027–5035. doi: 10.1128/jb.174.15.5027-5035.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jebbar M, Sohn-Bosser L, Bremer E, Bernard T, Blanco C. Ectoine-induced proteins in Sinorhizobium meliloti include an Ectoine ABC-type transporter involved in osmoprotection and ectoine catabolism. J Bacteriol. 2005;187:1293–1304. doi: 10.1128/JB.187.4.1293-1304.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanapathipillai M, Lentzen G, Sierks M, Park CB. Ectoine and hydroxyectoine inhibit aggregation and neurotoxicity of Alzheimer's β-amyloid. FEBS Lett. 2005;579:4775–4780. doi: 10.1016/j.febslet.2005.07.057. [DOI] [PubMed] [Google Scholar]
- Kennedy SP, Ng WV, Salzberg SL, Hood L, DasSarma S. Understanding the adaptation of Halobacterium species NRC-1 to its extreme environment through computational analysis of its genome sequence. Genome Res. 2001;11:1641–1650. doi: 10.1101/gr.190201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffas MA, Jung GY, Stephanopoulos G. Engineering metabolism and product formation in Corynebacterium glutamicum by coordinated gene overexpression. Metab Eng. 2003;5:32–41. doi: 10.1016/s1096-7176(03)00002-8. [DOI] [PubMed] [Google Scholar]
- Kolp S, Pietsch M, Galinski EA, Gutschow M. Compatible solutes as protectants for zymogens against proteolysis. Biochim Biophys Acta. 2006;1764:1234–1242. doi: 10.1016/j.bbapap.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Kuhlmann AU, Bremer E. Osmotically regulated synthesis of the compatible solute ectoine in Bacillus pasteurii and related Bacillus spp. Appl Environ Microbiol. 2002;68:772–783. doi: 10.1128/AEM.68.2.772-783.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann SI, Terwisscha van Scheltinga AC, Bienert R, Kunte HJ, Ziegler C. 1.55 A Structure of the Ectoine Binding Protein TeaA of the Osmoregulated TRAP-Transporter TeaABC from Halomonas elongata. Biochemistry. 2008;47:9475–9485. doi: 10.1021/bi8006719. [DOI] [PubMed] [Google Scholar]
- Kunte HJ. Transport and Accumulation of Osmoregulatory Solutes in Bacteria: Characterization of Transport-Systems and Their Regulatory Mechanisms in Halomonas Elongata, Marinococcus Halophilus, and Escherichia Coli. Bonn, Germany: Rheinische Friedrich-Wilhelms-Universität; 2004. [Google Scholar]
- Kunte HJ. Osmoregulation in bacteria: compatible solute accumulation and osmosensing. Environ Chem. 2006;3:94–99. [Google Scholar]
- Kunte HJ, Galinski EA. Transposon mutagenesis in halophilic eubacteria: conjugal transfer and insertion of transposon Tn5 and Tn1732 in Halomonas elongata. FEMS Microbiol Lett. 1995;128:293–299. doi: 10.1111/j.1574-6968.1995.tb07539.x. [DOI] [PubMed] [Google Scholar]
- Kunte HJ, Galinski EA, Trüper HG. A modified FMOC-method for the detection of amino acid-type osmolytes and tetrahydropyrimidines (ectoines) J Microbiol Methods. 1993;17:129–136. [Google Scholar]
- Kunte HJ, Galinski EA, Grammann K, Volke A, Bestvater T. Austria, Belgium, Switzerland/Lichtenstein, Cypres, Germany, Denmark, Spain, Finnland, France, Great Britain, Greece, Iralnd, Italy, Luxemburg, Monaco, The Netherlands, Portugal, Sweden, Turkey: Bitop Aktiengesellschaft Für Biochemische Optimierung. 2002. Verfahren zur Gewinnung von Wertstoffen aus Organismen; pp. 1–12. European Patent, Patent number EP1409707, pp. [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanyi JK. Salt-dependent properties of proteins from extremely halophilic bacteria. Bacteriol Rev. 1974;38:272–290. doi: 10.1128/br.38.3.272-290.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PI, Sydnes LK, Landfald B, Strøm AR. Osmoregulation in Escherichia coli by accumulation of organic osmolytes: betaines, glutamic acid, and trehalose. Arch Microbiol. 1987;147:1–7. doi: 10.1007/BF00492896. [DOI] [PubMed] [Google Scholar]
- Le Coq J, Pavlovsky A, Malik R, Sanishvili R, Xu C, Viola RE. Examination of the mechanism of human Brain aspartoacylase through the binding of an intermediate analogue. Biochemistry. 2008;47:3484–3492. doi: 10.1021/bi702400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Gralla JD. Osmo-regulation of bacterial transcription via poised RNA polymerase. Mol Cell. 2004;14:153–162. doi: 10.1016/s1097-2765(04)00202-3. [DOI] [PubMed] [Google Scholar]
- Lentzen G, Schwarz T. Extremolytes: natural compounds from extremophiles for versatile applications. Appl Microbiol Biotechnol. 2006;72:623–634. doi: 10.1007/s00253-006-0553-9. [DOI] [PubMed] [Google Scholar]
- Lin Y-H, Chang BCH, Chiang P-W, Tang S-L. Questionable 16S ribosomal RNA gene annotations are frequent in completed microbial genomes. Gene. 2008;416:44–47. doi: 10.1016/j.gene.2008.02.023. [DOI] [PubMed] [Google Scholar]
- Lippert K, Galinski EA. Enzyme stabilization by ectoine-type compatible solutes: protection against heating, freezing and drying. Appl Microbiol Biotechnol. 1992;37:61–65. [Google Scholar]
- Lo CC, Bonner CA, Xie G, D'Souza M, Jensen RA. Cohesion group approach for evolutionary analysis of aspartokinase, an enzyme that feeds a branched network of many biochemical pathways. Microbiol Mol Biol Rev. 2009;73:594–651. doi: 10.1128/MMBR.00024-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHardy AC, Goesmann A, Pühler A, Meyer F. Development of joint application strategies for two microbial gene finders. Bioinformatics. 2004;20:1622–1631. doi: 10.1093/bioinformatics/bth137. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Grishin NV. The Zn-peptidase superfamily: functional convergence after evolutionary divergence. J Mol Biol. 1999;292:11–17. doi: 10.1006/jmbi.1999.3059. [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Sanguino A, Torres NV, Mendoza ER, Oesterhelt D. Metabolic engineering with power-law and linear-logarithmic systems. Math Biosci. 2009;218:50–58. doi: 10.1016/j.mbs.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Marmur J. A procedure for the isolation of deoxyribonucleic acid from microorganisms. J Mol Biol. 1961;3:208–218. [Google Scholar]
- Márquez MC, Ventosa A. Marinobacter hydrocarbonoclasticus Gauthier et al. 1992 and Marinobacter aquaeolei Nguyen et al. 1999 are heterotypic synonyms. Int J Syst Evol Microbiol. 2005;55:1349–1351. doi: 10.1099/ijs.0.63591-0. [DOI] [PubMed] [Google Scholar]
- Maskow T, Babel W. Calorimetrically obtained information about the efficiency of ectoine synthesis from glucose in Halomonas elongata. Biochim Biophys Acta. 2001;1527:4–10. doi: 10.1016/s0304-4165(01)00115-5. [DOI] [PubMed] [Google Scholar]
- Meyer F, Goesmann A, McHardy AC, Bartels D, Bekel T, Clausen J, et al. GenDB – an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003;31:2187–2195. doi: 10.1093/nar/gkg312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothes G, Schubert T, Harms H, Maskow T. Biotechnological coproduction of compatible solutes and polyhydroxyalkanoates using the genus Halomonas. Eng Life Sci. 2008;8:658–662. [Google Scholar]
- Muller J, Szklarczyk D, Julien P, Letunic I, Roth A, Kuhn M, et al. eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 2010;38:D190–D195. doi: 10.1093/nar/gkp951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan C, Kelly DJ, Thomas GH. Tripartite ATP-Independent Periplasmic Transporters: application of a Relational Database for Genome-Wide Analysis of Transporter Gene Frequency and Organization. J Mol Microbiol Biotechnol. 2007;12:218–226. doi: 10.1159/000099643. [DOI] [PubMed] [Google Scholar]
- Nikerel IE, van Winden WA, Verheijen PJ, Heijnen JJ. Model reduction and a priori kinetic parameter identifiability analysis using metabolome time series for metabolic reaction networks with linlog kinetics. Metab Eng. 2009;11:20–30. doi: 10.1016/j.ymben.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Nyyssölä A, Keruvuo J, Kaukinen P, von Weymarn N, Reinikainen T. Extreme halophiles synthesize betaine from glycine by methylation. J Biol Chem. 2000;275:22196–22201. doi: 10.1074/jbc.M910111199. [DOI] [PubMed] [Google Scholar]
- Ono H, Sawada K, Khunajakr N, Tao T, Yamamoto M, Hiramoto M, et al. Characterization of biosynthetic enzymes for ectoine as a compatible solute in a moderately halophilic eubacterium, Halomonas elongata. J Bacteriol. 1999;181:91–99. doi: 10.1128/jb.181.1.91-99.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren A. Bioenergetic aspects of halophilism. Microbiol Mol Biol Rev. 1999;63:334–348. doi: 10.1128/mmbr.63.2.334-348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren A. Halophilic Microorganisms and Their Environments. Dordrecht, The Netherlands: Kluwer Academic Publishers; 2002. [Google Scholar]
- Oren A. Microbial life at high salt concentrations: phylogenetic and metabolic diversity. Saline Systems. 2008;4:2. doi: 10.1186/1746-1448-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters P, Galinski EA, Trüper HG. The biosynthesis of ectoine. FEMS Microbiol Lett. 1990;71:157–162. [Google Scholar]
- Pfeiffer F, Schuster SC, Broicher A, Falb M, Palm P, Rodewald K, et al. Evolution in the laboratory: the genome of Halobacterium salinarum strain R1 compared to that of strain NRC-1. Genomics. 2008a;91:335–346. doi: 10.1016/j.ygeno.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Pfeiffer F, Broicher A, Gillich T, Klee K, Mejia J, Rampp M, Oesterhelt D. Genome information management and integrated data analysis with HaloLex. Arch Microbiol. 2008b;190:281–299. doi: 10.1007/s00203-008-0389-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhu J, Schauwecker F, Grammel N, Keller U, Bernhard M. Functional expression of the ectoine hydroxylase gene (thpD) from Streptomyces chrysomallus in Halomonas elongata. Appl Environ Microbiol. 2004;70:3130–3132. doi: 10.1128/AEM.70.5.3130-3132.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick M, Yano H, Goldberg NR, Duan L, Beuming T, Shi L, et al. State-dependent conformations of the translocation pathway in the tyrosine transporter Tyt1, a novel neurotransmitter: sodium symporter from Fusobacterium nucleatum. J Biol Chem. 2006;281:26444–26454. doi: 10.1074/jbc.M602438200. [DOI] [PubMed] [Google Scholar]
- Racher KI, Voegele RT, Marshall EV, Culham DE, Wood JM, Jung H, et al. Purification and reconstitution of an osmosensor: transporter ProP of Escherichia coli senses and responds to osmotic shifts. Biochemistry. 1999;38:1676–1684. doi: 10.1021/bi981279n. [DOI] [PubMed] [Google Scholar]
- Rampp M, Soddemann T, Lederer H. The MIGenAS integrated bioinformatics toolkit for web-based sequence analysis. Nucleic Acids Res. 2006;34:W15–W19. doi: 10.1093/nar/gkl254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts MF. Organic compatible solutes of halotolerant and halophilic microorganisms. Saline Systems. 2006;1:1–30. doi: 10.1186/1746-1448-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory; 2001. [Google Scholar]
- Saum SH, Müller V. Growth phase-dependent switch in osmolyte strategy in a moderate halophile: ectoine is a minor osmolyte but major stationary phase solute in Halobacillus halophilus. Environ Microbiol. 2008;10:716–726. doi: 10.1111/j.1462-2920.2007.01494.x. [DOI] [PubMed] [Google Scholar]
- Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994;145:69–73. doi: 10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]
- Severin J, Wohlfarth A, Galinski EA. The predominant role of recently discovered tetrahydropyrimidines for the osmoadaptation of halophilic eubacteria. J Gen Microbiol. 1992;138:1629–1638. [Google Scholar]
- Simon R, Priefer U, Pühler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Biotechnology (NY) 1983;1:784–791. [Google Scholar]
- Sydlik U, Gallitz I, Albrecht C, Abel J, Krutmann J, Unfried K. The compatible solute ectoine protects against nanoparticle-induced neutrophilic lung inflammation. Am J Respir Crit Care Med. 2009;180:29–35. doi: 10.1164/rccm.200812-1911OC. [DOI] [PubMed] [Google Scholar]
- Tebbe A, Klein C, Bisle B, Siedler F, Scheffer B, Garcia-Rizo C, et al. Analysis of the cytosolic proteome of Halobacterium salinarum and its implication for genome annotation. Proteomics. 2005;5:168–179. doi: 10.1002/pmic.200400910. [DOI] [PubMed] [Google Scholar]
- Tetsch L, Kunte HJ. The substrate-binding protein TeaA of the osmoregulated ectoine transporter TeaABC from Halomonas elongata: purification and characterization of recombinant TeaA. FEMS Microbiol Lett. 2002;211:213–218. doi: 10.1111/j.1574-6968.2002.tb11227.x. [DOI] [PubMed] [Google Scholar]
- Thaw P, Sedelnikova SE, Muranova T, Wiese S, Ayora S, Alonso JC, et al. Structural insight into gene transcriptional regulation and effector binding by the Lrp/AsnC family. Nucleic Acids Res. 2006;34:1439–1449. doi: 10.1093/nar/gkl009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas C, Jebbar M, Carrasco R, Blanco C, Calderón MI, Iglesias-Guerra F, Nieto JJ. Ectoines as compatible solutes and carbon and energy sources for the halophilic bacterium Chromohalobacter salexigens. J Appl Microbiol. 2006;100:98–107. doi: 10.1111/j.1365-2672.2005.02757.x. [DOI] [PubMed] [Google Scholar]
- Vargas C, Argandoña M, Reina-Bueno M, Rodríguez-Moya J, Fernández-Aunión C, Nieto JJ. Unravelling the adaptation responses to osmotic and temperature stress in Chromohalobacter salexigens, a bacterium with broad salinity tolerance. Saline Systems. 2008;4:14. doi: 10.1186/1746-1448-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma A, Palsson BO. Metabolic Capabilities of Escherichia coli: I. Synthesis of Biosynthetic Precursors and Cofactors. J Theor Biol. 1993a;165:477–502. doi: 10.1006/jtbi.1993.1202. [DOI] [PubMed] [Google Scholar]
- Varma A, Palsson BO. Metabolic Capabilities of Escherichia coli II. Optimal Growth Patterns. J Theor Biol. 1993b;165:503–522. doi: 10.1006/jtbi.1993.1202. [DOI] [PubMed] [Google Scholar]
- Vreeland RH, Litchfield CD, Martin EL, Elliot E. Halomonas elongata, a new genus and species of extremely salt-tolerant bacteria. Int J Syst Bacteriol. 1980;30:485–495. [Google Scholar]
- Wei L, Wedeking A, Buttner R, Kalff JC, Tolba RH, van Echten-Deckert G. A natural tetrahydropyrimidine protects small bowel from cold ischemia and subsequent warm in vitro reperfusion injury. Pathobiology. 2009;76:212–220. doi: 10.1159/000218338. [DOI] [PubMed] [Google Scholar]
- Wohlfarth A, Severin J, Galinski EA. The spectrum of compatible solutes in heterotrophic halophilic eubacteria of the family Halomonadaceae. J Gen Microbiol. 1990;136:705–712. [Google Scholar]
- Wolf A, Krämer R, Morbach S. Three pathways for trehalose metabolism in Corynebacterium glutamicum ATCC13032 and their significance in response to osmotic stress. Mol Microbiol. 2003;49:1119–1134. doi: 10.1046/j.1365-2958.2003.03625.x. [DOI] [PubMed] [Google Scholar]
- Yi H, Lim YW, Chun J. Taxonomic evaluation of the genera Ruegeria and Silicibacter: a proposal to transfer the genus Silicibacter Petursdottir and Kristjansson 1999 to the genus Ruegeria Uchino et al. 1999. Int J Syst Evol Microbiol. 2007;57:815–819. doi: 10.1099/ijs.0.64568-0. [DOI] [PubMed] [Google Scholar]
- Zmasek CM, Eddy SR. ATV: display and manipulation of annotated phylogenetic trees. Bioinformatics. 2001;17:383–384. doi: 10.1093/bioinformatics/17.4.383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.