

Abstract
Although the systemic administration of a number of different gene products has been shown to result in the inhibition of angiogenesis and tumor growth in different animal tumor models, the relative potency of those gene products has not been studied rigorously. To address this issue, recombinant adenoviruses encoding angiostatin, endostatin, and the ligand-binding ectodomains of the vascular endothelial growth factor receptors Flk1, Flt1, and neuropilin were generated and used to systemically deliver the different gene products in several different preexisting murine tumor models. Single i.v. injections of viruses encoding soluble forms of Flk1 or Flt1 resulted in ≈80% inhibition of preexisting tumor growth in murine models involving both murine (Lewis lung carcinoma, T241 fibrosarcoma) and human (BxPC3 pancreatic carcinoma) tumors. In contrast, adenoviruses encoding angiostatin, endostatin, or neuropilin were significantly less effective. A strong correlation was observed between the effects of the different viruses on tumor growth and the activity of the viruses in the inhibition of corneal micropocket angiogenesis. These data underscore the need for comparative analyses of different therapeutic approaches that target tumor angiogenesis and provide a rationale for the selection of specific antiangiogenic gene products as lead candidates for use in gene therapy approaches aimed at the treatment of malignant and ocular disorders.
The central role of angiogenesis in the development of numerous pathologic conditions including cancer, diabetic retinopathy, and vascular malformations is now well appreciated (1). In the case of cancer, the concept of an “angiogenic switch” has been proposed by Hanahan and Folkman (2), wherein angiogenesis both precedes and is necessary for the development of frank tumorigenicity. Recent findings nevertheless have underscored the mechanistic complexity underlying the development of tumor blood supply including delayed angiogenesis into initially avascular tumor masses (3), the early cooption of vasculature from neighboring tissue (4), and the contribution of circulating endothelial stem cells (5).
Extensive data have implicated the vascular endothelial growth factor (VEGF) family and their receptors as critical mediators of physiologic and tumor blood vessel formation, and consequently these molecules have attracted particular attention as targets for antiangiogenic therapy by a variety of strategies (6–13). Recently, the administration of several tumor-derived circulating proteins have been proposed also as an alternative strategy for achieving the systemic inhibition of angiogenesis. In particular, both human and murine forms of angiostatin (AS), a proteolytic fragment of plasminogen, have been described to exert potent antiangiogenic and antitumor activities in a variety of murine tumor models, extending to frank regression of tumors (14, 15). Similarly, a C-terminal fragment of collagen XVIII, termed endostatin (ES), has been reported to exhibit antiangiogenic and tumor-regressing activities accompanied by a lack of acquired tumor resistance (16, 17).
Interestingly, despite the large number of previous studies that have demonstrated the antitumor activity of different gene products that inhibit angiogenesis via either VEGF-dependent or -independent pathways, a systematic comparison of the relative efficacy of the different gene products in the same tumor models has not been described. To begin to address this important issue, we have generated a series of recombinant adenoviral vectors encoding different antiangiogenic gene products and have used the viruses to deliver the different gene products in several different preexisting murine tumor models. Here, we present a comparative evaluation of the antitumor and antiangiogenic activity of those gene products.
Methods
Construction and Purification of Recombinant Adenoviruses.
The Flk1-Fc cDNA was a gift from T. Niederman (Children's Hospital, Boston) and contained the murine Flk1 cDNA sequence encoding the signal peptide and the ectodomain (to TIRRVRKEDGG, amino acid 731) fused to a murine IgG2α Fc fragment. The Flk1-Fc fusion gene was released with XbaI and BamHI and inserted in the polylinker of the adenovirus shuttle vector HIHG Add2 (J. Gray and R.C.M., unpublished data). In the resulting construct, Flk1-Fc expression is controlled by the human cytomegalovirus promoter and the rabbit β-globin intron and polyadenylation signal. The expression cassette is flanked by the adenovirus type 5 sequences encompassing nucleotides 1–459 and 3328–4619. The murine Flt1(1–3) cDNA was amplified by PCR from Flt-1 cDNA (S. Soker, Children's Hospital, Boston) resulting in amplification of the Flt-1 signal sequence, coding sequence with the first three Ig repeats to FNTSVHV, with an added C-terminal 6× His tag. The tagged cDNA then was ligated into HIHG Add2 as an EcoRI–SalI fragment.
For the control Fc fragment, a cDNA encoding the murine IgG2α Fc cDNA (Lexigen, Lexington, MA) was released with XhoI and XbaI and ligated into HIHG Add2. The human soluble neuropilin (sNRP) cDNA with signal peptide, ABC domains, and a C-terminal 6× His tag (S. Soker) was excised with BamHI and XbaI and cloned into HIHG Add2. A fragment comprising the human growth hormone leader peptide-encoding sequence fused to the human AS cDNA (Lys-97–Glu-458, kringle domains 1–4) was synthesized by PCR of human plasminogen cDNA. The PCR product was digested with BamHI and XhoI and cloned into the shuttle vector pAd-MDM, which differs from HIHG Add2 only by the plasmid backbone. A cDNA encoding the murine ES coding sequence (HTHQD… TSFSK) fused to the collagen XVIII signal peptide (B. Olsen, Harvard Medical School, Boston) was cloned into the HIHG Add2 shuttle vector to generate pAdd2 mu endo II. An alternative cDNA containing the same ES sequence fused to the human growth hormone signal sequence (MATGSRTSLLLAFGLLCLPWLQEGSA) was produced by PCR from murine collagen XVIII cDNA (B. Olsen), and the BamHI and XhoI-restricted product cloned into pAd-MDM to generate pAdd2 mu endo I. All the PCR-generated DNA fragments were sequenced on both strands to exclude PCR errors.
The HIHG pAdd2-derived recombinant shuttle vectors then were digested with PacI and MfeI to release a fragment where the transgene is flanked by 2.0 and 1.4 kb of homology with the adenovirus plasmid pAd-GM-CSF. The GM-CSF insert of the adenovirus plasmid is replaced with our transgene by homologous recombination in Escherichia coli (18). The Flk1-Fc, Flt1(1–3), Fc, sNRP, and mu endo II adeno vectors were rescued by transfection of the PacI-restricted adenovirus plasmids in 293 cells. Homologous recombination between the pAd-MDM-derived shuttle vectors and viral DNA in 293 cells (19) allowed the rescue of the AS and mu endo I recombinant Ad vectors. The viral vectors are propagated on 293 cells and purified by CsCl banding as described (18).
Protein Analysis of Virally Produced ES and Flt1(1–3).
C57BL/6 mice were injected with Ad mu endo II or Ad Flt1(1–3) [109 plaque-forming units (pfu) by tail vein]. After 3 days mice were bled, and the respective proteins were purified from plasma by using either heparin-Sepharose chromatography with NaCl elution (ES) or Ni-agarose chromatography with imidazole elution [Flt1(1–3)]. These purified proteins were transferred to poly(vinylidene difluoride) membrane and digested in situ with trypsin followed by N-terminal sequencing and mass spectroscopy.
ELISA Determination of Transgene Expression.
Plasma samples were obtained by retroorbital puncture with heparinized capillary tubes after anesthesia. Murine Flk1-Fc concentrations were determined by sandwich ELISA with anti-murine Flk1 primary (PharMingen) and anti-murine IgG2α Fc-horseradish peroxidase secondary (Jackson ImmunoResearch). Murine ES plasma levels were quantitated by competition ELISA (CytImmune Sciences, College Park, MD) and human AS plasma levels by sandwich ELISA (Entremed, Rockville, MD).
Western Blot Determination of Transgene Expression.
Plasma was analyzed by Western blot for Flk1-Fc (rat anti-murine Flk1, PharMingen, or goat anti-murine Fc, Jackson ImmunoResearch), Flt1(1–3) (rabbit anti-His, Santa Cruz Biotechnology), ES (rabbit anti-mouse ES, gift of K. Javaherian, Children's Hospital, Boston), AS (rabbit anti-human plasminogen, Accurate, Westbury, NY) or sNRP (rabbit anti-His, Santa Cruz Biotechnology). Flt(1–3) and sNRP levels were estimated by Western blot against purified standards. Development was performed with species-specific secondary Ab-horseradish peroxidase conjugates and chemiluminescence.
Tumor Cell Lines, Mice, and Adenoviral Injections.
Murine Lewis lung carcinoma (LLC) cells were passaged on the dorsal midline of C57BL/6 mice or in DMEM/10% FCS/penicillin/streptomycin (PNS)/L-glutamine. T241 murine fibrosarcoma was grown in DMEM/10% FCS/PNS/L-glutamine and human pancreatic BxPc3 adenocarcinoma in RPMI medium 1640/10% FCS/PNS. Tumor cells (106) were injected s.c. into the dorsal midline of C57BL/6 mice (8–10 weeks old) for murine tumors and severe combined immunodeficient (SCID) mice for human tumors, grown to 100–200 mm3 (typically 10–14 days) to demonstrate tumor take, and 109 pfu of antiangiogenic adenoviruses or the control adenovirus Ad Fc given by i.v. tail-vein injection. In Fig. 2B, seven Flt1 control animals received Ad GFP instead of Ad Fc, although we have not observed any differences in tumor inhibition with either control construct. Ad mu endo II was used in all ES experiments, except in Fig. 2B in which Ad mu endo I was used. Tumor size in mm3 was calculated by caliper measurements over a 10- to 14-day period by using the formula 0.52 × length (mm) × width (2) (mm), using width as the smaller dimension. P values were determined by using a two-tailed t test assuming unequal variances (Microsoft EXCEL).
Figure 2.
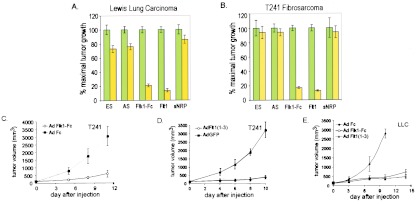
Inhibition of preexisting tumor growth by antiangiogenic adenoviruses. C57BL/6 mice were implanted s.c. with 106 cells of murine LLC (A) or murine T241 fibrosarcoma (B). At a tumor volume of 100–150 mm3, tumor-bearing mice received i.v. injection of 109 pfu of the control virus Ad Fc (green bars) or the appropriate antiangiogenic adenovirus (yellow bars), and tumor volume was calculated after 10–14 days. Tumor size is expressed as percentage of maximal tumor volume standardized to 100% for Ad Fc, which did not produce significant inhibition relative to PBS controls. Percentage of inhibition of animals receiving antiangiogenic adenoviruses relative to animals injected with the control virus Ad Fc is calculated. Error bars, ± 1 SEM; N, number of individual mice assayed with each adenovirus. For LLC, the number of animals was as follows for Fc and the treatment group: ES, n = 24,22; AS, n = 11,9; Flk1-Fc, n = 18,17; Flt1, n = 8,10; sNRP, n = 8,8. For T241, the number of animals was as follows for Fc and the treatment group: ES, n = 6,10; AS, n = 6,7; Flk1-Fc, n = 24,25; Flt1, n = 19,20; sNRP, n = 7,5. (C and D) Representative growth curves of T241 fibrosarcoma in C57BL/6 mice treated with Ad Flk1-Fc (n = 6) (C) or Ad Flt1(1–3; n = 7) (D). C57BL/6 mice bearing preexisting T241 tumors of 100–150 mm3 received 109 pfu i.v. of the appropriate adenoviruses, and tumor size was measured over time. Error bars, ± 1 SD. (E) Suppression of LLC growth by Ad Flk1-Fc. Mice with preexisting tumors of 150 mm3 received i.v. injections of 109 particles of Ad Fc (n = 4), Ad Flk1-Fc (n = 5), or Ad Flt1(1–3) (n = 5), and tumor growth was measured over time. Error bars, ± 1 SD.
Corneal Micropocket Assay.
C57BL/6 mice received 109 pfu i.v. of antiangiogenic adenoviruses or the control adenovirus Ad Fc 2 days before assay. Mice were anesthetized with avertin i.p. and the eye was treated with topical proparacaine⋅HCl (Allergan, Irvine, CA). Hydron/sucralfate pellets containing VEGF-A165 (R & D Systems) were implanted into a corneal micropocket at 1 mm from the limbus of both eyes under an operating microscope (Zeiss) followed by intrastomal linear keratotomy by using a microknife (Medtroni Xomed, Jacksonville, FL). A corneal micropocket was dissected toward the limbus with a von Graefe knife #3 (2 × 30 mm), followed by pellet implantation and application of topical erythromycin. After 5 days, neovascularization was quantitated by using a slit lamp biomicroscope and the formula 2π × (vessel length/10) × (clock hours). P values were determined by using a two-tailed t test assuming unequal variances (Microsoft EXCEL).
Immunohistochemistry.
C57BL/6 mice bearing LLC tumors on the dorsal midline at 50 mm3 received 109 pfu i.v. of Ad Fc, Ad Flk1-Fc, or Ad Flt1(1–3). After tumor growth to ≈200 mm3, tumors were harvested, fixed in formalin, and paraffin-embedded sections were stained for CD31 by using a biotin-streptavidin horseradish peroxidase system (Vectastain). Microvessel areas were quantitated by manual counting of hotspots in sections.
Results
Construction and Characterization of Adenoviruses Encoding Antiangiogenic Gene Products.
By using homologous recombination techniques in bacteria (18), DNA sequences encoding human AS, murine ES, and the ligand-binding ectodomains of the VEGF receptors Flk1, Flt1, and neuropilin were introduced into the E1 region of a standard E1-deleted adenoviral vector (Fig. 1A; see Methods for details of construction). Viruses encoding each of the gene products were generated after transfection of the different vector DNAs into 293 cells as described (18). In the case of each vector, particle titers of ≈1013/ml and infectious titers of ≈1011 pfu/ml were obtained routinely, with a particle/infectivity ratio of 40:60.
Figure 1.

Construction and characterization of antiangiogenic adenoviruses. (A) Schematic of insertion of various antiangiogenic cDNAs into the E1 region of E3-deleted adenovirus type 5. (B) Western blot analysis of adenovirus-expressed antiangiogenic proteins in mouse plasma. C57BL/6 mice received i.v. injection of 109 particles of the appropriate adenovirus, followed after 2–3 days by Western blot of 1 μl of plasma, except for Flk1-Fc which was taken at day 17 and was a 1:10 dilution. *, position of transgene products: Flk1-Fc (180 kDa), Flt1(1–3) (53 kDa), ES (20 kDa), AS (55 kDa), and sNRP-ABC (120 kDa). Levels in adjacent blots are not comparable because of different enhanced chemiluminescence exposure times. (C) Pharmacokinetics of expression from antiangiogenic adenoviruses. Plasma from mice infected i.v. with 109 pfu of the appropriate adenovirus was analyzed after the indicated times for expression by ELISA (Flk1-Fc, n = 4; ES, n = 4; AS, n = 3). See the text for further details. Error bars, ± 1 SD.
To evaluate the in vivo expression potential of the different viruses, 109 pfu of each virus was administered by i.v. or i.m. routes into immunocompetent C57BL/6 mice. Transgene expression was easily detectable in the plasma of infected mice by Western blotting (Fig. 1B). In the case of Flk-Fc, AS, and ES, plasma expression levels at different times after injection of virus were quantitated by sandwich ELISA (Fig. 1C). Ad Flk1-Fc virus provided very high levels of protein expression (2–8 mg/ml) compared with Ad AS (100–250 μg/ml) or Ad ES (>10 μg/ml), and the expression of all gene products declined progressively with time, consistent with the known transient nature of transgene expression afforded by first-generation adenoviral vectors (20). In the case of animals injected with viruses encoding Flt(1–3) or sNRP, Western blot analysis, in conjunction with purified protein standards, was used to estimate the serum concentration of each gene product. By this method, peak Flt(1–3) ectodomain plasma levels were 3–10 μg/ml, whereas peak sNRP levels were estimated to be >50 μg/ml (data not shown).
In vitro assays were used to confirm the functional activity of several of the adenovirus-expressed gene products. Vector encoded Flt1(1–3) and Flk1-Fc proteins both were shown to inhibit VEGF-induced human umbilical vein endothelial cells (HUVEC) proliferation in vitro, with IC50s of ≈5 and 100 ng/ml, respectively (data not shown), paralleling reports of the relative affinities of the two receptors for VEGF (21). Because endothelial proliferation assays take at least 3 days and migration assays through a Boyden chamber are demanding technically, we used a bioassay for virally encoded ES on the basis of the dispersion of endothelial cells from endothelial tubes in Matrigel in vitro. In this assay, we were able to show that the virus-encoded protein consistently inhibited endothelial migration in Matrigel cultures in a manner similar to that observed with recombinant ES produced in yeast, baculovirus, or myeloma cells (C.J.K., unpublished observations). In addition, as an additional biochemical measure of the structural integrity of the virally encoded ES, we were able to demonstrate by both mass spectroscopy and N-terminal sequencing analysis that the virally encoded ES purified from the serum of mice injected with ES-encoding virus possessed the expected protein sequence (K. Javaherian and C.J.K., unpublished data).
Systemic Inhibition of Tumor Growth by Antiangiogenic Adenoviruses.
The ability of each recombinant adenovirus vector to provide systemic inhibition of preestablished tumors was evaluated first in the aggressive LLC model in which recombinant AS and ES had been evaluated previously (14–17). LLC cells were implanted s.c. on the dorsum of C57BL/6 mice for 10–14 days to a size of 100–150 mm3, consistent with definitive tumor engraftment, followed by i.v. injection of 109 pfu of the various adenoviruses. Under these conditions, adenoviral infection occurs primarily in liver without significant intratumoral infection (data not shown); consequently, any inhibition of tumor growth on the dorsum from protein produced in a remote site (i.e., liver) would presumably occur by a systemic mechanism.
In mice bearing preexisting LLC tumors, i.v. injection of Ad Fc did not inhibit tumor growth, with animals often requiring sacrifice by 14 days after virus injection, and no significant difference was observed between tumor growth in Ad Fc- and PBS-treated animals (F.F., C.J.K., and R.C.M., unpublished observations). In contrast, after 10–14 days of treatment, tumors in either Ad Flk1-Fc- or Ad Flt1-injected mice exhibited ≈80% growth inhibition relative to controls, which was statistically significant compared with the Ad Fc control virus (P < 0.000001; Fig. 2 A and E). On the other hand, LLC growth was inhibited less strongly by Ad ES (27%, P = 0.004), Ad AS (24%, P = 0.001), or Ad neuropilin (14%, P = 0.15; Fig. 2A). The antitumor effects of both Ad Flk1-Fc and Ad Flt1 were dose-dependent, with the minimal day-3 plasma concentrations for effective systemic tumor suppression being approximately >1 mg/ml for Flk1-Fc and >2 μg/ml for Flt1(1–3) (F.F., E.Y., B.S., and C.K., unpublished data). In most cases, tumor growth eventually supervened after 3–4 weeks (data not shown). Although the studies do not rule out acquired endothelial and/or tumor resistance as the mechanism underlying the observed escape from inhibition, the rapid decline of vector-mediated gene expression over time most likely accounts for the observed results.
A similar relative efficacy of the different viruses was observed in a syngeneic murine T241 fibrosarcoma-C57BL/6 tumor model (Fig. 2 B–D) and in a xenogeneic BxPc3-SCID tumor model (Fig. 3 A and B). In the case of the T241 model, strong tumor suppression was exhibited again by Ad Flk1-Fc (83%, P < 0.000001) and Ad Flt1 (87%, P < 0.000001); yet in this model, little or no inhibition of tumor growth was achieved by Ad ES (6%, P = 0.71), Ad AS (6%, P = 0.86), or Ad neuropilin (6%, P = 0.77) (Fig. 2 B–D). In the case of the BxPc3 model, Ad Flk1-Fc produced a strong suppression of tumor growth (83%, P = 0.025), whereas Ad ES, Ad sNRP, or Ad AS did not inhibit growth of preestablished BxPC3 tumors significantly with <12% inhibition (P = 0.60–0.98) (Fig. 3 A and B). For these latter studies, the data for Ad Flt1-injected animals was not included because of the death of the animals before completion of the experiments (see Discussion). In a last series of experiments, Ad Flk-Fc was shown also to strongly inhibit tumor growth in another xenogenic tumor model involving LS174T human colon carcinoma and SCID mice (79%, P = 0.0003; Fig. 3C).
Figure 3.

Suppression of human tumor xenografts in SCID mice by Ad Flk1-Fc. (A) Treatment of BxPC3 human pancreatic carcinoma with Ad Flk1-Fc. CB17 SCID mice bearing preexisting tumors BxPC3 tumors of 60 mm3 received 109 pfu i.v. of the appropriate adenoviruses, and tumor size was measured over time. Error bars, ± 1 SD; Fc, n = 6; Flk1-Fc, n = 7. (B) Comparative inhibition of preexisting BxPC3 tumor growth by antiangiogenic adenoviruses. Ad Fc and Ad Flk1-Fc mice in A were compared with tumor-bearing mice in the same experiment that received Ad ES (n = 7), Ad AS (n = 7), or Ad sNRP (n = 6). Tumor size is expressed as percentage of maximal tumor volume standardized to 100% for Ad Fc, which did not produce significant inhibition relative to PBS controls. Error bars, ± 1 SEM; N, number of individual mice assayed with each adenovirus. (C) Treatment of human LS174T colon adenocarcinoma in SCID mice with Ad Flk1-Fc. n = 5 per group. Error bars, ± 1 SD.
Systemic Inhibition of Tumor Angiogenesis by Ad Flk1-Fc and Ad Flt1.
Microvessel density has been used extensively as a marker for tumor angiogenesis, tumor grade, and inhibition of microvessel density as a measure of antiangiogenic activity (22). To evaluate the mechanism for Ad Flk1-Fc and Ad Flt1 suppression of tumor growth, the microvessel density of treated vs. nontreated tumors was measured. LLC cells (106) were implanted s.c. in the dorsal midline of C57BL/6 mice, and tumors were allowed to grow to ≈50 mm3. The tumor-bearing mice then received i.v. injections of Ad Flk1-Fc, Ad Flt1(1–3), or Ad Fc followed by confirmation of expression levels by ELISA and were killed for histologic analysis after reaching a size of 200 mm3. Immunohistochemistry for the endothelial antigen CD31 demonstrated an ≈50% reduction of microvessel density in Flt1(1–3) and Flk1-Fc mice relative to Fc mice (Fig. 4). Parallel administration of Ad lacZ virus produced strong staining in liver and minor staining in lung but did not produce significant intratumoral β-galactosidase staining (data not shown).
Figure 4.

Decreased microvessel density in tumors treated with Ad Flk1-Fc or Ad Flt1(1–3). C57BL/6 mice bearing LLC tumors of ≈50 mm3 received i.v. injection of 109 pfu of Ad Fc, Ad Flk1-Fc, or Ad Flt1(1–3). Tumors were harvested at a size of 200 mm3 for CD31 immunohistochemistry, magnification, and manual quantitation of microvessel density. Error bars, ± 1 SD with four representative fields counted per condition.
Systemic Inhibition of VEGF-Stimulated Corneal Angiogenesis by Antiangiogenic Adenoviruses.
The ability of the different adenovirus-produced proteins to provide systemic inhibition of angiogenesis in vivo was evaluated also in a VEGF-dependent corneal neovascularization model. C57BL/6 mice received i.v. injections of the various adenoviruses followed after 2 days by implantation of hydron pellets containing human VEGF-A165 into the mouse cornea. Plasma expression of the appropriate transgene was confirmed by ELISA or Western blotting followed by quantitation of corneal neovascularization 5 days after pellet implantation. In mice receiving VEGF pellets, corneal neovascularization was inhibited strongly by Ad Flk1-Fc (74%, P < 0.0000001) or Ad Flt1 (80%, P < 0.0000001), which was statistically significant relative to the Ad Fc control virus (Fig. 5 A and B). VEGF-stimulated corneal angiogenesis was inhibited to a lesser degree by Ad ES (33%, P = 0.0001), Ad AS (23%, P = 0.002), or Ad neuropilin (35%, P = 0.027) Fig. 5 A and B).
Figure 5.

Systemic inhibition of corneal angiogenesis by antiangiogenic adenoviruses. (A) Comparative activity in VEGF corneal micropocket assays. C57BL/6 mice received i.v. injection of 109 pfu of the appropriate adenovirus, followed after 2 days by implantation of VEGF-A165-containing hydron pellets into the mouse cornea. Five days after pellet implantation, corneal neovascularization was quantitated by slit lamp examination. Results are presented as percentage of maximal neovascularization relative to the control virus Ad Fc, which was standardized at 100% and produced <5% inhibition relative to PBS. Error bars, ± 1 SEM. The number of eyes examined was as follows for Fc and the treatment group: ES, n = 13,18; AS, n = 13,14; Flk1-Fc, n = 16,15; Flt1, n = 21,25; sNRP, n = 10,8. (B) Systemic inhibition of corneal neovascularization by Ad Flk1-Fc or Ad Flt1(1–3). Representative corneas from experiments in A with preinjection of Ad Fc, Ad Flk1-Fc, or Ad Flt1(1–3) were photographed 5 days after pellet implantation. The position of the VEGF pellet is indicated by the arrow. Robust blood vessel ingrowth toward the pellet is noted in Ad Fc but not Ad Flk1-Fc or Ad Flt1(1–3) mice.
Discussion
The studies presented above provide important information regarding the relative potency of a number of antiangiogenic gene products previously shown to possess antitumor activity and specifically identify soluble forms of Flk1 and Flt1 as candidates for future gene therapy strategies. Our finding that soluble forms of Flk1 and Flt1 possessed significantly more potent antitumor activity than AS or ES when delivered via gene transfer was quite unexpected and is of particular interest in light of previous reports of the extremely potent antitumor effects of ES and AS delivered via conventional protein administration (14–16). The reasons for this important discrepancy are unclear currently. Although the serum levels of AS and ES achieved in the previous studies that reported frank tumor regression were not measured (14–17), it is highly likely that the levels of the proteins obtained after adenoviral-mediated gene transfer are far greater. In addition, although differences in protein structure, folding, or posttranslational processing between the conventionally produced molecules and those produced via gene transfer could account for differences in their bioactivity, at least in the case of vector-encoded ES, mass spectroscopy and N-terminal sequencing demonstrated that the expected protein structure was present in mouse serum after gene transfer. Moreover, in this regard, the adenovirus-produced ES exhibits motility-inhibiting properties comparable to that of recombinant ES produced in yeast, baculovirus, or myeloma cells in matrigel assays. Taken together, the data suggests that, at a minimum, ES or AS will not be as easily utilizable as soluble VEGF receptors in conventional single-injection adenoviral strategies aimed at the systemic delivery of protein and may require more innovative approaches with different vector systems, modified transgenes, or alternative routes of administration. Clearly, further studies aimed at understanding the discrepancy between our results and those involving the administration of conventionally produced ES and AS are warranted.
Although several previous reports also had documented the antitumor effects of vector-mediated delivery of AS, ES, soluble Flt1 ectodomains, and sNRP domains (13, 23–27), the ability of the gene products to provide for the potent inhibition of large (>100 mm3) aggressive preexisting tumors such as LLC had not been demonstrated previously. For example, although it has been shown that tumor lines stably transfected with AS cDNA exhibit impaired tumor growth, systemic gene therapy with AS has not been well documented to strongly suppress preexisting tumor growth (23, 25). Additionally, although several studies report the inhibition of tumor growth and metastases in mice after vector-mediated delivery of ES, no strong activity against preexisting tumors has been reported (24–27). In the case of soluble Flt-1 ectodomains, Kong et al. (28) have documented the efficacy of adenovirus vector-encoded Flt when delivered locally but not systemically, whereas Takayama et al. (13) have reported systemic antitumor efficacy of adenovirus Flt, but only against coinjected and not preexisting tumor burdens. In this latter case, the inability to observe significant activity against preexisting tumors may have resulted from insufficient production of Flt ectodomains, as our preliminary dosing studies suggest that high levels of gene product (>2 μg/ml) may be necessary for activity against preexisting tumors of >100 mm3. In the case of soluble forms of neuropilin (sNRP), previous studies have shown that a soluble form of neuropilin representing a naturally occurring spliced form of the gene product was able to inhibit the ability of rat prostatic carcinoma cell lines engineered to express the gene product to grow as tumors (29). The inability of our Ad sNRP to inhibit tumor growth could reflect either the stringency of the tumor models used in our study or the use of a suboptimal soluble form of NRP (the sNRP gene used in the current studies differs from that used in previous studies in that the “C” domain is included). It is noteworthy that sNRP binds to regions of VEGF encoded by exon 7 (30, 31), whereas Flk1 and Flt1 bind to more N-terminal domains of VEGF (32).
In addition to identifying candidate gene products of potential use in cancer therapy, our studies also represent the first comparative study of systemically administered antiangiogenic agents against ocular angiogenesis. Small molecule inhibitors of the Flk1/KDR kinase domain, direct intraocular injection of soluble VEGF receptors, or adenoviral production of soluble Flt-1 have been shown previously to inhibit experimental retinal vascularization (33–35). Potentially, a variety of conditions accompanied by pathologic eye angiogenesis, such as diabetic retinopathy, macular degeneration, retinal ischemia, and ocular melanomas (36, 37) could benefit from the sustained delivery afforded by single injection dosing of gene transfer vectors.
Lastly, although the comparative analysis we have presented is obviously imperfect in that we were not able to provide for the same level of each gene product in the circulation, the expression levels we have achieved likely represent a theoretical “maximum” that reflects the inherent pharmacokinetic properties governing the circulating levels of each protein that can be achieved via gene transfer. As such, the results provide important practical information regarding which antiangiogenic gene products are most likely to be therapeutically effective when delivered via gene therapy. In addition to the need to evaluate the use of vector systems that can provide for the sustained high level expression of genes in vivo such as the recently developed “gutless” adenoviral vectors (38), considerably more effort will need to be paid to the issue of the safety and long-term sequelae of systemic, soluble receptor-mediated VEGF inhibition in adult organisms. In this regard, we have observed that although non-tumor-bearing animals injected with Ad Flk1-Fc and viruses encoding ES, AS, and sNRP remained grossly asymptomatic for >1 year, ≈30% of animals injected with Ad Flt1(1–3) develop ascites after 22–28 days followed by frequent mortality despite a several log lower serum concentration of Flt than Flk1-Fc (unpublished results). Determination of whether the toxicity we have observed after injection of Ad Flt1 results from either excessive VEGF chelation by higher affinity Flt1 (21) or the distinct VEGF binding spectra of these receptors should aid the safety assessment of chronic VEGF-based antiangiogenic therapies.
Acknowledgments
We are indebted to C. Chartier for advice with adenovirus methods and comments on the manuscript, J.-S. Lee, T. Sheehan, R. Snyder, R. Bash, and S. Jungles for large-scale adenovirus preparation, O. Kisker and C. Becker for assistance with animal experimentation, and members of the Mulligan lab, M. Klagsbrun, S. Artandi, and H.-P. Biemann for many helpful discussions. We thank K. Javaherian for anti-murine ES polyclonal sera and for mass spectroscopy analysis, Entremed for human AS ELISA protocols, T. Niederman for generously providing Flk1-Fc cDNA, S. Soker for Flt-1 and sNRP cDNA, and B. Olsen for murine collagen XVIII cDNA. C.J.K. was supported by an Howard Hughes Medical Institute Physician-Scientist Fellowship. F.F. was supported by the Wenner-Gren Foundation and The Swedish Institute. R.C. was supported by the Swedish Children's Cancer Fund and Her Royal Highness Crown Princess Louisa's Fund for Child Care. R.A.S. and J.K. were supported by the Katharine Pappas Foundation. This work was supported by the National Institutes of Health, Howard Hughes Medical Institute, and generous grants from The Association for the Cure of Cancer of the Prostate (CaPCURE) and Radley Family Foundations.
Abbreviations
- VEGF
vascular endothelial growth factor
- sNRP
soluble neuropilin
- ES
endostatin
- AS
angiostatin
- pfu
plaque-forming unit
- LLC
Lewis lung carcinoma
- SCID
severe combined immunodeficient
References
- 1.Folkman J. N Engl J Med. 1995;333:1757–1763. doi: 10.1056/NEJM199512283332608. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Folkman J. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 3.Folkman J, D'Amore P A. Cell. 1996;87:1153–1155. doi: 10.1016/s0092-8674(00)81810-3. [DOI] [PubMed] [Google Scholar]
- 4.Holash J, Maisonpierre P C, Compton D, Boland P, Alexander C R, Zagzag D, Yancopoulos G D, Wiegand S J. Science. 1999;284:1994–1998. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- 5.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner J M. Circ Res. 1999;85:221–228. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 6.Kim K J, Li B, Winer J, Armanini M, Gillett N, Phillips H S, Ferrara N. Nature (London) 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 7.Presta L G, Chen H, O'Connor S J, Chisholm V, Meng Y G, Krummen L, Winkler M, Ferrara N. Cancer Res. 1997;57:4593–4599. [PubMed] [Google Scholar]
- 8.Lin P, Sankar S, Shan S, Dewhirst M W, Polverini P J, Quinn T Q, Peters K G. Cell Growth Differ. 1998;9:49–58. [PubMed] [Google Scholar]
- 9.Ferrara N, Alitalo K. Nat Med. 1999;5:1359–1364. doi: 10.1038/70928. [DOI] [PubMed] [Google Scholar]
- 10.Asano M, Yukita A, Suzuki H. Jpn J Cancer Res. 1999;90:93–100. doi: 10.1111/j.1349-7006.1999.tb00671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fong T A, Shawver L K, Sun L, Tang C, App H, Powell T J, Kim Y H, Schreck R, Wang X, Risau W, et al. Cancer Res. 1999;59:99–106. [PubMed] [Google Scholar]
- 12.Wedge S R, Ogilvie D J, Dukes M, Kendrew J, Curwen J O, Hennequin L F, Thomas A P, Stokes E S, Curry B, Richmond G H, Wadsworth P F. Cancer Res. 2000;60:970–975. [PubMed] [Google Scholar]
- 13.Takayama K, Ueno H, Nakanishi Y, Sakamoto T, Inoue K, Shimizu K, Oohashi H, Hara N. Cancer Res. 2000;60:2169–2177. [PubMed] [Google Scholar]
- 14.O'Reilly M S, Holmgren L, Shing Y, Chen C, Rosenthal R A, Moses M, Lane W S, Cao Y, Sage E H, Folkman J. Cell. 1994;79:315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 15.O'Reilly M S, Holmgren L, Chen C, Folkman J. Nat Med. 1996;2:689–692. doi: 10.1038/nm0696-689. [DOI] [PubMed] [Google Scholar]
- 16.O'Reilly M S, Boehm T, Shing Y, Fukai N, Vasios G, Lane W S, Flynn E, Birkhead J R, Olsen B R, Folkman J. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 17.Boehm T, Folkman J, Browder T, O'Reilly M S. Nature (London) 1997;390:404–407. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- 18.Chartier C, Degryse E, Gantzer M, Dieterle A, Pavirani A, Mehtali M. J Virol. 1996;70:4805–4810. doi: 10.1128/jvi.70.7.4805-4810.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berkner K L. Curr Top Microbiol Immunol. 1992;158:39–66. doi: 10.1007/978-3-642-75608-5_3. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Nunes F A, Berencsi K, Furth E E, Gonczol E, Wilson J M. Proc Natl Acad Sci USA. 1994;91:4407–4411. doi: 10.1073/pnas.91.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin C H. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 22.Weidner N. Am J Pathol. 1995;147:9–19. [PMC free article] [PubMed] [Google Scholar]
- 23.Griscelli F, Li H, Bennaceur-Griscelli A, Soria J, Opolon P, Soria C, Perricaudet M, Yeh P, Lu H. Proc Natl Acad Sci USA. 1998;95:6367–6372. doi: 10.1073/pnas.95.11.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blezinger P, Wang J, Gondo M, Quezada A, Mehrens D, French M, Singhal A, Sullivan S, Rolland A, Ralston R, Min W. Nat Biotechnol. 1999;17:343–348. doi: 10.1038/7895. [DOI] [PubMed] [Google Scholar]
- 25.Chen Q R, Kumar D, Stass S A, Mixson A J. Cancer Res. 1999;59:3308–3312. [PubMed] [Google Scholar]
- 26.Sauter B V, Martinet O, Zhang W J, Mandeli J, Woo S L. Proc Natl Acad Sci USA. 2000;97:4802–4807. doi: 10.1073/pnas.090065597. . (First Published April 11, 2000; 10.1073/pnas.090065597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feldman A L, Restifo N P, Alexander H R, Bartlett D L, Hwu P, Seth P, Libutti S K. Cancer Res. 2000;60:1503–1506. [PMC free article] [PubMed] [Google Scholar]
- 28.Kong H L, Hecht D, Song W, Kovesdi I, Hackett N R, Yayon A, Crystal R G. Hum Gene Ther. 1998;9:823–833. doi: 10.1089/hum.1998.9.6-823. [DOI] [PubMed] [Google Scholar]
- 29.Gagnon M L, Bielenberg D R, Gechtman Z, Miao H Q, Takashima S, Soker S, Klagsbrun M. Proc Natl Acad Sci USA. 2000;97:2573–2578. doi: 10.1073/pnas.040337597. . (First Published February 25, 2000; 10.1073/pnas.040337597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soker S, Takashima S, Miao H Q, Neufeld G, Klagsbrun M. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- 31.Soker S, Fidder H, Neufeld G, Klagsbrun M. J Biol Chem. 1996;271:5761–5767. doi: 10.1074/jbc.271.10.5761. [DOI] [PubMed] [Google Scholar]
- 32.Keyt B A, Nguyen H V, Berleau L T, Duarte C M, Park J, Chen H, Ferrara N. J Biol Chem. 1996;271:5638–5646. doi: 10.1074/jbc.271.10.5638. [DOI] [PubMed] [Google Scholar]
- 33.Aiello L P, Pierce E A, Foley E D, Takagi H, Chen H, Riddle L, Ferrara N, King G L, Smith L E. Proc Natl Acad Sci USA. 1995;92:10457–10461. doi: 10.1073/pnas.92.23.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Honda M, Sakamoto T, Ishibashi T, Inomata H, Ueno H. Gene Ther. 2000;7:978–985. doi: 10.1038/sj.gt.3301203. [DOI] [PubMed] [Google Scholar]
- 35.Ozaki H, Seo M S, Ozaki K, Yamada H, Yamada E, Okamoto N, Hofmann F, Wood J M, Campochiaro P A. Am J Pathol. 2000;156:697–707. doi: 10.1016/S0002-9440(10)64773-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aiello L P. Ophthalmic Res. 1997;29:354–362. doi: 10.1159/000268033. [DOI] [PubMed] [Google Scholar]
- 37.Aiello L P. Curr Opin Ophthalmol. 1997;8:19–31. doi: 10.1097/00055735-199706000-00005. [DOI] [PubMed] [Google Scholar]
- 38.Mountain A. Trends Biotechnol. 2000;18:119–128. doi: 10.1016/s0167-7799(99)01416-x. [DOI] [PubMed] [Google Scholar]