

Abstract
The P2Y12 receptor on platelets with which ADP interacts has an important role in promoting platelet function and thereby platelet involvement in both haemostasis and thrombosis. Agents that act as antagonists at this receptor are thus likely to provide effective antithrombotic therapy, provided that there are no adverse effects on haemostasis. Here we describe the ADP receptor antagonists that are available and in development. We also consider their mode of action and ask whether there are additional mechanisms through which they exert their inhibitory effects on platelet function.
Keywords: anti-platelet, cangrelor, clopidogrel, platelet, P2Y12, prasugrel, ticagrelor
Introduction
Adenosine diphosphate (ADP) interacts with two different purinergic receptors on platelets, known as P2Y1 and P2Y12 (Figure 1). These are two receptors of the seven transmembrane (7TM) class of receptors [1]. Interaction with P2Y1 receptors initiates the platelet response while interaction with P2Y12 receptors promotes the response. Blockade of the effects of ADP at either of these receptors results in a marked reduction in the overall effect of ADP on platelet function [2, 3].
Figure 1.
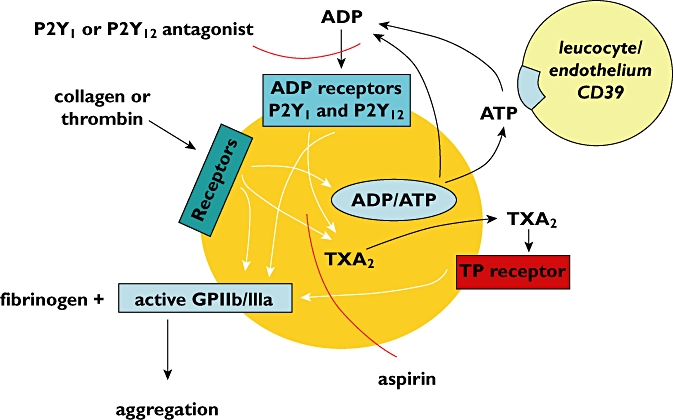
Some of the receptors and pathways involved in platelet aggregation. The points at which P2Y1 or P2Y12 antagonists and aspirin inhibit platelet aggregation are shown
The initial response to ADP is a change in the shape of the platelet whereby disc shaped cells convert into a spherical form from which pseudopodia emerge. This change, which is mediated by the P2Y1 receptor, involves Ca2+ influx, intracellular Ca2+ mobilization and actin polymerization. Interaction of ADP with the P2Y12 receptor results in inhibition of adenylate cyclase, which is accompanied by platelet aggregation [4]. The latter is mediated by a change in conformation of glycoprotein complexes on the platelet surface known as GPIIb/IIIa complexes, such that they become receptors for fibrinogen (Figure 1). Fibrinogen is a bivalent molecule present in high concentrations in blood plasma. Once bound to some of the 50 000–100 000 GPIIb/IIIa complexes on a single platelet, fibrinogen links the GPIIb/IIIa on adjacent platelets together. The result is that individual single platelets are transformed into a large mass in which many hundreds of thousands of platelets have aggregated together [5, 6].
ADP appears along with adenosine triphosphate (ATP) in the circulation consequent to cell and tissue damage. ADP can also be produced from ATP by the action of CD39 on endothelium and white cells (Figure 1) [7, 8]. ATP itself can also promote platelet function under certain conditions through interaction with another purinergic receptor, the P2X1 receptor [9]. Other important agents that result in platelet activation and platelet aggregation are collagen and thrombin. In addition to causing platelet shape change and aggregation, collagen and thrombin promote secretion by platelets of the contents of storage granules which include ADP and ATP. The ADP, acting via P2Y1 and P2Y12 receptors on the platelet surface amplifies the effects of the primary stimulus provided by the collagen or thrombin [10].
Other platelet functional responses that are amplified by released ADP include synthesis of thromboxane A2 (TXA2), which also promotes platelet aggregation acting via TP receptors (Figure 1) [11]. ADP also promotes secretion of other materials from intracellular granules including P-selectin which is the prime mediator of platelet interactions with leucocytes [12]. In addition, platelet microparticles are formed which provide procoagulant surfaces that accelerate the coagulation cascade [13]. Collectively all these mechanisms contribute to haemostasis, thrombosis and inflammation.
ADP is thus an important contributor to platelet function through the P2Y1 and P2Y12 receptors. Agents that reduce the effects of ADP at either of these could provide effective antithrombotic therapy.
P2Y1 antagonists
Studies in P2Y1 knockout mice have shown that absence of the P2Y1 receptor leads to impaired platelet aggregation in response to ADP and resistance to thromboembolism [14, 15]. A number of pharmacological agents have been identified that act as platelet P2Y1 antagonists [16–21]. These have been investigated experimentally and clear inhibition of ADP-induced platelet function has been shown. However, given the ubiquitous expression of P2Y1 receptors in other tissues of the body, the clinical potential for the P2Y1 receptor to be used as an antithrombotic target is questionable [22]. As far as we are aware, there have been no clinical studies evaluating the use of P2Y1 antagonists as antithrombotic agents in human subjects. Consequently this review will focus on P2Y12 receptor antagonists.
P2Y12 antagonists
There are several agents that interact with the P2Y12 receptor on platelets, thereby reducing platelet function. Some are already in use as antithrombotic agents and others are in development. The drugs we will discuss here are clopidogrel, prasugrel, ticagrelor, cangrelor and elinogrel. These drugs are routinely used in conjunction with aspirin which inhibits TXA2 synthesis in platelets and thus provides an additional means of reducing platelet function.
Clopidogrel
The P2Y12 antagonist that is currently most widely used as an antithrombotic agent is clopidogrel. It is recommended in current clinical guidelines for prevention of further thrombotic events following acute coronary syndrome and ischaemic stroke [23]. Clopidogrel is a thienopyridine and succeeded another thienopyridine, ticlopidine, which has been withdrawn gradually from clinical use due to a high incidence of haematological side effects including thrombotic thrombocytopenic purpura, neutropenia and aplastic anaemia [24].
Clopidogrel is a prodrug and its effects on platelets following oral administration derive from generation in vivo of an active metabolite [25]. This binds to P2Y12 receptors irreversibly, rendering the receptor unable to respond to ADP, thus reducing platelet function. The irreversible effect at the P2Y12 receptor is consequent to covalent binding to cysteine sulphydryl residues within the receptor. Its effect on platelet function lasts for the lifetime of the affected platelet, which is between 7 and 10 days.
Although it is clear that clopidogrel provides significant protection against thrombotic episodes when administered to patients at risk of thrombosis (e.g. the CAPRIE study [26]) and especially when used in combination with aspirin (e.g. the CURE study [27]) the drug does have some drawbacks. Its onset of action as an inhibitor of platelet function is rather slow following initiation of standard therapy, taking several days for a full effect to be realized, and also its inhibitory effects on platelet function are variable in different people with some patients continuing to have high platelet reactivity despite treatment.
Multiple factors may contribute to this high on-clopidogrel platelet reactivity. Genetic factors include polymorphisms of the ABCB1 gene affecting intestinal absorption [28], and polymorphisms of hepatic cytochrome P450 enzymes including CYP2C9 and CYP2C19 that are involved in the generation of the clopidogrel active metabolite [29]. Recently, variation in another enzyme, paraoxonase-1, has been shown to affect clopidogrel metabolism [30]. Other factors that could affect clopidogrel efficacy include body mass index, gender, ethnicity, and comorbidities such as liver disease and insulin resistance. A number of drug–drug interactions have also been implicated [29]. As well as inter-individual variability, it has recently been shown that there is also large intra-individual variability in the response to clopidogrel during long term therapy [31].
High on-clopidogrel platelet reactivity has been shown to be an important predictor of adverse thrombotic outcomes [32]. Hence it is important that this issue is adequately addressed in clinical practice. Unfortunately, despite some genetic associations, it is not easy to predict how an individual will respond to clopidogrel due to the multiple factors involved. Platelet function testing methods are now becoming available that enable the degree of antiplatelet effect to be determined [33–36].
Recent clinical trials have looked at the effects of increasing the dose of clopidogrel in patients with inadequate inhibition of platelet function on standard dose treatment. In the recently published GRAVITAS trial [37], a point of care platelet function test was used to identify patients with high on-clopidogrel reactivity. In these patients, a modest improvement in platelet inhibition was noted when randomized to high dose clopidogrel treatment compared with a comparison group receiving standard doses. Despite this, no difference in clinical outcome between the groups was seen. It must be noted that patients received different doses of aspirin in this trial, but there were no data on whether these were equally randomized between the groups. It is also possible that the low event rate in the GRAVITAS trial may have masked any difference in outcome. Also, as with previously published data [38], some patients continued to have very high platelet reactivity on higher doses of clopidogrel in this study [37], which may have skewed the outcome data.
Further work is needed to evaluate the potential use of platelet function testing clinically. Questions have been raised about defining the cut off for high platelet reactivity, the optimal time of measuring platelet function on treatment, and whether serial platelet function measurements may be useful [39]. Patients with high on-clopidogrel platelet reactivity may particularly benefit from newer, more effective drugs. Encouraging results to this effect have been seen in an earlier study where the use of intensified platelet inhibition with the GPIIb/IIIa inhibitor tirofiban was shown to improve significantly outcomes in patients who had poor responsiveness to clopidogrel as determined using point-of-care platelet function testing [40]. Further clinical trials are underway evaluating the potential role of personalized antiplatelet therapy [39].
There is also an economic decision to be made when deciding on antiplatelet treatment, because clopidogrel is now widely available as various generic salt formulations that are relatively inexpensive [41], whereas other more effective antiplatelet agents and the newer P2Y12 inhibitors (see below) would cost the health service much more to administer. However despite introduction to the market, there are very little data comparing the newer, generic clopidogrel salts with the original clopidogrel bisulphate. Some limited evidence from small studies on healthy subjects have shown absence of an overall difference in bioequivalence or antiplatelet effect of clopidogrel besylate compared with clopidogrel bisulphate [42–44]. However in one cross-over study it was shown that there was marked inter- and intra-individual variability between the two different clopidogrel salts [44], and there is a general lack of patient data. We have been unable to find any peer reviewed publications on other commercially available clopidogrel salts, such as clopidogrel hydrochloride. The limited availability of data about formulations of a drug that already shows notable variation in patient response in its original form, is concerning. This also highlights the potential importance of platelet function testing and monitoring of antiplatelet therapy.
In the future it is likely that platelet function testing will become obligatory if only to ascertain which patients are receiving adequate treatment on clopidogrel, which patients in whom higher doses of clopidogrel might be tried, and which patients would be best receiving a newer, more expensive antiplatelet agent. Using platelet function testing to tailor therapy according to each individual patient's response may offer a cost-effective strategy in future antiplatelet therapy.
Prasugrel
Prasugrel is also a thienopyridine and prodrug which, like clopidogrel, needs to be converted to an active metabolite to effect inhibition of platelet function [45]. As with clopidogrel, the prasugrel active metabolite binds to cysteine sulphydryl groups in the P2Y12 receptors irreversibly, rendering the receptors unable to respond to ADP and producing inhibition of platelet function for the lifetime of the affected platelet. Prasugrel differs from clopidogrel, however, in that it has a more rapid onset of action after oral administration, it achieves greater and more consistent platelet inhibition in individual patients [46, 47] and thus its antiplatelet effects are much more predictable. This is achieved mainly because the metabolism of the drug differs from that of clopidogrel and far greater and more predictable amounts of active metabolite are produced. For example, polymorphisms in CYP2C9 and CYP2C19 genes do not affect prasugrel metabolism, unlike the metabolism of clopidogrel described above [48].
All this translates into a more effective anti-thrombotic therapy as compared with clopidogrel as revealed in the TRITON-TIMI study [49]. This was a head-to-head comparison of prasugrel and clopidogrel in patients with acute coronary syndromes with scheduled percutaneous coronary intervention (PCI). The group treated with prasugrel had a significantly better outcome in terms of a composite of adverse events comprising death from cardiovascular causes, nonfatal myocardial infarction and nonfatal stroke (hazard ratio 0.81, 95% confidence interval 0.73, 0.90, P < 0.001). There was also a significant reduction in stent thrombosis compared with the clopidogrel group, although this was achieved at the expense of an increased incidence of bleeding. The clinical benefit of prasugrel over clopidogrel was particularly marked in patients with diabetes. It was noted that risks of bleeding outweighed the clinical benefit in patients with a previous history of stroke, the elderly (≥75 years), and those with a body weight of less than 60 kg. A higher incidence of adverse events related to colon cancer was also noted among patients treated with prasugrel in this study. The reasons for this are unclear, but it is suggested that this may be due to possible earlier recognition of otherwise subclinical lesions because of the higher incidence of bleeding secondary to prasugrel.
Despite its predictable and more effective action compared with clopidogrel, a factor limiting the widespread use of prasugrel clinically is its cost. At present, the retail price of prasugrel is about 10 times higher than generic clopidogrel. In the United Kingdom, prasugrel is currently recommended as the preferred antiplatelet agent in patients with low risk of bleeding who are undergoing primary percutaneous coronary intervention for ST segment elevation myocardial infarction [50].
Ticagrelor
Ticagelor (also known as AZD6140) is a cyclopentyl-triazolo-pyrimidine [51] and is a very different molecule to clopidogrel and prasugrel. Ticagrelor is also metabolized and forms an active metabolite, ARC124910XX, but this has very similar pharmacokinetics to the parent compound. Like the thienopyridines ticagrelor interacts with P2Y12 receptors on platelets rendering them unable to interact with ADP [52], but in this case the drug does not necessarily require metabolic conversion to interact with the receptor [53]. This is likely to at least partly account for the higher efficacy of ticagrelor over clopidogrel in patients irrespective of genetic differences in hepatic enzyme activity [54].
The mode of action of ticagrelor and its metabolite is reversible, in contrast to the irreversible effect of thienopyridines. Consequently, ticagrelor can no longer inhibit platelet function once its concentration in the blood is reduced on clearance of the drug from the circulation. The onset of platelet inhibition is rapid and predictable as ticagrelor is rapidly absorbed following oral administration [55]. Its off-rate is surprisingly quite slow despite its reversible mode of action. Although residual platelet inhibition is less than 50% at 24 h after a dose, approximately 20% inhibition remains at 3 days [56]. This may be because ticagrelor binds to plasma proteins in the circulation [57], which can reduce its rate of clearance from the blood. Like clopidogrel, ticagrelor is also a CYP450 substrate and hence there is the potential for drug–drug interactions [58].
Ticagrelor has been shown to produce a more effective anti-thrombotic effect compared with clopidogrel. The PLATO trial [59] was a head-to-head comparison of ticagrelor and clopidogrel in patients admitted to hospital with an acute coronary syndrome. The end result was a significant reduction in the composite of cardiovascular death, non-fatal MI and stroke with ticagrelor (hazard ratio 0.84, 95% confidence interval 0.77, 0.92, P < 0.001), without any difference in the overall incidence of fatal bleeding or major bleeding as defined by PLATO criteria as well as using TIMI (Thrombolysis In Myocardial Infarction) bleeding scores [60]. There was, however, an increase in major bleeding not related to coronary artery bypass grafting. Of particular interest, for the first time with a new agent, a significant reduction in the overall death rate was noted using ticagrelor in place of clopidogrel. The PLATO PlateLET substudy demonstrated that antiplatelet effects were greater and more consistent with ticagrelor over clopidogrel both at initiation of treatment and during maintenance therapy [61]. An unwanted side-effect was an increased incidence of dyspnoea in the ticagrelor-treated group compared with the clopidogrel-treated group (discussed later in this review).
Another potential problem was a geographical variation in the results of the PLATO trial. The benefits of ticagrelor appeared to be attenuated in patients enrolled from North America. The reasons for this difference are unclear. This may, of course, have just been a result of the play of chance, but it is also possible that geographical confounding factors may have been involved. It is of note that higher doses of aspirin were used in the North American patients compared with the rest of the world. High doses of aspirin may lead to inhibition of synthesis of natural modulators of platelet function such as the vascular prostaglandins, PGI2, PGD2 and PGE2, while recent work (see below) has shown that P2Y12 antagonists can promote the inhibitory effects of these same natural modulators of platelet function [62]. The possibility that this interaction with high doses of aspirin could explain the reduced antithrombotic benefits seen in the North American subgroup warrants careful consideration.
Ticagrelor has recently been approved for use in Europe for prevention of atherothrombotic events in patients with acute coronary syndrome [63], and is currently under review by the FDA (Food and Drugs Administration) regarding approval in the United States.
Cangrelor
Cangrelor (also known as AR-C69931MX) is an ATP analogue and is another direct acting inhibitor of platelet function acting via the P2Y12 receptor. Cangrelor also acts in a reversible manner [64]. Unlike ticagrelor, cangrelor cannot be administered orally, but is being developed for intravenous use. The half-life of cangrelor is very short and its effects on platelet function disappear within minutes following infusion [65]. This agent may therefore be a useful therapeutic tool for achieving targeted antiplatelet cover for defined periods of time according to clinical need [66].
If cangrelor is to be used in this way, intravenously for short periods of time, caution will need to be applied when switching treatment from cangrelor to another P2Y12 antagonist. This stems from the different modes of action of different antagonists at P2Y12 receptors. For example, it has been shown that cangrelor, while in contact with the P2Y12 receptor, limits the ability of the clopidogrel active metabolite or the prasugrel active metabolite to act as irreversible inhibitors of platelet function [67, 68]. This could have important clinical implications when switching treatment from cangrelor infusion to oral clopidogrel or prasugrel. If oral therapy is initiated early, cangrelor would still be occupying the P2Y12 receptor at the time that the active metabolite of clopidogrel or prasugrel appeared in the blood, and irreversible P2Y12 blockade would not be achieved. Once the cangrelor was withdrawn the platelets would then become uninhibited. If, alternatively, administration of the oral drug is delayed until after cangrelor is withdrawn this too could lead to a period of absent P2Y12 blockade between clearance of the reversible agent and availability of active metabolites from the oral agent. Such a period of incomplete platelet inhibition could potentially increase the risk of thrombosis during this period. Further work is needed to determine the optimal timing and sequence when switching between different P2Y12 antagonists.
The definitive trials needed to demonstrate the superiority of cangrelor over other P2Y12 antagonists have not yet been completed or published. Two studies in which this was attempted had disappointing outcomes [69, 70] that may have resulted from inappropriate trials designs. The CHAMPION PHOENIX trial is currently underway, evaluating the efficacy and safety profile of cangrelor compared with clopidogrel, in patients who require percutaneous coronary intervention [71].
Elinogrel
A newer P2Y12 receptor blocker currently undergoing phase II clinical trials is elinogrel (PRT060128), a sulphonylurea derivative that too is a direct acting, reversible P2Y12 antagonist. It is the first P2Y12 antagonist being developed that can be used both as an oral and intravenous agent [72, 73].
This drug is at a relatively early stage in its development. In the new phase II trial, INNOVATE PCI, it was reported that both the intravenous and oral forms of elinogrel produced greater and more rapid antiplatelet effects than clopidogrel in stable cardiac patients undergoing percutaneous coronary intervention. This trial, however, did not demonstrate any difference in the clinical efficacy of elinogrel over clopidogrel, which may be due to insufficient statistical power in the study for efficacy outcomes. It is encouraging to note that there was no excess in minor or major bleeding with elinogrel, although there was a dose-dependent increase in minor bleeds when it was used during the peri-procedural phase [74].
Further considerations regarding the mode of action of P2Y12 antagonists
We have seen above that all the drugs discussed have a common mechanism of action in that they interact with the P2Y12 receptor on platelets, reduce the ability of ADP to interact with that receptor, and thereby reduce platelet function. Thus there is marked inhibition of platelet function induced by ADP itself, and also by agents such as collagen and thrombin, where ADP secreted from platelets in response to stimulation by these agents contributes to the overall functional response. P2Y12 antagonists reduce platelet aggregation, platelet secretion, platelet-leucocyte interaction and the contribution of platelets to coagulation. P2Y12 antagonists also reduce TXA2 synthesis by platelets and thereby the amplification of platelet functions mediated by this molecule [75].
But is this the only means through which such drugs exert their antiplatelet and antithrombotic effects? Also, are there any additional mechanisms through which particular drugs might affect platelet function differently compared with other P2Y12 antagonists?
Are there additional mechanisms through which P2Y12 antagonists affect platelet function?
It is interesting to consider whether adenosine production is relevant to the effects of P2Y12 antagonists. ADP can be rapidly broken down to AMP and then to adenosine in blood plasma via ecto-ADPases such as CD39 and CD73 (Figure 2) [76, 77]. Adenosine is an inhibitor of platelet function acting mainly via A2A receptors on platelets through which there is stimulation of adenylate cyclase and an increase in intracellular cAMP. In a recent communication [78] it was clearly demonstrated that adenosine derived from ADP breakdown in blood plasma inhibits platelet function and that adenosine is particularly effective as an inhibitor of platelet function in the presence of a P2Y12 antagonist [78]. P2Y12 antagonists reduce inhibition of adenylate cyclase (Figure 2), so perhaps it is not surprising that an agent that increases cAMP does so more readily in the presence of a P2Y12 antagonist and this does prove to be the case. So might part of the action of a P2Y12 antagonist, and indeed the clinical benefit to be derived from their use, be mediated via adenosine?
Figure 2.
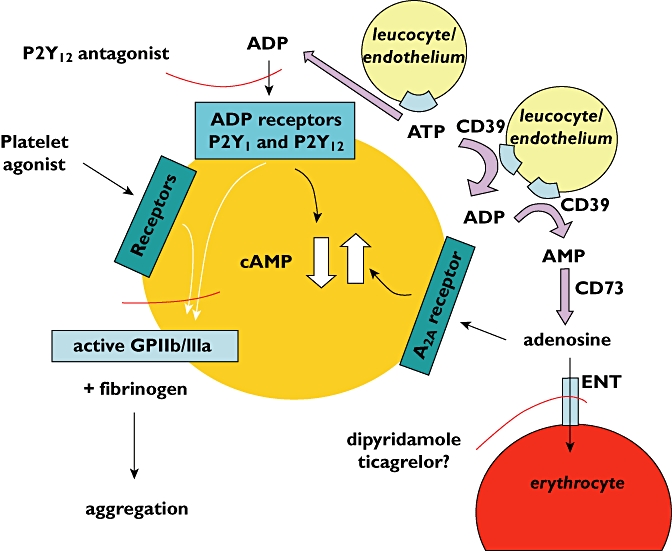
Mechanisms involved in adenosine production and removal, and the way in which adenosine interacts with platelets to increase cAMP and inhibit platelet aggregation. The points at which a P2Y12 antagonist and dipyridamole influence the pathways are shown. Recent research does not support an effect of ticagrelor at the equilibrative nucleoside transporter (ENT) [78]
An important consideration is that adenosine is rapidly transported into red cells via the equilibrative nucleoside transporter and this makes the adenosine unavailable to interact with platelets (Figure 2). Thus experiments performed in whole blood (i.e. with red cells present) provided very different results to those obtained in platelet-rich plasma (i.e. with red cells absent). In whole blood any inhibitory effects of adenosine produced from ADP could not be seen. Inhibition of platelet aggregation could only be seen in whole blood when the action of the equilibrative nucleoside transporter was blocked using dipyridamole [78]. There is still the possibility, though, that adenosine could be important in vivo, possibly at sites of thrombus formation or haemostasis where, for haemodynamic reasons, red cells may be excluded.
In considering a possible role for adenosine in determining the effects of P2Y12 antagonists on platelet function, it is pertinent to consider separately the situation with ticagrelor. This drug has been reported to inhibit the equilibrative nucleoside transporter in addition to its ability to act as a P2Y12 antagonist (Figure 2) [79]. Indeed, this is believed to be a possible explanation of the occurrence of dyspnoea observed in clinical trials involving this drug, a condition that is thought could be a consequence of raised adenosine concentrations in the circulation [80]. So, in theory, by both blocking the equilibrative nucleoside transporter and by acting as a P2Y12 antagonist ticagrelor should provide more effective inhibition of platelet aggregation in whole blood than other P2Y12 antagonists. However, when this was looked for [71], it was found that the effects of ticagrelor on platelet aggregation in both platelet rich plasma and in whole blood were exactly the same as for cangrelor and the prasugrel active metabolite. For all three P2Y12 antagonists studied, adenosine produced from ADP inhibited platelet aggregation in platelet-rich plasma but not in whole blood. This finding does not support an additional mode of action of ticagrelor on platelet function via inhibition of adenosine uptake.
P2Y12 antagonists may also promote the inhibitory potency of some other natural modulators of platelet function in addition to adenosine (Figure 3). In this regard, it has already been shown that prostaglandin E1 (PGE1) is a better inhibitor of platelet aggregation in the presence of cangrelor or clopidogrel than in the absence of one of these P2Y12 antagonists [81]. Also prostaglandin I2 (PGI2) acting through the IP receptor has been shown to be a better inhibitor of platelet aggregation in the presence of cangrelor [82]. More recently further studies were performed on the effects of cangrelor on the ability of various prostaglandins, PGD2, PGE1, PGE2, PGE3, PGI2 and the PGI2 mimetic iloprost to inhibit platelet aggregation. In all cases cangrelor potentiated the dose-dependent inhibition of platelet aggregation by some 3–10-fold, meaning that much lower concentrations of the prostaglandins were needed to inhibit the aggregation response [62]. This effect was not limited to cangrelor since ticagrelor and the prasugrel active metabolite produced similar potentiating effects. Interestingly, potentiation of the effects of these natural prostaglandins in the presence of a P2Y12 antagonist outweighed the ability of a P2Y12 antagonist used alone to inhibit platelet aggregation. This may be a major finding regarding the mechanism through which P2Y12 antagonists bring about clinical benefit, and requires further investigation. We have already speculated (above) on the possible implications of this finding for the geographically different outcomes of the PLATO study.
Figure 3.
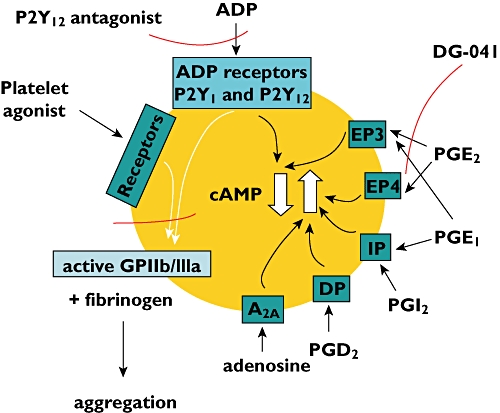
The receptors with which some natural modulators of platelet function interact to either increase or decrease cAMP and thus inhibit or promote platelet aggregation. The points at which a P2Y12 antagonist and the EP3 antagonist DG-041 influence the pathways are shown
Another interesting finding in the study described above [62] was the ability of the EP3 receptor antagonist DG-041 to potentiate the inhibitory effects a P2Y12 antagonist used in combination with either PGE1 or PGE2. Previous observations on the ability of DG-041 to modulate the effects of PGE2 on platelet function and thrombus formation have suggested that this EP3 antagonist may produce a useful additional therapy for atherothrombosis [83–85].
There are a number of potential additional effects of particular P2Y12 antagonists that have received consideration and investigation, but so far there is no certainty that any such additional effects exist or have any bearing on the effectiveness or otherwise on the outcomes of therapy.
The mode of action of clopidogrel and prasugrel at P2Y12 receptors could mean that these drugs also affect other receptors in addition to P2Y12 receptors. Other receptors in the 7TM class have cysteine residues within them that could interact with the active metabolites of these two drugs [86]. Effects at such receptors could either promote or inhibit their function. This possibility prompted a study [87] to look for any evidence of effects of the prasugrel active metabolite at a number of G-receptors on platelets and to compare the results with those for ticagrelor and cangrelor. The authors looked for any ability of the P2Y12 antagonists to modify the effects on platelet aggregation of a number of agonists and antagonists known to act at a variety of such receptors, all under circumstances where the role of P2Y12 receptors were diminished by working under conditions where any ADP or metabolites of ADP were purposely removed. However, no effects of any of the P2Y12 antagonists at receptors other than P2Y12 receptors could be detected. The receptors that were unaffected by the P2Y12 antagonists were the PAR1 receptor, the TP receptor, the IP receptor, the A2A receptor and the EP receptors. These mediate the effects on platelets of thrombin, TXA2, PGI2, adenosine and PGE2, respectively,
The lack of effects of cangrelor at receptors other than P2Y12 is also worthy of note because it had been reported previously that cangrelor was able to inhibit platelet aggregation through interaction with an unidentified Gs-coupled receptor leading to increased cAMP concentrations [88]. Indeed these authors argued that this may be the main mode of action of cangrelor rather than through P2Y12 blockade. However Iyu et al. [87] were unable to replicate the marked inhibition of aggregation induced by agents other than ADP or the increases in cAMP observed previously and thereby found little evidence for a direct effect of cangrelor at receptors other than P2Y12 receptors on platelets. They concluded that large quantities of ADP in the platelet preparations may have lead to misleading interpretations of the data obtained previously.
Conclusions
The P2Y12 receptor antagonists clopidogrel, prasugrel and ticagrelor are already in clinical use as antithrombotic agents. Variability in antiplatelet effectiveness translating into poor clinical outcomes is a particular problem with clopidogrel, the most widely used and economical ADP receptor antagonist available at present. This makes it crucial to be able to measure the antiplatelet response in individual patients. Platelet function tests are becoming available for clinical use, and these together with the choice of different ADP receptor antagonists available, may allow tailored antiplatelet therapy to be given to individual patients for optimal effect to reduce thrombotic risk. Newer agents with distinct properties are in development and may offer additional therapeutic options. Research is ongoing to gain more information on interactions between P2Y12 receptor antagonists and natural agents that may contribute to inhibition of platelet function, and also to look for any differences in mechanism of action between the different agents that are already available and in development.
Competing Interests
Stan Heptinstall, on behalf of the University of Nottingham, has received a grant for a platelet function workshop from Lilly and a grant for research from AstraZeneca. He is also director of Platelet Solutions Ltd, a new company that is developing simple to use kits for platelet function testing. Yanushi Dullewe Wijeyeratne has no competing interests.
REFERENCES
- 1.Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Storey RF, Newby LJ, Heptinstall S. Effects of P2Y(1) and P2Y(12) receptor antagonists on platelet aggregation induced by different agonists in human whole blood. Platelets. 2001;12:443–7. doi: 10.1080/09537100120085450. [DOI] [PubMed] [Google Scholar]
- 3.Gachet C. P2 receptors, platelet function and pharmacological implications. Thromb Haemost. 2008;99:466–72. doi: 10.1160/TH07-11-0673. [DOI] [PubMed] [Google Scholar]
- 4.Gachet C, Léon C, Hechler B. The platelet P2 receptors in arterial thrombosis. Blood Cells Mol Dis. 2006;36:223–7. doi: 10.1016/j.bcmd.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 5.Bennett JS. Platelet-fibrinogen interactions. Ann NY Acad Sci. 2001;936:340–54. doi: 10.1111/j.1749-6632.2001.tb03521.x. [DOI] [PubMed] [Google Scholar]
- 6.Jennings LK. Mechanisms of platelet activation: need for new strategies to protect against platelet-mediated atherothrombosis. Thromb Haemost. 2009;102:248–57. doi: 10.1160/TH09-03-0192. [DOI] [PubMed] [Google Scholar]
- 7.Marcus AJ, Broekman MJ, Drosopoulos JH, Olson KE, Islam N, Pinsky DJ, Levi R. Role of CD39 (NTPDase-1) in thromboregulation, cerebroprotection, and cardioprotection. Semin Thromb Hemost. 2005;31:234–46. doi: 10.1055/s-2005-869528. [DOI] [PubMed] [Google Scholar]
- 8.Glenn JR, White AE, Johnson A, Fox SC, Behan MW, Dolan G, Heptinstall S. Leukocyte count and leukocyte ecto-nucleotidase are major determinants of the effects of adenosine triphosphate and adenosine diphosphate on platelet aggregation in human blood. Platelets. 2005;16:159–70. doi: 10.1080/09537100500063889. [DOI] [PubMed] [Google Scholar]
- 9.Hu H, Hoylaerts MF. The P2X1 ion channel in platelet function. Platelets. 2010;21:153–66. doi: 10.3109/09537101003599549. [DOI] [PubMed] [Google Scholar]
- 10.Li Z, Delaney MK, O'Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30:2341–9. doi: 10.1161/ATVBAHA.110.207522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin J, Quinton TM, Zhang J, Rittenhouse SE, Kunapuli SP. Adenosine diphosphate (ADP)-induced thromboxane A(2) generation in human platelets requires coordinated signaling through integrin alpha(IIb)beta(3) and ADP receptors. Blood. 2002;99:193–8. doi: 10.1182/blood.v99.1.193. [DOI] [PubMed] [Google Scholar]
- 12.Storey RF, Judge HM, Wilcox RG, Heptinstall S. Inhibition of ADP-induced P-selectin expression and platelet-leukocyte conjugate formation by clopidogrel and the P2Y12 receptor antagonist AR-C69931MX but not aspirin. Thromb Haemost. 2002;88:488–94. [PubMed] [Google Scholar]
- 13.Kahner BN, Dorsam RT, Kunapuli SP. Role of P2Y receptor subtypes in platelet-derived microparticle generation. Front Biosci. 2008;13:433–9. doi: 10.2741/2690. [DOI] [PubMed] [Google Scholar]
- 14.Fabre JE, Nguyen M, Latour A, Keifer JA, Audoly LP, Coffman TM, Koller BH. Decreased platelet aggregation, increased bleeding time and resistance to thromboembolism in P2Y1-deficient mice. Nat Med. 1999;5:1199–202. doi: 10.1038/13522. [DOI] [PubMed] [Google Scholar]
- 15.Léon C, Freund M, Ravanat C, Baurand A, Cazenave JP, Gachet C. Key role of the P2Y(1) receptor in tissue factor-induced thrombin-dependent acute thromboembolism: studies in P2Y(1)-knockout mice and mice treated with a P2Y(1) antagonist. Circulation. 2001;103:718–23. doi: 10.1161/01.cir.103.5.718. [DOI] [PubMed] [Google Scholar]
- 16.Baurand A, Raboisson P, Freund M, Léon C, Cazenave JP, Bourguignon JJ, Gachet C. Inhibition of platelet function by administration of MRS2179, a P2Y1 receptor antagonist. Eur J Pharmacol. 2001;412:213–21. doi: 10.1016/s0014-2999(01)00733-6. [DOI] [PubMed] [Google Scholar]
- 17.Boyer JL, Adams M, Ravi RG, Jacobson KA, Harden TK. 2-Chloro N(6)-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate is a selective high affinity P2Y(1) receptor antagonist. Br J Pharmacol. 2002;135:2004–10. doi: 10.1038/sj.bjp.0704673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hechler B, Nonne C, Roh EJ, Cattaneo M, Cazenave JP, Lanza F, Jacobson KA, Gachet C. MRS2500 [2-iodo-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate], a potent, selective, and stable antagonist of the platelet P2Y1 receptor with strong antithrombotic activity in mice. J Pharmacol Exp Ther. 2006;316:556–63. doi: 10.1124/jpet.105.094037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manolopoulos P, Glenn JR, Fox SC, May JA, Dovlatova NL, Tang SW, Thomas NR, Ralevic V, Heptinstall S. Acyl derivatives of coenzyme A inhibit platelet function via antagonism at P2Y1 and P2Y12 receptors: a new finding that may influence the design of anti-thrombotic agents. Platelets. 2008;19:134–45. doi: 10.1080/09537100701708498. [DOI] [PubMed] [Google Scholar]
- 20.Pfefferkorn JA, Choi C, Winters T, Kennedy R, Chi L, Perrin LA, Lu G, Ping YW, McClanahan T, Schroeder R, Leininger MT, Geyer A, Schefzick S, Atherton J. P2Y1 receptor antagonists as novel antithrombotic agents. Bioorg Med Chem Lett. 2008;18:3338–43. doi: 10.1016/j.bmcl.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 21.Thalji RK, Aiyar N, Davenport EA, Erhardt JA, Kallal LA, Morrow DM, Senadhi S, Burns-Kurtis CL, Marino JP., Jr Benzofuran-substituted urea derivatives as novel P2Y(1) receptor antagonists. Bioorg Med Chem Lett. 2010;20:4104–7. doi: 10.1016/j.bmcl.2010.05.072. [DOI] [PubMed] [Google Scholar]
- 22.Kunapuli SP, Ding Z, Dorsam RT, Kim S, Murugappan S, Quinton TM. ADP receptors-targets for developing antithrombotic agents. Curr Pharm Des. 2003;9:2303–16. doi: 10.2174/1381612033453947. [DOI] [PubMed] [Google Scholar]
- 23.NICE Guidelines. Clopidogrel and modified-release dipyridamole for the prevention of occlusive vascular events. 2010. NICE Technology Appraisal Guidance 210 [Online] NICE 2010. Available at http://www.nice.org.uk/nicemedia/live/13285/52030/52030.pdf [last accessed 31 March 2011] [DOI] [PubMed]
- 24.US National Library of Medicine. Ticlopidine hydrochloride. 2009. Current Medication Information [Online] NLM 2009. Available at http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?id = 11822 [last accessed 31 March 2011]
- 25.Savi P, Pereillo JM, Uzabiaga MF, Combalbert J, Picard C, Maffrand JP, Pascal M, Herbert JM. Identification and biological activity of the active metabolite of clopidogrel. Thromb Haemost. 2000;84:891–6. [PubMed] [Google Scholar]
- 26.CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE) Lancet. 1996;348:1329–39. doi: 10.1016/s0140-6736(96)09457-3. [DOI] [PubMed] [Google Scholar]
- 27.Fox KA, Mehta SR, Peters R, Zhao F, Lakkis N, Gersh BJ, Yusuf S. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non-ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation. 2004;110:1202–8. doi: 10.1161/01.CIR.0000140675.85342.1B. [DOI] [PubMed] [Google Scholar]
- 28.Peters BJ, Harmsze AM, Ten Berg JM, Maitland-van der Zee AH, Tjoeng MM, de Boer A, Deneer VH. CYP2C19 and ABCB1 genes and individualized treatment with clopidogrel. Pharmacogenomics. 2011;12:141–4. doi: 10.2217/pgs.10.211. [DOI] [PubMed] [Google Scholar]
- 29.Xie HG, Zou JJ, Hu ZY, Zhang JJ, Ye F, Chen SL. Individual variability in the disposition of and response to clopidogrel: pharmacogenomics and beyond. Pharmacol Ther. 2011;129:267–89. doi: 10.1016/j.pharmthera.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 30.Bouman HJ, Schömig E, van Werkum JW, Velder J, Hackeng CM, Hirschhäuser C, Waldmann C, Schmalz HG, ten Berg JM, Taubert D. Paraoxonase-1 is a major determinant of clopidogrel efficacy. Nat Med. 2011;17:110–6. doi: 10.1038/nm.2281. [DOI] [PubMed] [Google Scholar]
- 31.Arméro S, Camoin Jau L, Omar Aït Mokhtar O, Mancini J, Burignat-Bonello C, Tahirou I, Arques S, Dignat-George F, Paganelli F, Bonello L. Intra-individual variability in clopidogrel responsiveness in coronary artery disease patients under long term therapy. Platelets. 2010;21:503–7. doi: 10.3109/09537104.2010.499483. [DOI] [PubMed] [Google Scholar]
- 32.Sofi F, Marcucci R, Gori AM, Giusti B, Abbate R, Gensini GF. Clopidogrel non-responsiveness and risk of cardiovascular morbidity. An updated meta-analysis. Thromb Haemost. 2010;103:841–8. doi: 10.1160/TH09-06-0418. [DOI] [PubMed] [Google Scholar]
- 33.Price MJ. Bedside evaluation of thienopyridine antiplatelet therapy. Circulation. 2009;119:2625–32. doi: 10.1161/CIRCULATIONAHA.107.696732. [DOI] [PubMed] [Google Scholar]
- 34.Fox SC, May JA, Shah A, Neubert U, Heptinstall S. Measurement of platelet P-selectin for remote testing of platelet function during treatment with clopidogrel and/or aspirin. Platelets. 2009;20:250–9. doi: 10.1080/09537100902912451. [DOI] [PubMed] [Google Scholar]
- 35.Ko YG, Suh JW, Kim BH, Lee CJ, Kim JS, Choi D, Hong MK, Seo MK, Youn TJ, Chae IH, Choi DJ, Jang Y. Comparison of 2 point-of-care platelet function tests, VerifyNow Assay and Multiple Electrode Platelet Aggregometry, for predicting early clinical outcomes in patients undergoing percutaneous coronary intervention. Am Heart J. 2011;161:383–90. doi: 10.1016/j.ahj.2010.10.036. [DOI] [PubMed] [Google Scholar]
- 36.Koessler J, Kobsar AL, Rajkovic MS, Schafer A, Flierl U, Pfoertsch S, Bauersachs J, Steigerwald U, Rechner AR, Walter U. The new INNOVANCE® PFA P2Y cartridge is sensitive to the detection of the P2Y12 receptor inhibition. Platelets. 2011;22:19–25. doi: 10.3109/09537104.2010.514967. [DOI] [PubMed] [Google Scholar]
- 37.Price MJ, Berger PB, Teirstein PS, Tanguay JF, Angiolillo DJ, Spriggs D, Puri S, Robbins M, Garratt KN, Bertrand OF, Stillablower ME, Aragon JR, Kandzari DE, Stinis CT, Lee MS, Manoukian SV, Cannon CP, Schork NJ, Topol EJ, GRAVITAS Investigators Standard- vs high-dose clopidogrel based on platelet function testing after percutaneous coronary intervention: the GRAVITAS randomized trial. JAMA. 2011;305:1097–105. doi: 10.1001/jama.2011.290. [DOI] [PubMed] [Google Scholar]
- 38.Mehta SR, Tanguay JF, Eikelboom JW, Jolly SS, Joyner CD, Granger CB, Faxon DP, Rupprecht HJ, Budaj A, Avezum A, Widimsky P, Steg PG, Bassand JP, Montalescot G, Macaya C, Di Pasquale G, Niemela K, Ajani AE, White HD, Chrolavicius S, Gao P, Fox KA, Yusuf S. CURRENT-OASIS 7 trial investigators. Double-dose versus standard-dose clopidogrel and high-dose versus low-dose aspirin in individuals undergoing percutaneous coronary intervention for acute coronary syndromes (CURRENT-OASIS 7): a randomised factorial trial. Lancet. 2010;376:1233–43. doi: 10.1016/S0140-6736(10)61088-4. [DOI] [PubMed] [Google Scholar]
- 39.Gurbel PA, Tantry US. An initial experiment with personalized antiplatelet therapy: the GRAVITAS trial. JAMA. 2011;305:1136–7. doi: 10.1001/jama.2011.332. [DOI] [PubMed] [Google Scholar]
- 40.Valgimigli M, Campo G, de Cesare N, Meliga E, Vranckx P, Furgieri A, Angiolillo DJ, Sabatè M, Hamon M, Repetto A, Colangelo S, Brugaletta S, Parrinello G, Percoco G, Ferrari R. Tailoring Treatment With Tirofiban in Patients Showing Resistance to Aspirin and/or Resistance to Clopidogrel (3T/2R) Investigators. Intensifying platelet inhibition with tirofiban in poor responders to aspirin, clopidogrel, or both agents undergoing elective coronary intervention: results from the double-blind, prospective, randomized Tailoring Treatment with Tirofiban in Patients Showing Resistance to Aspirin and/or Resistance to Clopidogrel study. Circulation. 2009;119:3215–22. doi: 10.1161/CIRCULATIONAHA.108.833236. [DOI] [PubMed] [Google Scholar]
- 41.British Medical Association and the Royal Pharmaceutical Society. British National Formulary: clopidogrel. Available at http://bnf.org/bnf/bnf/61/213738.htm#_213738 [last accessed 31 March 2011]
- 42.Kim SD, Kang W, Lee HW, Park DJ, Ahn JH, Kim MJ, Kim EY, Kim SW, Nam HS, Na HJ, Yoon YR. Bioequivalence and tolerability of two clopidogrel salt preparations, besylate and bisulfate: a randomized, open-label, crossover study in healthy Korean male subjects. Clin Ther. 2009;31:793–803. doi: 10.1016/j.clinthera.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 43.Neubauer H, Krüger JC, Lask S, Endres HG, Pepinghege F, Engelhardt A, Bulut D, Mügge A. Comparing the antiplatelet effect of clopidogrel hydrogensulfate and clopidogrel besylate: a crossover study. Clin Res Cardiol. 2009;98:533–40. doi: 10.1007/s00392-009-0033-1. [DOI] [PubMed] [Google Scholar]
- 44.Park JY, Kim KA, Ryu JH, Lee GH, Jeon SH, Kim JS. Pharmacokinetics and the antiplatelet effect of a new clopidogrel formulation, clopidogrel besylate, in healthy subjects. Int J Clin Pharmacol Ther. 2010;48:259–69. doi: 10.5414/cpp48259. [DOI] [PubMed] [Google Scholar]
- 45.Jakubowski JA, Winters KJ, Naganuma H, Wallentin L. Prasugrel: a novel thienopyridine antiplatelet agent. A review of preclinical and clinical studies and the mechanistic basis for its distinct antiplatelet profile. Cardiovasc Drug Rev. 2007;25:357–74. doi: 10.1111/j.1527-3466.2007.00027.x. [DOI] [PubMed] [Google Scholar]
- 46.Wallentin L. P2Y(12) inhibitors: differences in properties and mechanisms of action and potential consequences for clinical use. Eur Heart J. 2009;30:1964–77. doi: 10.1093/eurheartj/ehp296. [DOI] [PubMed] [Google Scholar]
- 47.Michelson AD. New P2Y12 antagonists. Curr Opin Hematol. 2009;16:371–7. doi: 10.1097/MOH.0b013e32832ea2f2. [DOI] [PubMed] [Google Scholar]
- 48.Brandt JT, Close SL, Iturria SJ, Payne CD, Farid NA, Ernest CS, 2nd, Lachno DR, Salazar D, Winters KJ. Common polymorphisms of CYP2C19 and CYP2C9 affect the pharmacokinetic and pharmacodynamic response to clopidogrel but not prasugrel. J Thromb Haemost. 2007;5:2429–36. doi: 10.1111/j.1538-7836.2007.02775.x. [DOI] [PubMed] [Google Scholar]
- 49.Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, Neumann FJ, Ardissino D, De Servi S, Murphy SA, Riesmeyer J, Weerakkody G, Gibson CM, Antman EM, TRITON-TIMI 38 Investigators Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001–15. doi: 10.1056/NEJMoa0706482. [DOI] [PubMed] [Google Scholar]
- 50.NICE Guidelines. Prasugrel for the treatment of acute coronary syndromes with percutaneous coronary intervention. 2009. NICE Technology Appraisal Guidance 182 [Online] NICE 2009. Available at http://www.nice.org.uk/nicemedia/live/12324/45849/45849.pdf [last accessed 12 April 2011]
- 51.Husted S, van Giezen JJ. Ticagrelor: the first reversibly binding oral P2Y12 receptor antagonist. Cardiovasc Ther. 2009;27:259–74. doi: 10.1111/j.1755-5922.2009.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Giezen JJ, Nilsson L, Berntsson P, Wissing BM, Giordanetto F, Tomlinson W, Greasley PJ. Ticagrelor binds to P2Y12 receptors independently from ADP but antagonizes ADP-induced receptor signalling and platelet aggregation. J Thromb Haemost. 2009;7:1556–65. doi: 10.1111/j.1538-7836.2009.03527.x. [DOI] [PubMed] [Google Scholar]
- 53.Kowalczyk M, Banach M, Mikhailidis DP, Hannam S, Rysz J. Ticagrelor – a new platelet aggregation inhibitor in patients with acute coronary syndromes. An improvement of other inhibitors? Med Sci Monit. 2009;15:MS24–30. [PubMed] [Google Scholar]
- 54.Wallentin L, James S, Storey RF, Armstrong M, Barratt BJ, Horrow J, Husted S, Katus H, Steg PG, Shah SH, Becker RC, PLATO investigators Effect of CYP2C19 and ABCB1 single nucleotide polymorphisms on outcomes of treatment with ticagrelor versus clopidogrel for acute coronary syndromes: a genetic substudy of the PLATO trial. Lancet. 2010;376:1320–8. doi: 10.1016/S0140-6736(10)61274-3. [DOI] [PubMed] [Google Scholar]
- 55.Teng R, Butler K. Pharmacokinetics, pharmacodynamics, tolerability and safety of single ascending doses of ticagrelor, a reversibly binding oral P2Y(12) receptor antagonist, in healthy subjects. Eur J Clin Pharmacol. 2010;66:487–96. doi: 10.1007/s00228-009-0778-5. [DOI] [PubMed] [Google Scholar]
- 56.Gurbel PA, Bliden KP, Butler K, Tantry US, Gesheff T, Wei C, Teng R, Antonino MJ, Patil SB, Karunakaran A, Kereiakes DJ, Parris C, Purdy D, Wilson V, Ledley GS, Storey RF. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation. 2009;120:2577–85. doi: 10.1161/CIRCULATIONAHA.109.912550. [DOI] [PubMed] [Google Scholar]
- 57.Butler K, Teng R. Pharmacokinetics, pharmacodynamics, and safety of ticagrelor in volunteers with mild hepatic impairment. J Clin Pharmacol. 2010 doi: 10.1177/0091270010379409. [Epub ahead of print] doi: 10.1177/0091270010379409. [DOI] [PubMed] [Google Scholar]
- 58.Nawarskas JJ, Clark SM. Ticagrelor: a novel reversible oral antiplatelet agent. Cardiol Rev. 2011;19:95–100. doi: 10.1097/CRD.0b013e3182099d86. [DOI] [PubMed] [Google Scholar]
- 59.Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, Horrow J, Husted S, James S, Katus H, Mahaffey KW, Scirica BM, Skene A, Steg PG, Storey RF, Harrington RA, PLATO Investigators. Freij A, Thorsén M. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045–57. doi: 10.1056/NEJMoa0904327. [DOI] [PubMed] [Google Scholar]
- 60.Rao AK, Pratt C, Berke A, Jaffe A, Ockene I, Schreiber TL, Bell WR, Knatterud G, Robertson TL, Terrin ML. Thrombolysis in Myocardial Infarction (TIMI) Trial-phase I: hemorrhagic manifestations and changes in plasma fibrinogen and the fibrinolytic system in patients treated with recombinant tissue plasminogen activator and streptokinase. J Am Coll Cardiol. 1988;11:1–11. doi: 10.1016/0735-1097(88)90158-1. [DOI] [PubMed] [Google Scholar]
- 61.Storey RF, Angiolillo DJ, Patil SB, Desai B, Ecob R, Husted S, Emanuelsson H, Cannon CP, Becker RC, Wallentin L. Inhibitory effects of ticagrelor compared with clopidogrel on platelet function in patients with acute coronary syndromes: the PLATO (PLATelet inhibition and patient Outcomes) PLATELET substudy. J Am Coll Cardiol. 2010;56:1456–62. doi: 10.1016/j.jacc.2010.03.100. [DOI] [PubMed] [Google Scholar]
- 62.Iyú D, Glenn JR, White AE, Fox SC, Dovlatova N, Heptinstall S. P2Y12 and EP3 antagonists promote the inhibitory effects of natural modulators of platelet aggregation that act via cAMP. Platelets. 2011 doi: 10.3109/09537104.2011.576284. [Epub ahead of print] doi: 10.3109/09537104.2011.576284. [DOI] [PubMed] [Google Scholar]
- 63.European Medicines Agency Summary of Opinion. Ticagleror [Online] EMA/536945/2010. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_-_Initial_authorisation/human/001241/WC500097028.pdf [last accessed 12 April 2011]
- 64.Ingall AH, Dixon J, Bailey A, Coombs ME, Cox D, McInally JI, Hunt SF, Kindon ND, Teobald BJ, Willis PA, Humphries RG, Leff P, Clegg JA, Smith JA, Tomlinson W. Antagonists of the platelet P2T receptor: a novel approach to antithrombotic therapy. J Med Chem. 1999;42:213–20. doi: 10.1021/jm981072s. [DOI] [PubMed] [Google Scholar]
- 65.Storey RF, Oldroyd KG, Wilcox RG. Open multicentre study of the P2T receptor antagonist ARC69931MX assessing safety, tolerability and activity in patients with acute coronary syndromes. Thromb Haemost. 2001;85:401–7. [PubMed] [Google Scholar]
- 66.Ueno M, Ferreiro JL, Angiolillo DJ. Update on the clinical development of cangrelor. Expert Rev Cardiovasc Ther. 2010;8:1069–77. doi: 10.1586/erc.10.90. [DOI] [PubMed] [Google Scholar]
- 67.Steinhubl SR, Oh JJ, Oestreich JH, Ferraris S, Charnigo R, Akers WS. Transitioning patients from cangrelor to clopidogrel: pharmacodynamic evidence of a competitive effect. Thromb Res. 2008;121:527–34. doi: 10.1016/j.thromres.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 68.Dovlatova NL, Jakubowski JA, Sugidachi A, Heptinstall S. The reversible P2Y antagonist cangrelor influences the ability of the active metabolites of clopidogrel and prasugrel to produce irreversible inhibition of platelet function. J Thromb Haemost. 2008;6:1153–9. doi: 10.1111/j.1538-7836.2008.03020.x. [DOI] [PubMed] [Google Scholar]
- 69.Bhatt DL, Lincoff AM, Gibson CM, Stone GW, McNulty S, Montalescot G, Kleiman NS, Goodman SG, White HD, Mahaffey KW, Pollack CV, Jr, Manoukian SV, Widimsky P, Chew DP, Cura F, Manukov I, Tousek F, Jafar MZ, Arneja J, Skerjanec S, Harrington RA, CHAMPION PLATFORM Investigators Intravenous platelet blockade with cangrelor during PCI. N Engl J Med. 2009;361:2330–41. doi: 10.1056/NEJMoa0908629. [DOI] [PubMed] [Google Scholar]
- 70.Harrington RA, Stone GW, McNulty S, White HD, Lincoff AM, Gibson CM, Pollack CV, Jr, Montalescot G, Mahaffey KW, Kleiman NS, Goodman SG, Amine M, Angiolillo DJ, Becker RC, Chew DP, French WJ, Leisch F, Parikh KH, Skerjanec S, Bhatt DL. Platelet inhibition with cangrelor in patients undergoing PCI. N Engl J Med. 2009;361:2318–29. doi: 10.1056/NEJMoa0908628. [DOI] [PubMed] [Google Scholar]
- 71.US National Institute of Health. A Clinical Trial Comparing Cangrelor to Clopidogrel Standard Therapy in Subjects Who Require Percutaneous Coronary Intervention (PCI) (CHAMPION PHOENIX) [Online] Clinicaltrials.gov 2010. Available at http://clinicaltrials.gov/ct2/show/NCT01156571 [last Accessed 12 April 2011]
- 72.Oestreich JH. Elinogrel, a reversible P2Y12 receptor antagonist for the treatment of acute coronary syndrome and prevention of secondary thrombotic events. Curr Opin Investig Drugs. 2010;11:340–8. [PubMed] [Google Scholar]
- 73.Ueno M, Rao SV, Angiolillo DJ. Elinogrel: pharmacological principles, preclinical and early phase clinical testing. Future Cardiol. 2010;6:445–53. doi: 10.2217/fca.10.67. [DOI] [PubMed] [Google Scholar]
- 74.Rao SV, Zeymer U, Thompson V, Huber K, Kochman J, McClure MW, Gretler DD, Bhatt DL, Gibson CM, Madan M, Berdan L, Paynter G, Leonardi S, Harrington R, Welsh R. INNOVATE PCI: a Phase II Safety and Efficacy Study of PRT060128 (Elinogrel), a Novel Intravenous and Oral P2Y12 Inhibitor, in Non-Urgent PCI. In: ESC (European Society of Cardiology), ESC Congress 2010, Stockholm, Sweden 28 Aug – 1 Sep 2010.
- 75.Kim S, Kunapuli SP. P2Y12 receptor in platelet activation. Platelets. 2011;22:54–8. doi: 10.3109/09537104.2010.497231. [DOI] [PubMed] [Google Scholar]
- 76.Heptinstall S, Johnson A, Glenn JR, White AE. Adenine nucleotide metabolism in human blood – important roles for leukocytes and erythrocytes. J Thromb Haemost. 2005;3:2331–9. doi: 10.1111/j.1538-7836.2005.01489.x. [DOI] [PubMed] [Google Scholar]
- 77.Jones S, Evans RJ, Mahaut-Smith MP. Extracellular Ca(2+) modulates ADP-evoked aggregation through altered agonist degradation: implications for conditions used to study P2Y receptor activation. Br J Haematol. 2011;153:83–91. doi: 10.1111/j.1365-2141.2010.08499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Iyú D, Glenn JR, White AE, Fox SC, Heptinstall S. Adenosine derived from ADP can contribute to inhibition of platelet aggregation in the presence of a P2Y12 antagonist. Arterioscler Thromb Vasc Biol. 2011;31:416–22. doi: 10.1161/ATVBAHA.110.219501. [DOI] [PubMed] [Google Scholar]
- 79.Bjorkman JA, Kirk I, van Giezen JJ. AZD6140 inhibits adenosine uptake into erythrocytes and enhances coronary blood flow after local ischemia or intracoronary adenosine infusion. Circulation. 2007;116(Suppl) II-28. [Abstract 245] [Google Scholar]
- 80.Storey RF, Bliden KP, Patil SB, Karunakaran A, Ecob R, Butler K, Teng R, Wei C, Tantry US, Gurbel PA. ONSET/OFFSET Investigators. Incidence of dyspnea and assessment of cardiac and pulmonary function in patients with stable coronary artery disease receiving ticagrelor, clopidogrel, or placebo in the ONSET/OFFSET study. J Am Coll Cardiol. 2010;56:185–93. doi: 10.1016/j.jacc.2010.01.062. [DOI] [PubMed] [Google Scholar]
- 81.Fox SC, Behan MW, Heptinstall S. Inhibition of ADP-induced intracellular Ca2+ responses and platelet aggregation by the P2Y12 receptor antagonists AR-C69931MX and clopidogrel is enhanced by prostaglandin E1. Cell Calcium. 2004;35:39–46. doi: 10.1016/s0143-4160(03)00170-2. [DOI] [PubMed] [Google Scholar]
- 82.Cattaneo M, Lecchi A. Inhibition of P2Y12 receptor by adenosine diphosphate potentiates the antiplatelet effects of prostacyclin. J Thromb Haemost. 2007;5:577–82. doi: 10.1111/j.1538-7836.2007.02356.x. [DOI] [PubMed] [Google Scholar]
- 83.Heptinstall S, Espinosa DI, Manolopoulos P, Glenn JR, White AE, Johnson A, Dovlatova N, Fox SC, May JA, Hermann D, Magnusson O, Stefansson K, Hartman D, Gurney M. DG-041 inhibits the EP3 prostanoid receptor: a new target for inhibition of platelet function in atherothrombotic disease. Platelets. 2008;19:605–13. doi: 10.1080/09537100802351073. [DOI] [PubMed] [Google Scholar]
- 84.Singh J, Zeller W, Zhou N, Hategen G, Mishra R, Polozov A, Yu P, Onua E, Zhang J, Zembower D, Kiselyov A, Ramírez JL, Sigthorsson G, Bjornsson JM, Thorsteinsdottir M, Andrésson T, Bjarnadottir M, Magnusson O, Fabre JE, Stefansson K, Gurney ME. Antagonists of the EP3 receptor for prostaglandin E2 are novel antiplatelet agents that do not prolong bleeding. ACS Chem Biol. 2009;4:115–26. doi: 10.1021/cb8002094. [DOI] [PubMed] [Google Scholar]
- 85.Fabre JE, Gurney ME. Limitations of current therapies to prevent thrombosis: a need for novel strategies. Mol Biosyst. 2010;6:305–15. doi: 10.1039/b914375k. [DOI] [PubMed] [Google Scholar]
- 86.Dohlman HG, Thorner J, Caron MG, Lefkowitz RJ. Model systems for the study of seven-transmembrane-segment receptors. Annu Rev Biochem. 1991;60:653–88. doi: 10.1146/annurev.bi.60.070191.003253. [DOI] [PubMed] [Google Scholar]
- 87.Iyú D, Glenn JR, White AE, Fox SC, van Giezen H, Nylander S, Heptinstall S. Mode of action of P2Y(12) antagonists as inhibitors of platelet function. Thromb Haemost. 2011;105:96–106. doi: 10.1160/TH10-07-0482. [DOI] [PubMed] [Google Scholar]
- 88.Srinivasan S, Mir F, Huang JS, Khasawneh FT, Lam SC, Le Breton GC. The P2Y12 antagonists, 2-methylthioadenosine 5′-monophosphate triethylammonium salt and cangrelor (ARC69931MX), can inhibit human platelet aggregation through a Gi-independent increase in cAMP levels. J Biol Chem. 2009;284:16108–17. doi: 10.1074/jbc.M809780200. [DOI] [PMC free article] [PubMed] [Google Scholar]