

Abstract
This case study describes early phase purification process development for a recombinant anticancer minibody produced in mammalian cell culture. The minibody did not bind to protein A. Cation-exchange, anion-exchange, hydrophobic-interaction, and hydroxyapatite (eluted by phosphate gradient) chromatographic methods were scouted, but the minibody coeluted with BSA to a substantial degree on each. Hydroxyapatite eluted with a sodium chloride gradient separated BSA and also removed a dimeric contaminant, but BSA consumed so much binding capacity that this proved impractical as a capture tool. Capto MMC media proved capable of supporting adequate capture and significant dimer removal, although both loading and elution selectivity varied dramatically with the amount of supernatant applied to the column. An anion-exchange step was included to fortify overall virus and DNA removal. These results illustrate the value of multimodal chromatography methods when affinity chromatography methods are lacking and conventional alternatives prove inadequate.
Keywords: Minibodies, multimodal chromatography methods, phase 1 purification, in vivo diagnostic imaging
Small, genetically engineered immunological constructs are being developed industry-wide for a growing range of in vivo applications. Examples include Fab, F(ab')2, single-chain (sc) Fv, bis-scFV, diabodies, minibodies, and single-domain antibodies (1). Their small size potentially gives them access to tissues that are poorly accessible by intact antibodies; rapid clearance from blood and nontargeted tissues; lower immunogenic response; and eye-drop, inhalant, or oral administration.
We report here on purification of an affinity-matured, humanized, antiprostate stem-cell antigen (PSCA) minibody for first-in-human clinical studies. This minibody and an earlier chimeric version have demonstrated excellent high-contrast microPET imaging for PSCA-positive human prostate, pancreatic, and bladder cancers in animal models (Figure 1) (2). The overall structure is reminiscent of IgG, bivalent but only half the size. The “Fc region” is reduced to the Cγ3 domains and “Fab” to the variable domains (Figure 2). Although beneficial for effectiveness of the application, this architecture lacks the binding sites for protein A. Thus, the principal enabling tool for IgG platform purification is inapplicable, thereby presenting a greater challenge to process developers. As with antibodies, conventional chromatography methods may provide an effective alternative in some cases, but it is unrealistic to expect them to do so for most products and production systems.
Figure 1.
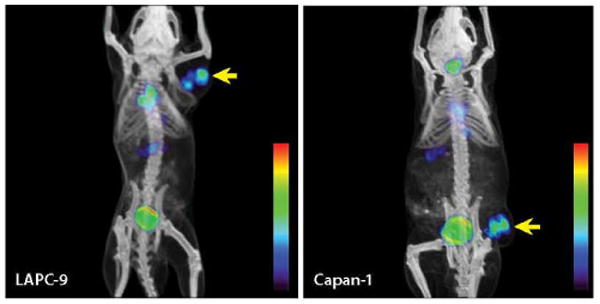
Coregistered microPET/CT scans of mice bearing LAPC-9 or Capan-1 xenografts, 21 hours after injection of 124I-labeled A11 minibody (100–150 μCi); LAPC-9 is an androgen-dependent human prostate cancer cell line, and Capan-1 is a human pancreatic cancer cell line. Tumor locations are indicated by yellow arrows. Response scale is red (high) to blue (low).
Figure 2.
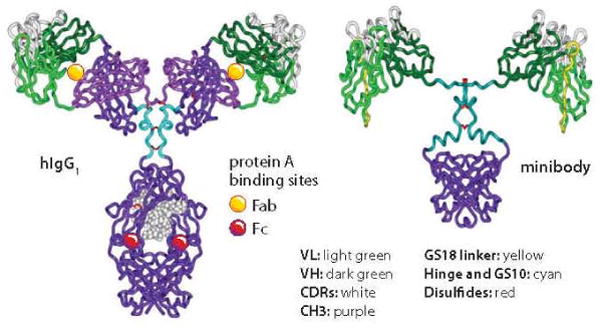
Structural comparison of a human IgG1 and an anti-PSCA minibody; references 2, 28, and 29 provide more information about the minibody's development, structure, characterization, and applications.
Multimodal (mixed-mode) chromatography methods have existed since the 1950s (3). Hydroxyapatite (HA) is the archetype of multimodal chromatography methods, combining cation exchange and metal affinity. (4). It is also a prime example of the barriers that need to be overcome for mixed modes to become mainstream tools. HA's selectivity was recognized as unique from its introduction, but a lack of practical knowledge concerning its binding mechanisms long delayed the development of scouting pathways that fully revealed its abilities. That discouraged process developers who might have benefited from its capabilities. As those pathways were defined, it became possible to control each binding mechanism, and HA has emerged as the most broadly capable process option for removing fragments and high levels of aggregates from antibody preparations (5–9).
Successes in recombinant immunotherapy have stimulated introduction of other mixed-mode media. Charged-hydrophobic mixed modes began to appear in the 1980s, mostly as hopeful protein A replacements. They included products such as T-gel, ABx, Avid-AL, and MEP Hypercel media (10–21). More recent entries such as AcroSep HEA, AcroSep PPA, Capto MMC, and Capto adhere media have been applied for capture but focus more on aggregate removal (22–26).
Such products arrive at a propitious moment: HA's successes have made process developers willing to confront the complexity of mixed modes and provided a conceptual framework for exploring multimodal interactions. High-throughput screening and statistical design of experiments (DoE) enable rapid accumulation of process-pertinent data (7, 25, 26). And the well-defined chemical structures of mixed-mode ligands provide valuable guidance on the types of eluting agents that promise useful results (27).
Materials and Methods
Cell Culture
Anti-PSCA minibody (clone A11.1 2C3) was grown in NS0 cells using media supplemented with 2% fetal bovine serum. This minibody is about 80 kDa and has an isoelectric point (pI) of about 7.3. Initial purification process development was conducted with minibody produced in T flasks, and product concentration was ∼50 mg/L. Later material produced in hollow-fiber bioreactors was at 1–2 g/L. All preparations contained a subpopulation of dimers created by noncovalent association of variable regions (28, 29).
Media Conditioning
We obtained Dowex AG1×8 (cholestyramine) media — a particulate, microporous, strong anion exchanger on a hydrophobic styrene divinyl benzene backbone — from Bio-Rad Laboratories (www.bio-rad.com). It was added to harvested supernatant at a proportion of 5% (v/v) and incubated with gentle mixing overnight at 4 °C (29), then removed by membrane filtration at 0.22 μm. This method has been shown to remove cell debris, DNA, lipopolysaccharide, phospholipids, fatty acids, steroids, and pH indicator dyes (27), which constitute most of the primary foulants of chromatography media. It is effective even at physiological pH and conductivity levels, at which its affinity for IgG and minibodies is nil. Used media can be discarded.
Purification
All chromatography experiments were conducted using an ÄKTA 100 Explorer system from GE Healthcare (www.gelifesciences.com). Buffers and salts came from Sigma Chemical Company (www.sigmaaldrich.com) except for arginine from Ajinomoto Aminoscience (www.ajiaminoscience.com). Initial cation exchange (CX) and anion exchange (AX) scouting were conducted on 334-μL CIM SO3 and CIM QA monoliths (12 mm diameter × 3 mm height) from BIA Separations (www.biaseparations.com).
These experiments were conducted at a linear flow rate of 300 cm/hr (4-mL/min volumetric flow rate). The CX sample was prepared by 5× dilution of filtered cell culture supernatant (CCS) with 20 mM MES at pH 6. The AX sample was prepared by 5× dilution of CCS with 20 mM Tris at pH 8. Initial scouting was performed with 5-min (60 column volume) conductivity gradients to 500 mM sodium chloride (NaCl). We used 20 mM MES for buffering at pH 6, 20 mM Hepes at pH 7, and 20 mM Tris at pH 8. CX experiments (pH 4.5) were conducted in 20 mM sodium acetate. Later-stage AX and CX applications were conducted with UNOsphere Q or UNOsphere S media from Bio-Rad at a 300-cm/h linear flow rate.
For hydrophobic-interaction chromatography (HIC) we used ToyoPearl Phenyl 600M from Tosoh BioScience (www.tosohbioscience.com) packed into an HR 5/5 column (5 × 50 mm) from GE Healthcare. Sample was prepared by 3× dilution of CCS with 4 M NaCl. The column was equilibrated with 20 mM sodium phosphate and 1.5 M ammonium sulfate at pH 7 and eluted with a linear gradient to 20 mM phosphate at pH 7.0. Linear flow rate was 300 cm/h (1 mL/min).
CHT type I ceramic hydroxyapatite (40 μm) from Bio-Rad was packed into a range of MediaScout Minichrom columns by ATOLL GmbH (www.atoll-bio.com). We ran experiments at a linear flow rate of 300 cm/h and applied CCS undiluted. For an initial scouting run, the column was equilibrated to 10 mM sodium phosphate at pH 6.5, eluted in a linear gradient to 250 mM phosphate, and cleaned with 500 mM phosphate. A second run was conducted by equilibrating to 20 mM sodium phosphate at pH 6.5, eluting with a linear gradient to 20 mM phosphate and 1 M NaCl, then cleaning with 500 mM phosphate.
We obtained Capto MMC media, 1-mL and 5-mL HiTrap columns, and bulk media from GE Healthcare and conducted scouting on a 1-mL HiTrap column (7 × 25 mm) at a linear flow rate of 150 cm/h (1 mL/min). Sample was prepared by 1:1 dilution of CCS with 50 mM MES at pH 6.0. The column was equilibrated with the same buffer, eluted with a linear gradient to 50 mM MES and 500 mM arginine, then cleaned with 2 M guanidine at pH 5.5. We used the higher MES concentration to compensate for buffer capacity of the MMC carboxyl group.
Analysis
We ran reduced and nonreduced sodium-dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on precast Ready Gel 10% Tris-HCl Ready gels from Bio-Rad. We calibrated protein size using SeeBlue Plus2 prestained standards from Invitrogen (www.invitrogen.com) and detected sample proteins by either Bio-Safe Coomassie stain from Bio-Rad or Western blot with affinity-purified goat antihuman Fc polyclonal antibody from Jackson Immunoresearch Laboratories (www.jacksonimmuno.com). Analytical size-exclusion chromatography (SEC) was run on Superdex 75 HR 10/30 columns (10 × 300 mm) from GE Healthcare in PBS from Irvine Scientific (www.irvinesci.com) at a linear flow rate of 37.5 cm/h (0.5 mL/min). We calibrated protein size using gel filtration molecular weight standards from Bio-Rad.
Results and Discussion
First-pass scouting results were revealing but disappointing. The minibody eluted from AX at about physiological conductivity (15.6 mS/cm) at pH 8 but only about 6 mS at pH 7.0. It eluted slightly in advance of bovine serum albumin (BSA) with small injections and extended linear gradients but as a leading shoulder on the BSA peak with larger loads. Some BSA bound at pH 6.0, but the minibody did not. It bound very strongly to CX at pH 4.5, requiring 48 mS/cm conductivity for elution, which corresponds to ∼0.5 M NaCl. Binding was much weaker at pH 6.0, with the minibody eluting at ∼9 mS/cm. It eluted slightly after BSA with small injections but as a trailing shoulder with larger column loads. Both the minibody and most BSA failed to bind CX at pH 7.0. HIC supported slightly better separation than AX or CX, but with BSA still trailing through the later-eluting minibody peak.
HA with phosphate gradients has been used successfully for purification of Fab (30), anticarcinoembryonic antigen diabody and minibody (29), but the anti-PSCA minibody coeluted on center with BSA (Figure 3). More recent work with HA describing the relative behavior of IgG, fragments, and BSA has shown that although BSA and Fc binding are dominated by calcium affinity, HA binding of Fab is dominated by cation exchange (31, 32). This suggested that NaCl gradient elution might support more effective BSA removal, which it did (Figure 4). This was the first “eureka moment” in the development process, not only for BSA removal, but also because such gradients have been shown to remove >3 logs of DNA, >4 logs of endotoxin, and 4 logs of murine leukemia virus from IgG preparations (8, 33). Chloride gradients typically support the most effective removal of IgG aggregates (8). Indeed, when we ran an extended postgradient hold at 1.0 M NaCl, a second product peak eluted (Figure 5) predominantly populated by product dimer.
Figure 3.
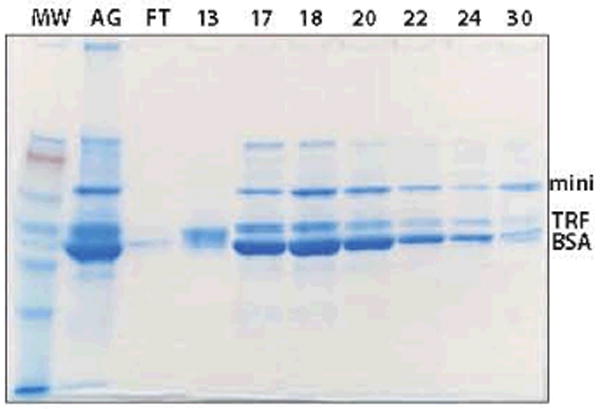
Nonreduced SDS-PAGE of HA scouting with a phosphate gradient; AG indicates supernatant after conditioning with AG1×8, FT flow-through, TRF transferrin, and numbers are elution fractions throughout the gradient.
Figure 4.
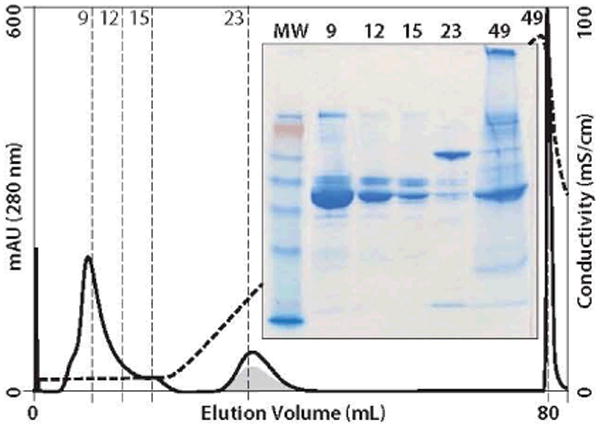
HA fractionation of minibody supernatant with NaCl gradient; chromatogram and nonreduced SDS-PAGE; gray area approximates minibody distribution; abbreviations are as in Figure 2. Most of the minibody elutes in fraction 23 relatively free of BSA and TRF.
Figure 5.
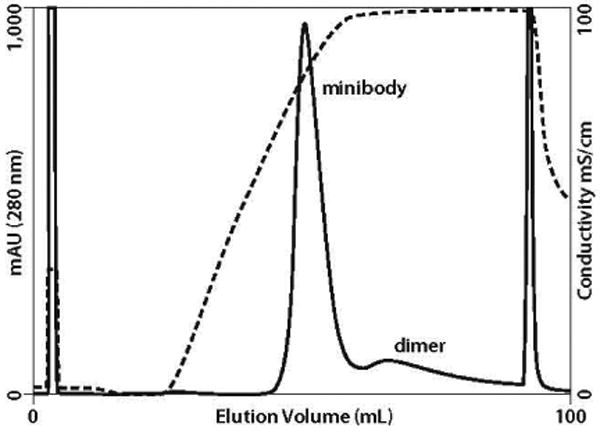
HA fractionation of minibody and dimer following MMC and AX
We hoped that HA would also support high-capacity capture, but minibody began to break through after loading only 12 mL of CCS per mL of HA (∼600 μg minibody/mL HA). We attributed this mainly to competition from BSA. CX offered marginally more effective capture but required titration of the feed stream to pH 4.5 (UNOsphere S). Breakthrough was still observed after application of only 15 mL CCS, and the minibody was only ∼20% pure, with BSA as the primary contaminant. Sample application to AX required CCS titration to pH 8 and 10-fold supernatant dilution. Still, breakthrough occurred after application of <20 mL diluted CCS, and purity was <10% (UNOsphere Q).
We did not consider HIC as a capture candidate because precipitation tended to occur under loading conditions — and because very large quantities of salt would have been required to bind significant amounts of the low-concentration product. However, because the minibody eluted after BSA on HIC (as with CX), we thought a chromatography support combining both mechanisms might achieve better fractionation than either one alone. The Capto MMC ligand includes both a phenyl group and a weak cation-exchange group (26). Our first attempt with MMC was the second “eureka moment” of the development process. After 1:1 dilution with 50 mM MES at pH 6, we could load 180 mL of diluted CCS without significant breakthrough (∼4.5 mg minibody/mL MMC). Although not impressive by protein A standards, this represented >5× the capacity of CX. Purity was also improved 2.5–fold over CX (to ∼50%). BSA was still the primary contaminant, enriched on the leading side of the peak with minibody eluting on the trailing side.
We ran an experiment with 5 mL diluted CCS, expecting to confirm these results, but we were surprised to observe that purity of the eluted minibody diminished to <20% (Figure 6). We ran additional experiments with 5 mL and 180 mL of diluted CCS, reproducing our initial results with both. From a mechanistic perspective, the data suggested competition for binding substrate between the minibody and BSA, with the stronger-binding minibody displacing BSA — up to a point — over the course of column loading. From a practical perspective, the results showed that binding selectivity depended on loading.
Figure 6.
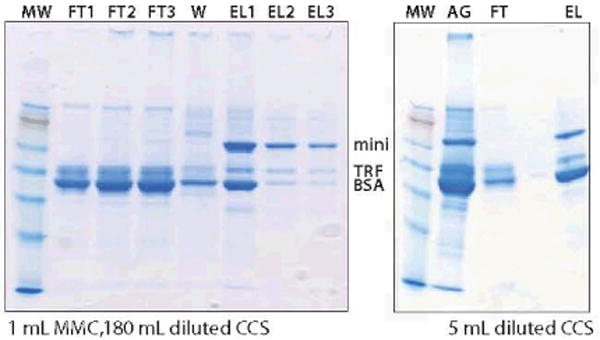
Column loading's effect on purification performance of MMC; nonreduced SDS-PAGE; note the difference between the ratio of minibody to BSA in EL1 with the larger load (left) and the ratio with the smaller load (right, EL). FT refers to flow-through fractions, EL to elution fractions; other abbreviations are as in Figure 3.
This is unattractive in a capture step because initial sample composition is typically the most variable feature of a purification process. Variations are common in product concentration and the product/contaminant ratio. If such variation ripples through the capture step, it can affect the performance of subsequent purification steps and quality of a final product. We therefore evaluated a series of wash conditions in the hope of improving both purity and reproducibility of the MMC elution.
These experiments were conducted with 50-mL loads of diluted CCS to conserve sample, and indeed we developed conditions that increased purity up to ∼75%. However, when the load increased to 180 mL of diluted CCS, most of the minibody eluted in the wash. This was even more undesirable than the original problem because it showed that not only binding selectivity, but also elution selectivity depended on column loading. That left us with the sole option of setting load specifications based on minibody concentration in the CCS — with the inherent risk that purification performance could still vary with the product/contaminant ratio. Downstream purification steps would need to accommodate this variation.
Given that an unoptimized scouting run on HA had already demonstrated the highest single-step purification potential, it seemed an obvious place to seek further improvements. We evaluated the effects of different phosphate concentrations on binding selectivity. Contaminant binding diminished progressively up to 25 mM phosphate. Minibody began to break through at higher concentrations. Previous experience has shown that HA binding capacity is inversely proportional to phosphate concentration ≥5 mM, which is required to maintain the stability of HA itself (6, 34). We also determined that the most effective removal of contaminants occurred in a NaCl gradient conducted at 10 mM phosphate.
Accordingly, we equilibrated the sample by adding phosphate to a final concentration of 5 mM. The column was initially equilibrated with 25 mM phosphate at pH 7. Sample application reequilibrated the column to 5 mM phosphate for maximum binding capacity. Washing with 25 mM phosphate removed a suite of minor contaminants, including transferrin. Phosphate concentration was then reduced to 10 mM, and the column was eluted with a gradient to 1.0 M NaCl (10 mM phosphate at pH 7). The minibody eluted at ∼800 mM NaCl. This sequence reduced buffer volume and process time from the more conventional approach — which would have required equilibration, loading, and a first wash, all at 5 mM phosphate, followed by a wash with 25 mM phosphate, then reequilibration to 10 mM phosphate before elution.
We added an AX step to ensure adequate virus removal. We used monoliths to scout and model conditions because they produce data much faster than other formats (<10 minutes from one run to the next rather than ∼45 min for conventional media). Monoliths support ∼1.5 logs higher DNA capacity and 2 logs higher virus capacity than porous particle media and have twice the capacity of membrane anion exchangers (35, 36). Had AX been the final step in our process, we would likely have continued with monoliths, but that would have required a diafiltration step following the high-salt HA elution.
We chose instead to place AX before HA, which created a different compromise. Monoliths have relatively low binding capacity for small proteins, which probably would have been stressed by the ∼50% BSA load coming from the MMC step. Competition from bound BSA might in turn have compromised the efficiency of DNA and virus removal. We therefore decided to use a high-capacity porous-particle anion exchanger and chose UNOsphere Q media, with a dynamic binding capacity of ∼180 mg/mL BSA (37).
We initially evaluated the AX step in a flow-through format and achieved adequate minibody recovery at pH 7.0 and 12-mS/cm conductivity. These conditions fall within a range demonstrated to support effective reduction of nonenveloped retrovirus (38), but we ultimately chose to run AX as a bind–elute step to eliminate contaminants that bound more weakly than the minibody — to further enhance virus removal and improve reproducibility. This decision was driven mainly by the variability of loading and elution selectivity at the MMC step. But the bind–elute format also contributed to overall process economy by concentrating product and reducing sample volume going into the HA step.
Finally, we returned to the MMC step and evaluated alternative elution procedures. Although we were precluded from exploring conditions that would affect the level of contaminants eluting in advance of the minibody, latitude remained to reduce levels eluting after it and thereby minimize the contaminant load going into the AX step. In short, we discovered that eluting with NaCl significantly reduced dimer content (Figure 7), so we modified the process accordingly. Table 1 summarizes product and contaminant distribution throughout the process. Figure 8 shows PAGE results, Figure 9 shows analytical SEC results, and Table 2 summarizes the recovery.
Figure 7.
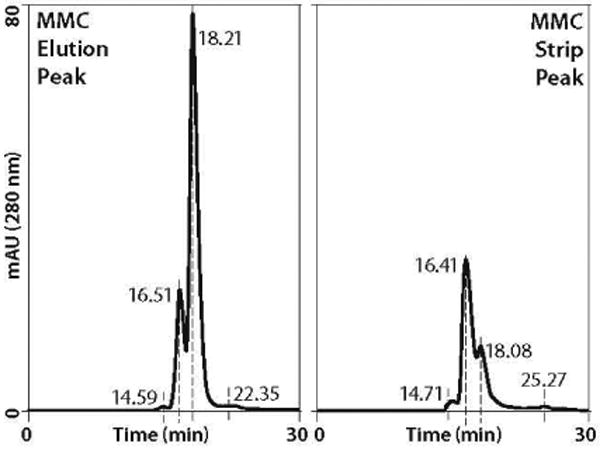
Dimer and aggregate distribution in MMC fractions; (left) a Superdex 75 profile of the MMC eluate with dimer peak at 16.51 minutes, higher aggregates at 14.59 minutes; (right) the SEC profile of a 1 M NaCl strip dominated by dimer and aggregates. Note that the minibody apparently coelutes on center with contaminating BSA at about 18.2 minutes. Compare with Figure 9.
Table 1. Minibody/contaminant distribution throughout purification.
| Stage | Unbound | Elute | Clean |
|---|---|---|---|
| AG | mini, HCP | — | DNA, ETX, lipid, dye |
| MMC | HCP, DNA | mini, dimer, HCP | dimer, HCP |
| AX | HCP | mini, dimer, HCP | HCP, DNA, ETX, virus |
| HA | HCP | mini | dimer, HCP, DNA, ETX, virus |
This table indicates trends and is not intended to suggest quantitative distribution. Lipid includes phospholipids, fatty acids, and steroids. Dye refers to pH indicator dyes used in cell culture. HCP = host cell protein; ETX = endotoxin.
Figure 8.
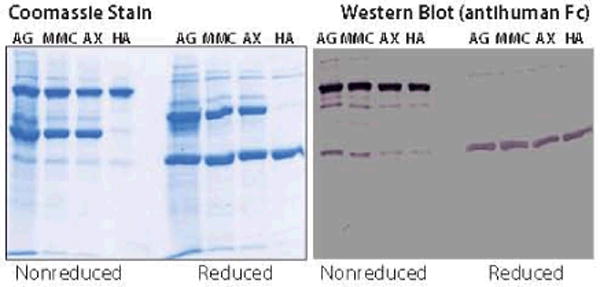
SDS-PAGE of purification process stages
Figure 9.
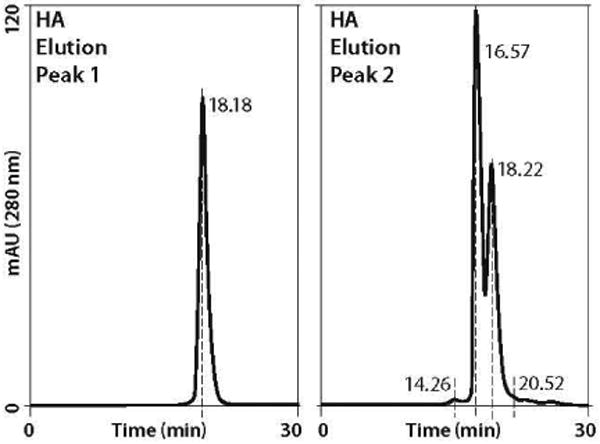
Dimer and aggregate removal by HA (chromatogram in Figure 4); the first HA peak is essentially all nonaggregated minibody; the second is dominated by dimers and aggregates. Peak 2 was concentrated before application to SEC. Compare with Figure 7; refer to Table 2 for relative amounts of minibody in the respective fractions.
Table 2. Recovery summary for purification of anti-PSCA minibody.
| Stage | μg/mL | mL | mg | Recovery |
|---|---|---|---|---|
| AG | 462 | 19.7 | 9.1 | 100% |
| MMC elution | 362 | 22 | 8.0 | 88% |
| MMC strip | 189 | 5.5 | 1.0 | 11%* |
| AX elution | 249 | 21 | 5.2 | 57% |
| AX strip | 117 | 7.4 | 0.9 | 10% |
| HA elution 1 | 361 | 11 | 4.0 | 44% |
| HA elution 2 | 120 | 11 | 1.3 | 14%* |
| HA strip | 76 | 3.8 | 0.3 | 3%** |
Predominantly dimer
Higher aggregates
Most of all, we were gratified that MMC delivered the same purification performance we had observed with CCS but containing 10-fold less product. This greatly lessened our concerns about lot-to-lot feed stream variability rippling through the process. Dimers and aggregates were essentially absent from the final product, as expected. We were initially concerned by an ordered series of lower molecular weight bands on nonreduced PAGE gels, but they were revealed by Western blots to be product-related. This phenomenon also occurs frequently with hIgG1 and hIgG4, and it appears to be a PAGE artifact attributable to disulfide scrambling during sample preparation, even in the absence of reducing agents (39).
Future Directions
This study highlights some key challenges with early phase purification process development. Cell lines and culture conditions often are not fully optimized. Product concentrations are frequently low (sometimes very low), and serum supplementation is common with mammalian cells. Despite these burdens, a product's surface chemistry and interactions with various chromatography media can be presumed to be consistent across platforms. Results of elementary scouting experiments can thus provide valuable guidance concerning the fractionation potential of the methods surveyed, reveal relative complementarity among them, and suggest process sequences that preclude extra processing steps such as concentration or diafiltration. These results also provide a preliminary indication of significant ranges for important process variables to support meaningful DoE optimization campaigns. Finally, early CCS in most respects represents the worst-case feed stream a purification process will encounter. Development of an effective process at this stage thereby reasonably ensures later success.
Pending favorable clinical results, we anticipate that cell culture conditions will be modified to eliminate serum supplementation. We suspect that the loading-related variability of MMC binding and elution selectivity is an unfortunate coincidence of this particular minibody sharing similar adsorption/desorption isotherms with BSA. Our interpretation is consistent with the inability of AX and CX to provide substantial fractionation of these solutes. Without BSA, we hope that MMC capacity will roughly double, and reproducibility issues will cease to be a concern. Refinement of elution conditions should then support much higher purity and perhaps further dimer reduction.
We anticipate conserving the AX step by virtue of its regulatory recognition for DNA and virus removal, although we may convert it to flow-through mode on a monolith if the contaminant load following MMC is reduced as expected. We also anticipate conserving the HA step unchanged except for conversion to a step gradient, mainly for its ability to remove aggregates, but also for removing multiple logs of DNA, endotoxins, and viruses. Given the orthogonal mechanistic relationship of HA to AX, their combination promises to be especially effective for virus removal.
Does this procedure offer platform potential? Bacterial, yeast, and mammalian cell cultures each represent different challenges. It is reasonable to expect that chemical differences among minibodies will compound those and other differences. Preliminary results with another minibody, diabody, Fab, and F(ab')2 nevertheless suggest that MMC has broad capture potential for small immunological constructs. HA has meanwhile demonstrated utility for aggregate removal from immunological constructs ranging from minibodies to IgM (8, 9) while supporting parallel removal of DNA, endotoxins, and viruses, so it seems reasonable to expect the same with other constructs. The ability of AX to remove DNA and viruses seems likely to translate well across constructs. Taken together, these points suggest that the present procedure may indeed have platform potential.
Results from this study also suggest reassessment of the role of multimodal methods in process chromatography. Mixed modes have frequently solved purification problems that traditional methods could not, but typically not until after extensive time-consuming efforts with traditional methods have proven futile. A growing number of presentations and publications (including this one) suggest that process developers could use their limited time and resources more effectively by including mixed modes at the earliest development stages (8, 22, 23, 25, 40). The broad success of HA for aggregate removal recommends initial scouting with NaCl gradients at low phosphate concentrations.
The growing number of capture applications on charged-hydrophobic–hydrogen bonding mixed modes like MMC recommends initial scouting with arginine gradients. Arginine simultaneously affects all three binding mechanisms. If arginine results are promising, then follow-up evaluation can include attempted elution with nonionic eluants such as propylene glycol (which principally affects hydrophobic interactions) or urea (which strongly affects hydrogen bonds too) or pH and salts such as NaCl (which principally affect the electrostatic component of binding) (41). Even if these agents fail to elute the product of interest, they may support washes that significantly improve overall process performance.
These suggestions accurately imply that development of mixed-mode methods is more complex than traditional methods. But they also reveal that mixed modes extend purification capabilities into dimensions beyond the scope of traditional methods. With emerging product classes diversifying rapidly beyond antibodies, these capabilities should receive an enthusiastic welcome.
Phase 1 Purification Process.
Capture on MMC
Dilute filtered supernatant 1:1 with 50 mM MES at pH 6. Equlibrate column with 50 mM MES at pH 6. Load supernatant containing 5 mg minibody per mL of MMC. Wash with 50 mM MES at pH 6. Elute with a step to 20 mM Tris and 75 mM NaCl at pH 8.5. Clean with 2 M guanidine at pH 5.5. Sanitize with 1 M NaOH. Store in 20% ethanol. Note that the high elution pH has two purposes: to weaken cation-exchange interactions and reduce the salt concentration required for elution, and to condition the sample pH for a subsequent AX step.
Intermediate Purification with UNOsphere Q
Dilute MMC eluate 1:3.5 with 20 mM Tris at pH 8.5. Equilibrate column with 20 mM Tris at pH 8.5. Load sample. Wash with 20 mM Tris at pH 8.5. Elute: a 10-CV linear gradient to 20 mM Tris and 225 mM NaCl at pH 8.5. Clean with 1 M NaCl at pH 8.5. Sanitize with 1 M NaOH. Store in 20% ethanol.
Polishing with HA
To AX eluate, add NaPO4 to a final concentration of 5 mM. Optionally adjust pH to about 7. Equilibrate column with 25 mM NaPO4 at pH 7. Load sample. Wash with 25 mM NaPO4 at pH 7. Reequilibrate with 10 mM NaPO4 at pH 7. Elute with a 10-CV linear gradient to 10 mM NaPO4 and 1 M NaCl at pH 7. Clean with 500 mM NaPO4 and pH 7. Wash with 1 CV water. Sanitize with 1 M NaOH. Store in 20% ethanol and 10 mM NaPO4 at pH 7.
Acknowledgments
Thanks to Felix Bergara for generating the minibody cell line, to Sarah McCaig for producing cell culture harvests, and to Alexander Brinkmann for helpful insights concerning disulfide scrambling. Some of this research was supported by NCI Grants CA43904 and CA92131. Some data discussed herein were presented at the Fifth International Symposium on Hydroxyapatite (42).
Footnotes
Product Focus: Recombinant proteins, antibody alternatives
Process Focus: Downstream processing (purification)
Who Should Read: Process development and manufacturing
Level: Intermediate
References
- 1.Holliger P, Hudson P. Engineered Antibody Fragments and the Rise of Single Domains. Nature Biotechnol. 2005;23(9):1126–1136. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 2.Leyton J, et al. Humanized Radioiodinated Minibody for Imaging of Prostate Stem Cell Antigen-Expressing Tumors. Clin Cancer Res. 2008;14(22):7488–7496. doi: 10.1158/1078-0432.CCR-07-5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tiselius A, Hjerten S, Levin O. Protein Chromatography on Calcium Phosphate Columns. Arch Biochem Biophys. 1956;65:132–155. doi: 10.1016/0003-9861(56)90183-7. [DOI] [PubMed] [Google Scholar]
- 4.Gorbunoff M, Timasheff S. The Interaction of Proteins with Hydroxyapatite. III. Mechanisms. Analyt Biochem. 1984;136:440–445. doi: 10.1016/0003-2697(84)90241-0. [DOI] [PubMed] [Google Scholar]
- 5.Guerrier L, Flayeux I, Boschetti E. A Dual-Mode Approach to the Selective Separation of Antibodies and Their Fragments. J Chromatogr B. 2001;755:37–46. doi: 10.1016/s0378-4347(00)00598-3. [DOI] [PubMed] [Google Scholar]
- 6.Gagnon P, et al. A Ceramic Hydroxypatite Based Purification Platform: Simultaneous Removal of Leached Protein A, Aggregates, DNA, and Endotoxins. BioProcess Int. 2006;4(2):50–60. [Google Scholar]
- 7.Wensel D, Kelly B, Coffman J. High-Throughput Screening of Chromatographic Separations: III. Monoclonal Antibodies on Ceramic Hydroxyapatite. Biotechnol Bioeng. 2008;100:839–854. doi: 10.1002/bit.21906. [DOI] [PubMed] [Google Scholar]
- 8.Gagnon P, Beam K. Antibody Aggregate Removal By Hydroxyapatite. Curr Pharmaceut Biotechnol. 2009;10(4):440–446. doi: 10.2174/138920109788488833. [DOI] [PubMed] [Google Scholar]
- 9.Gagnon P. Improved Aggregate Removal By Hydroxyapatite Chromatography in the Presence of Polyethylene Glycol. J Immunol Meth. 2009;342:115–118. doi: 10.1016/j.jim.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Porath J, Maisano F, Belew M. Thiophilic Adsorption: A New Method for Protein Fractionation. FEBS Lett. 1985;185(2):306–310. doi: 10.1016/0014-5793(85)80928-5. [DOI] [PubMed] [Google Scholar]
- 11.Porath J. Salt-Promoted Adsorption: Recent Developments. J Chromatogr A. 1986;376:331–341. doi: 10.1016/s0378-4347(00)80850-6. [DOI] [PubMed] [Google Scholar]
- 12.Hutchins T, Porath J. Thiophilic Adsorption of Immunoglobulins: Analysis of Conditions Optimal for Selective Immobilization and Purification. Anal Biochem. 1986;159:217–226. doi: 10.1016/0003-2697(86)90331-3. [DOI] [PubMed] [Google Scholar]
- 13.Belew M, et al. A One-Step Purification Method for Monoclonal Antibodies Based on Salt Promoted Adsorption on a “Thiophilic” Adsorbent. J Immunol Meth. 1987;102:173–182. doi: 10.1016/0022-1759(87)90074-3. [DOI] [PubMed] [Google Scholar]
- 14.Berna P, et al. Comparison of Protein Adsorption Selectivity of Salt-Promoted Agarose-Based Sorbents: Hydrophobic, Thiophilic, and Electron Donor–Acceptor Adsorbents. J Chromatogr A. 1998;800:151–159. doi: 10.1016/s0021-9673(97)01130-8. [DOI] [PubMed] [Google Scholar]
- 15.Ngo T, Khatter N. Chemistry and Preparation of Affinity Ligands Useful in Immunoglobulin Isolation and Serum Protein Separation. J Chromatogr A. 1990;510:281–291. doi: 10.1016/s0021-9673(01)93762-8. [DOI] [PubMed] [Google Scholar]
- 16.Ngo T, Khatter N, Avid AL. A Synthetic Ligand Affinity Gel Mimicking Immobilized Bacterial Antibody Receptor for Purification of Immunoglobulin. J Chromatogr A. 1992;597:101–109. doi: 10.1016/0021-9673(92)80100-9. [DOI] [PubMed] [Google Scholar]
- 17.Burton S, Harding D. Hydrophobic Charge Induction Chromatography: Salt Independent Protein Adsorption and Facile Elution with Aqueous Buffers. J Chromatogr A. 1998;814:71–81. doi: 10.1016/s0021-9673(98)00436-1. [DOI] [PubMed] [Google Scholar]
- 18.Guerrier G, Schwartz W, Boschetti E. New Method for the Selective Capture of Antibodies Under Physiological Conditions. Bioseparation. 2000;9:211–221. doi: 10.1023/a:1008170226665. [DOI] [PubMed] [Google Scholar]
- 19.Schwartz W, et al. Comparison of HCIC with Affinity Chromatography on Protein A for Harvest and Purification of Antibodies. J Chromatogr A. 2001;908:251–263. doi: 10.1016/s0021-9673(00)01013-x. [DOI] [PubMed] [Google Scholar]
- 20.Ghose S, Hubbard B, Cramer S. Evaluation and Comparison of Alternatives to Protein A Chromatography Mimetic and Hydrophobic Charge Induction Chromatography. J Chromatogr A. 2006;1122:144–152. doi: 10.1016/j.chroma.2006.04.083. [DOI] [PubMed] [Google Scholar]
- 21.Roque A, Silva C, Taipa M. Affinity-Based Methodologies and Ligands for Antibody Purification: Advances and Perspectives. J Chromatogr A. 2007;1160:44–55. doi: 10.1016/j.chroma.2007.05.109. [DOI] [PubMed] [Google Scholar]
- 22.Kaleas K, Schmelzer C, Pizarro S. Industrial Case Study: Evaluation of a Mixed Mode Resin for Selective Capture of a Human Growth Factor Recombinantly Expressed in E. coli. J Chromatogr A. doi: 10.1016/j.chroma.2009.07.023. in press. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, et al. The Distinctive Separation Attributes of Mixed Mode Resins and Their Application in Monoclonal Antibody Downstream Process. J Chromatogr A. doi: 10.1016/j.chroma.2009.09.047. in press. [DOI] [PubMed] [Google Scholar]
- 24.Selective Removal of Aggregates with Capto Adhere. GE Healthcare; 2007. Application Note 28-9078-93 AA. www1.gelifesciences.com/aptrix/upp00919.nsf/Content/B03659F218A8E649C1257628001D34C1/$file/28907893AA.pdf. [Google Scholar]
- 25.Eriksson K, et al. MAb Contaminant Removal with a Multimodal Anion Exchanger: A Platform Step to Follow Protein A. BioProcess Int. 2009;7(2):52–56. [Google Scholar]
- 26.Optimizing Elution Conditions on Capto MMC Using Design of Experiments. GE Healthcare; 2007. Application Note 11-0035-48 AB. www6.gelifesciences.com/aptrix/upp00919.nsf/Content/D3B413A001F26C75C1257628001D2014/$file/11003548AB.pdf. [Google Scholar]
- 27.Gagnon P. Purification Tools for Monoclonal Antibodies. Validated Biosystems; Tucson, AZ: 1996. pp. 225–228. [Google Scholar]
- 28.Wu A, et al. Multimerization of a Chimeric Anti-CD20 Single-Chain Fv-Fc Fusion Protein is Mediated Through Variable Domain Exchange. Protein Eng. 2001;14(12):1025–1033. doi: 10.1093/protein/14.12.1025. [DOI] [PubMed] [Google Scholar]
- 29.Yazaki P, et al. Mammalian Expression and Hollow Fiber Bioreactor Production of Recombinant Anti-CEA Diabody and Minibody for Clinical Applications. J Immunol Meth. 2001;253:195–208. doi: 10.1016/s0022-1759(01)00388-x. [DOI] [PubMed] [Google Scholar]
- 30.Bowles M, et al. Large Scale Production and Purification of Paraquat and Desipramine Monoclonal Antibodies and Their Fab Fragments. Int J Immunopharmaceut. 1988;10(5):537–545. doi: 10.1016/0192-0561(88)90071-9. [DOI] [PubMed] [Google Scholar]
- 31.Gagnon P, Cheung CW, Yazaki P. Reverse Calcium Affinity Purification of Fab with Calcium-Derivatized Hydroxyapatite. J Immunol Meth. 2009;342:115–118. doi: 10.1016/j.jim.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gagnon P, Cheung CW, Yazaki P. Cooperative Multimodal Retention of IgG, Fragments, and Aggregates on Hydroxyapatite. J Sep Sci. doi: 10.1002/jssc.200900055. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ng P, Cohen A, Gagnon P. Monoclonal Antibody Purification with CHT. Gen Eng News. 2006;26(14) www.genengnews.com/articles/chtitem.aspx?tid=1833.
- 34.CHT Ceramic Hydroxyapatite Instruction Manual. Bio-Rad Laboratories; Hercules, CA: 2007. LIT611 Rev E. [Google Scholar]
- 35.Gagnon P, et al. A Comparison of Microparticulate, Membrane, and Monolithic Anion Exchangers for Polishing Applications in the Purification of IgG Monoclonal Antibodies. Poster at BioProcess International Conference and Exposition; Boston, MA. 1–4 October 2007; www.validated.com/revalbio/pdffiles/IBCBOS07a.pdf. [Google Scholar]
- 36.Maurer E. Purification of Live Replication Deficient Influenza Virus. Presentation at Third International Symposium on Monoliths; Portoroz, Slovenia. 30 May–4 June 2008. [Google Scholar]
- 37.UNOsphere Q & S Ion Exchange Media Instruction Manual. Bio-Rad Laboratories; Hercules, CA: 2004. LIT4110109 Rev B. [Google Scholar]
- 38.Curtis S, et al. Generic/Matrix Evaluation of SV40 Clearance By Anion Exchange Chromatography in Flow-Through Mode. Biotechnol Bioeng. 2003;84(2):179–186. doi: 10.1002/bit.10746. [DOI] [PubMed] [Google Scholar]
- 39.Liu H, et al. Characterization of Lower Molecular Weight Artifact Bands of Recombinant Monoclonal IgG1 Antibodies on Non-Reducing SDS-PAGE. Biotechnol Lett. 2007;29(11):1611–1622. doi: 10.1007/s10529-007-9449-8. [DOI] [PubMed] [Google Scholar]
- 40.Gagnon P. Purification of Monoclonal Antibodies By Mixed-Mode Chromatography. In: Gottschalk U, editor. Process Scale Purification of Antibodies. John Wiley and Sons; New York, NY: 2009. pp. 125–144. [Google Scholar]
- 41.Gagnon P. Dissection of the Separation Mechanisms, and Enhancement of IgG Aggregate Removal By Charged-Hydrophobic Mixed-Mode Chromatography. Poster at Sixth International HIC/RPC Bioseparation Conference; Napa, CA. 15–19 March 2009; www.validated.com/revalbio/pdffiles/HICRPC09.pdf. [DOI] [PubMed] [Google Scholar]
- 42.Gagnon P, Cheung CW, Yazaki P. A Case Study of Early Phase Purification Process Development for an Anti-Cancer Minibody. Presentation at Fifth International Symposium on Hydroxyapatite; Rottach-Egern, Germany. 11–14 October 2009; www.validated.com/revalbio/pdffiles/CHT09.pdf. [Google Scholar]