

Abstract
Nrf2, a member of the “cap ‘n collar” group of transcription factors, is important for protecting cells against oxidative damage. We investigated its role in the detoxification of acetaminophen [N-acetyl-p-aminophenol (APAP)]-induced hepatotoxicity. When Nrf2 knockout (Nrf2−/−) and wild-type mice were given APAP by i.p. injection, the Nrf2−/− mice were highly susceptible to APAP treatment. With doses of APAP that were tolerated by wild-type mice, the Nrf2−/− mice died of liver failure. When hepatic glutathione was depleted after a dose of 400 mg/kg of APAP, the wild-type mice were able to compensate and regain the normal glutathione level. In contrast, the glutathione level in the Nrf2−/− mice was not compensated and remained low. This was because of the decrease in the gene expression of gcsH and gcsL as well as gss in the livers of the Nrf2−/− mice. In addition, the expression of ugt1a6 and gstpi that detoxify APAP by conjugation was also decreased. This increased susceptibility of the Nrf2−/− mice to APAP, because of an impaired capacity to replenish their glutathione stores, compounded with a decreased detoxification capability, highlights the importance of Nrf2 in the regulation of glutathione synthesis and cellular detoxification processes.
Keywords: CNC transcription factor, gene knockout, antioxidant response element, γ-glutamylcysteine synthetase
The transcription factor Nrf2 is a member of the “cap ‘n’ collar” family of basic leucine zipper transcription factors. The family is made up of six members: p45-Nfe2, Nrf1, Nrf2, Nrf3, Bach1, and Bach2 (1–7). The first three were originally cloned during the search for proteins that bind to the NFE2-AP1 motif, the core element on hypersensitive site 2 of the locus control region of the human β-globin gene cluster (8–10).
The NFE2-AP1 motif was later determined to share high sequence homology to the antioxidant response element (11–14), and Nrf1 and Nrf2 were shown to play important roles in the cellular detoxification process by several lines of evidence. Nrf1 and Nrf2 transactivate reporter genes linked to antioxidant response elements, and their sites of expression coincide with those of many of the phase II detoxifying genes (15–17). Nrf2 mediates the induction of detoxifying genes, NAD(P)H quinone oxidoreductase 1 and glutathione S-transferases (GSTs), by the phenolic antioxidant butylated hydoxyanisole (18). Nrf2 was also found to be essential for the protection against butylated hydroxytoluene-induced pulmonary injury (19). Nrf2 regulates the expression of other detoxifying genes such as heme oxygenase 1 (20), catalase, superoxide dismutase 1, UDP-glucuronosyltransferase [(UGT)1a6], and γ-glutamylcysteine synthetase (GCS), as evidenced by a lower expression of these genes in the Nrf2−/− mice (19). In addition, the regulation of GCS, the rate-limiting enzyme in the synthetic pathway of glutathione (GSH), by Nrf2 was recently demonstrated in Nrf2−/− fibroblasts, hepatocytes, and HepG2 cells (21–23).
Acetaminophen [N-acetyl-p-aminophenol (APAP)] is one of the most widely used analgesic and antipyretic drugs worldwide, and as such, its biochemical properties has been extensively studied (24). It is marketed under different brand names and is also incorporated in formulation with other drugs. It is generally considered a safe drug at normal therapeutic dose levels but is a major cause of liver failure and causes death when taken in excess. Under normal conditions, APAP is primarily metabolized in the liver by glucuronidation catalyzed by UGTs and sulfation by sulfotransferases. A small amount of the drug is metabolized by several of the cytochrome p450 enzymes into the reactive intermediate N-acetyl-p-benzoquinoneimine (NAPQI), which is normally detoxified by GSH both nonenzymatically and enzymatically in a reaction catalyzed by GSTs. In overdose, sulfation and glucuronidation become saturated, and GSH is depleted by NAPQI. Excess NAPQI causes oxidative stress and binds covalently to liver proteins. Although the precise mechanism by which APAP or its metabolites cause cell injury is still unknown, cell death and organ failure most likely result from the cumulative and additive effects from oxidative damage, depressed mitochondrial functions, and disruptions of calcium homeostasis and redox balances (24).
In light of the role of Nrf2 in the regulation of expression of GCS, GSTs, and UGT1a6, we used Nrf2−/− mice to investigate whether lack of this transcription factor renders the mice more vulnerable to APAP poisoning. We report here that i.p. injection of APAP at doses well tolerated by wild-type mice results in a high mortality rate in Nrf2−/− mice, and that the de novo synthesis of hepatic GSH, on depletion by APAP, is impeded because of down-regulation of GCS and glutathione synthetase (GSS) in Nrf2−/− mice.
Materials and Methods
Chemicals.
APAP and N-acetylcysteine (NAC) were purchased from Sigma. APAP was dissolved in warm buffered saline at concentrations of 10–40 mg/ml and delivered at doses ranging from 100 mg/kg to 800 mg/kg by injecting 0.1–0.20 ml per 10 g of body weight. NAC was dissolved in sterile saline (pH adjusted to 7.4) at a concentration of 1,200 mg/ml and delivered at doses of 1,200 mg/kg by injecting 0.1 ml per 10 g body weight.
Animals.
Wild-type and Nrf2−/− mice were obtained by breeding from our stock of C57/SV129 mice. Adult male and female mice (10–15 weeks old) were used for this study.
Survival and Hepatotoxicity Studies.
For survival studies, 8 each of wild-type or Nrf2−/− male or female mice were given 100–800 mg/kg APAP i.p. and observed for survival for 48 h. To evaluate hepatotoxicity, female wild-type and Nrf2−/− mice were given APAP i.p. at 100, 200, and 300 mg/kg body weight with 10 mice per dose. Blood was collected by retroorbital bleeding at 24 h for measurement of the serum alanine aminotransferase (ALT). To test the mechanism of toxicity, female mice were given one dose of NAC (an antidote to APAP poisoning) 1,200 mg/kg body weight, together with APAP at 300, 400 or 600, and 800 mg/kg body weight with 8 mice per dose of APAP. Blood was collected at 24 h for serum ALT measurement. To measure GSH level and gene expression, 9 female mice of each genotype were injected with 100 or 400 mg/kg APAP, and 3 mice were killed at 90, 180, and 270 min after injection to collect blood for ALT measurements and liver tissues for histology, GSH assays, and gene expression studies. Serum ALT activities, expressed in Sigma–Frankel (SF) units per milliliter, were assayed in duplicate by using a kit (Sigma), and samples were diluted in sterile saline when values obtained were above the maximum threshold detectable by the kit, as recommended by the manufacturer.
Histology and Northern Blots.
Mice were killed by cervical dislocation, and the liver was collected and rinsed in PBS. One lobe of the liver was fixed in buffered formalin solution and paraffin embedded. Sections (7 μm) were stained with hematoxylin and eosin. One small piece of the liver was flash frozen in a preweighted tube by immersing into liquid nitrogen. GSH levels in livers were measured by using a total GSH kit from Oxis International (Portland, OR). Briefly, the piece of frozen liver was homogenized in 20 × volume to weight of 7.5% trichloroacetic acid, the homogenate cleared by centrifugation, and 100 μl of cleared lysate was assayed according to the manufacturer's protocol. The rest of the liver was stored in RNAlater (Ambion, Austin, TX) for RNA extraction. Northern blots were performed according to standard procedures. Total RNA was extracted from liver by using Ultraspec RNA (Bio-Tex, Edmonton, AL, Canada). The radioactivities of the Northern blots were quantified with Fuji imaging plates and an FLA-2000 image analyzer (Fuji). The activities of the Nrf2−/− bands, and wild-type bands were normalized with those of the glyceraldehyde-3-phosphate dehydrogenase bands. Gene expression was calculated relative to values for the untreated wild-type mice expressed as 100%.
Probes Used in Gene Expression Studies.
Primer pairs were designed from sequences from GenBank. First-strand cDNAs were reverse transcribed from total RNA with random hexamers by using Omniscript from Qiagen (Chatsworth, CA). The sequences of primers used to amplify the cDNAs are shown in Table 1. The amplified cDNA fragments were then subcloned and sequenced to confirm their identities.
Table 1.
Primers for amplification of cDNA fragments used for probes
| Gene | Sense primer | Antisense primer |
|---|---|---|
| GCSH | ACAAGCACCCCCGCTTCGG | CTCCAGGCCTCTCTCCTC |
| GCSL | ATGTTTTGGAATGCACCATGTCC | TGAGCTGGAGTTAAGAGCCC |
| GSS | GCGGTGGTGCTACTGATTGC | CACTGGACCACTTGGGCAGG |
| GST pi | TTTGGGGCTTTATGGGAAAA | ACATAGGCAGAGAGCAGGGG |
| UGT1a6 | CTTCCTGCAGGGTTTCTCTTCC | CAACGATGCCATGCTCCCC |
| GAPDH | CAGTGCCAGCCTCGTCCCG | CCAAGATGCCCTTCAGTGGG |
Results
Survival of Mice with APAP Challenge.
APAP treatment by i.p. injection generated dose-response curves that were genotype and gender dependent. Nrf2−/− mice were more susceptible than wild-type mice, and male mice of either genotype were more susceptible than their female counterparts (Fig. 1). Furthermore, the Nrf2−/− mice died rapidly between 6 and 12 h after injection, whereas the wild-type mice died over a more extended period, with the last deaths occurring between 36 and 48 h. The LD50 was 235 and 320 mg/kg for male and female Nrf2−/− mice, respectively, and 400 and 540 mg/kg for the male and female wild-type mice, respectively. Starting with a cohort of 8 animals per dose, 1 male wild-type animal survived dosages of 500 or 600 mg/kg, and 1 female wild-type animal overcame a dosage of 600 mg/kg. A dosage of 700 mg/kg or higher resulted in 100% mortality for both sexes of wild-type animals.
Figure 1.

Survival of Nrf2−/− and wild-type mice injected i.p. with increasing doses of APAP. Percent survival at 48 h is plotted as a function of dose. LD50 values are indicated with arrows. Eight mice were used for each dose.
Serum ALT Level.
In this experiment, only female mice were used. Baseline serum ALT activities in untreated mice of either genotype ranged between 13 and 48 SF units/ml. APAP treatment at the dose of 100 mg/kg body weight did not elevate serum ALT activities in the wild-type mice. Nrf2−/− mice exposed to the same dose showed a wide variation in serum ALT activities that ranged from 27 to 4,200 SF units/ml. At doses of 200 and 300 mg/kg of APAP, both Nrf2−/− and wild-type mice exhibited a wide range of ALT levels from 33 to 13,000 SF units/ml and 25 to 8,400 SF units/ml, respectively (Fig. 2A). Because of the large spread of values, there were no significant statistical differences in serum ALT activities between the genotypes (P(100) = 0.056, P(200) = 0.102, P(300) not calculated). However, at 300 mg/kg APAP, two Nrf2−/− mice died before 24 h, and two others died at 48 h, whereas no death occurred in the wild-type controls.
Figure 2.

Individual serum ALT values of mice at 24 h after injection with different doses of APAP. (A) Ten wild-type (WT) and nrf2 knockout (KO) mice were injected with doses of 100, 200, and 300 mg/kg APAP; individual serum ALT values are plotted (P(100) = 0.056, P(200) = 0.102; P(300) was not calculated because two data points were lost when two Nrf2−/− mice died before blood collection). (B) Eight wild-type and nrf2 knockout mice were injected with 1,200 mg/kg NAC (+N) in addition to APAP at 300, 400 or 600, and 800 mg/kg APAP.
Rescue from APAP Toxicity by NAC.
NAC, the commonly used antidote to APAP poisoning, was coadministered with different doses of APAP. A single dose of NAC at 1,200 mg/kg body weight i.p. abrogated the elevation of serum ALT activities seen when 300 mg/kg dose of APAP was injected by itself in animals of both genotypes (Fig. 2B). When LD100 of APAP were used, NAC rescued 100% of the wild-type mice treated with a 600 mg/kg dose of APAP, lowering the measured serum ALT levels to between 31 and 195 SF units/ml, comparable to the level obtained in untreated controls. NAC partially alleviated the toxicity of APAP in the Nrf2−/− mice treated with the LD100 dose of 400 mg/kg, as only 2 of the 8 Nrf2−/− mice treated with NAC died. The serum ALT of the surviving mice ranged between 110 and 1,440 SF units/ml, higher than the levels obtained from the wild-type mice treated with NAC and 600 mg/kg APAP, none of which died (Fig. 2B). When treated with 800 mg/kg APAP, a dose that killed 100% of mice of either genotype, wild-type mice benefited most from NAC antidote, and serum ALT values at 24 h ranged from 31 to 3,900 SF units/ml. The one wild-type animal with the highest ALT level of 3,900 SF units/ml died between 36 and 48 h. NAC treatment did not protect the Nrf2−/− mice as effectively as the wild-type mice. At this dose of APAP and NAC, only 2 of the Nrf2−/− mice (25%) survived beyond 24 h, and only 1 single animal (12.5%) survived beyond 48 h. The 48-h survival of mice of either genotype treated with APAP with or without NAC is presented in Table 2.
Table 2.
Effect of NAC on female mice survival (%) at 48 hours with different doses of APAP
| Mice | 300 mg/Kg | 400 mg/Kg | 600 mg/Kg | 800 mg/Kg |
|---|---|---|---|---|
| WT | 100 | 100 | 12.5 | 0 |
| WT + NAC | 100 | — | 100 | 87.5 |
| KO | 60 | 0 | 0 | 0 |
| KO + NAC | 100 | 75 | — | 12.5 |
GSH Levels.
For the measurement of GSH levels and the expression of genes involved in synthesis of GSH and detoxification of APAP metabolites, we used one nonlethal and one lethal dose to maximize the difference in the response to APAP between wild-type and Nrf2−/− mice. Three time points at 90-min intervals were chosen because Nrf2−/− mice were observed to begin dying by 5.5–6 h after injection. APAP at 100 mg/kg body weight caused only a slight drop in the GSH levels of the wild-type mice but caused a larger drop in the Nrf2−/− mice at 90 min (Fig. 3A). At 180 min, in both genotypes, the GSH contents were back to baseline level and surpassed the baseline level at 270 min. The ALT activities for these mice were all within normal limits (data not shown).
Figure 3.

Liver GSH content and gene expression after APAP injection. Mice were injected with 100 (open symbols) or 400 mg/kg (closed symbols) APAP and killed after 90, 180, and 270 min. (A) Liver GSH expressed as micromol/gram of liver as a function of time after injection. (B) Gene expression calculated from Northern blots and expressed as a percentage of the level found in liver of untreated wild-type mice after normalization with glyceraldehyde-3-phosphate dehydrogenase.
A dose of 400 mg/kg of APAP caused a precipitous drop of the GSH levels in both genotypes at 90 min to 5 and 4% of the untreated levels for wild-type and Nrf2−/− mice, respectively. In the wild-type mice, the GSH levels recovered to 35% of baseline at 180 min and 74% at 270 min, but in the Nrf2−/− mice, the GSH levels remained low, at 6 and 8% of baseline, respectively (Fig. 3A). The ALT activities for all these mice at 90 and 180 min were within those of the untreated mice. The ALT activities at 270 min were elevated to 600, 3,000, and 8,800 SF units/ml for the Nrf2−/− mice, and 160, 210, and 2,300 SF units/ml for the wild-type mice.
Gene Expressions.
Northern blot analysis of liver RNAs revealed that the genes for both the light and heavy chain subunits of GCSH and GCSL were expressed at 58 and 65% of wild-type control, respectively, in the untreated Nrf2−/− mice. On APAP treatment, GCSH and GCSL expression was induced in wild-type mice, resulting in the de novo synthesis of GSH we observed in the liver. Also, the induction of GCSH, the catalytic subunit, was much higher than that of GCSL. In contrast, both GCSH and GCSL compensated poorly in Nrf2−/− animals. GSS expression in the untreated Nrf2−/− mice was 80% of the level in wild-type mice, and its response to APAP treatment was also blunted in the Nrf2−/− mice. GSTpi and UGT1a6 expressions were also significantly lower in the Nrf2−/− mice at 45 and 31%, respectively, of the wild-type baseline levels, and their response to APAP was also diminished (Fig. 3B).
To study the role Nrf2 plays in the change of expression for these enzymes, we examined the gene expression of Nrf2 in wild-type animals in response to APAP. We found that, whereas at a low dose of APAP Nrf2 was induced transiently, at a higher dose (400 mg/kg), the increase of Nrf2 was more protracted (Fig. 4). It is interesting to note that the pattern of expression for Nrf2 at 400 mg/kg APAP closely paralleled those of the enzymes described in Fig. 3B. Expression of Nrf1 was not affected by APAP in the liver of mice of either genotype (data not shown).
Figure 4.

Nrf2 gene expression after 100 and 400 mg/kg APAP injection.
Pathologic Changes.
To document the sequence of liver injury, we examined the liver histology in the mice of both genotypes treated with 100 and 400 mg/kg of APAP, as described above. No hepatic damage was seen at 100 mg/kg in mice of either genotype, as the drop in GSH levels was compensated for, and GSH levels were maintained. In contrast, at 90 min after 400 mg/kg APAP, overt hepatic injury was noted in the Nrf2−/− mice, whereas minimal changes were seen in wild-type mice (Fig. 5). At 180 min, liver injury could be seen in mice of both genotypes. Thus histological changes preceded the rise in ALT levels, which was not observed until 270 min.
Figure 5.
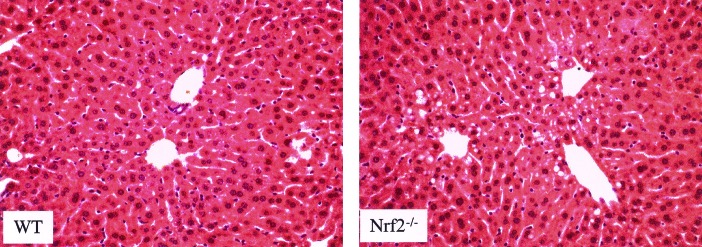
Liver injury found in mice at 90 min after injection of 400 mg/kg APAP. Moderate injury can be observed by the presence of cytoplasmic vacuoles in centrilobular hepatocytes in the Nrf2−/− livers (hematoxylin/eosin). (×184.)
Discussion
These studies show that Nrf2 is an important component of the cellular detoxification apparatus in response to oxidative stress. Large doses of the common analgesic APAP cause liver injury by oxidative stress, and mice lacking the Nrf2 transcription factor are more susceptible to the common analgesic acetaminophen than their wild-type counterparts. Nrf2−/− mice died sooner and at lower doses of APAP. For both genotypes, male mice were more sensitive than female mice to APAP. Whereas NAC, a GSH precursor used as an antidote for APAP overdose, can rescue LD100 doses given to wild-type mice, its rescue of Nrf2−/− mice is incomplete and is limited to lower APAP doses. Measurement of serum ALT activities at 24 h appears not to be a good indicator of the hepatic injury affecting the mice for several reasons. First, interanimal variability prevented statistical differentiation between the genotypes even though mortality differences were clearly observable. Second, moderate injury found on histological examination at 90 and 180 min preceded the rise in the serum ALT levels (Fig. 5).
GSH is a molecule that plays important roles in critical physiological functions and protection against oxidative damage (25). It functions by detoxifying electrophiles and scavenging free radicals. GSH exists in high concentration in cells, and its homeostasis is maintained by a balance between the rate of synthesis and the combined rate of utilization and loss through efflux. The biosynthesis of GSH is first catalyzed by GCS and then by GSS. The rate of synthesis depends critically on the availability of GCS, a holoenzyme composed of a catalytic heavy subunit GCSH and a regulatory light subunit GCSL. Nrf2−/− mice have been shown to have slight decreased basal hepatic GSH level and decreased expression of GCSH and GCSL (21). They can nonetheless compensate for a 57% drop in liver GSH on treatment with 100 mg/kg APAP. In contrast, the GSH level declined only 10% in the wild-type mice. We attribute this larger drop in the Nrf2−/− mice to a deficit in the rate of GSH synthesis compared with the combined rate of utilization and efflux. At 400 mg/kg APAP, the Nrf2−/− mice succumbed because of an inability to replenish their GSH stores (Fig. 3A).
The Nrf2−/− mice had difficulty in detoxifying APAP and its metabolites because of the lower level of hepatic GSH. Their baseline expression of GCSH and GCSL was about 65 and 58% of the wild type. On challenge with 100 and 400 mg/kg of APAP, the expression of these enzymes rose only slightly. Likewise, the GSS response to APAP was also blunted in the Nrf2−/− mice. In contrast, on APAP treatment, gene expression for both subunits was induced in wild-type mice, GCSH induction was up more than 400%, whereas that of GCSL was more modest at 175% the level of that of untreated mice (Fig. 3B). The lower level of expression of these enzymes that are responsible for the synthesis of GSH accounts for the less effective protection by the GSH precursor to APAP challenge in the Nrf2−/− mice. An additional factor that contributes to the higher sensitivity of Nrf2−/− mice to APAP is the intrinsic lower expression of detoxifying enzymes such as UGT1a6 and GSTpi, which are important for detoxifying the toxic metabolite N-acetyl-p-benzoquinoneimine by conjugation. In the Nrf2−/− mice, these two enzymes display a similar profile of expression to that of GCS in that their inherent expression is low, and their inductions were also lower than those observed in wild-type mice. It is interesting to note that the pattern of Nrf2 expression on induction by APAP (Fig. 4) closely parallels those of this group of enzymes in vivo in the liver (Fig. 3B). This indicates that in vivo, Nrf2 plays an important role in the control of the expression of these genes. In contrast, in in vitro experiments by using HepG2 cells line, GCS gene induction involving Nrf2 appeared to be contingent not on increase of Nrf2 transcript or protein (23) but on Nrf2 release from sequestration by Keap1 (26).
These experiments showed that Nrf2 plays a major role in the defense against APAP toxicity through the GSH synthesis pathway. It is possible that the residual GSH response in the liver of Nrf2−/− mice is mediated by Nrf1, as Nrf1−/− fibroblasts have been shown to have lower GSH levels (17). However, expression of Nrf1 in the absence of Nrf2 in vivo cannot provide adequate protection against APAP toxicity. Hence Nrf2 must have important functions in fighting acute oxidative stress in the liver as well as previously shown in the lung (19). It remains to be seen whether lack of Nrf2 and prolonged or chronic exposure to oxidative stress lead to DNA damage and genetic changes. If so, mutations in the Nrf2 gene may also result in cancer predisposition.
Acknowledgments
We thank Dr. Timothy Davern for reviewing the manuscript and Dr. Jefferson Chan for helpful suggestions. The work is supported in part by National Institutes of Health Grants DK16666, DK07636, and DK50267. Y.W.K. is an Investigator of the Howard Hughes Medical Institute.
Abbreviations
- GST
glutathione S-transferase
- GCS
γ-glutamylcysteine synthetase
- GSH
glutathione
- APAP
N-acetyl-p-aminophenol
- UGT
UDP-glucuronosyltransferase
- NAC
N-acetylcysteine
- ALT
alanine aminotransferase
- SF
Sigma–Frankel
- GSS
glutathione synthetase
References
- 1.Andrews N C, Erdjument-Bromage H, Davidson M B, Tempst P, Orkin S H. Nature (London) 1993;362:722–728. doi: 10.1038/362722a0. [DOI] [PubMed] [Google Scholar]
- 2.Chan J Y, Han X L, Kan Y W. Proc Natl Acad Sci USA. 1993;90:11371–11375. doi: 10.1073/pnas.90.23.11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan J Y, Han X L, Kan Y W. Proc Natl Acad Sci USA. 1993;90:11366–11370. doi: 10.1073/pnas.90.23.11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moi P, Chan K, Asunis I, Cao A, Kan Y W. Proc Natl Acad Sci USA. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caterina J J, Donze D, Sun C W, Ciavatta D J, Townes T M. Nucleic Acids Res. 1994;22:2383–2391. doi: 10.1093/nar/22.12.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kobayashi A, Ito E, Toki T, Kogame K, Takahashi S, Igarashi K, Hayashi N, Yamamoto M. J Biol Chem. 1999;274:6443–6452. doi: 10.1074/jbc.274.10.6443. [DOI] [PubMed] [Google Scholar]
- 7.Oyake T, Itoh K, Motohashi H, Hayashi N, Hoshino H, Nishizawa M, Yamamoto M, Igarashi K. Mol Cell Biol. 1996;16:6083–6095. doi: 10.1128/mcb.16.11.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tuan D, Solomon W, Li Q, London I M. Proc Natl Acad Sci USA. 1985;82:6384–6388. doi: 10.1073/pnas.82.19.6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forrester W C, Thompson C, Elder J T, Groudine M. Proc Natl Acad Sci USA. 1986;83:1359–1363. doi: 10.1073/pnas.83.5.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forrester W C, Takegawa S, Papayannopoulou T, Stamatoyannopoulos G, Groudine M. Nucleic Acids Res. 1987;15:10159–10177. doi: 10.1093/nar/15.24.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rushmore T H, Morton M R, Pickett C B. J Biol Chem. 1991;266:11632–11639. [PubMed] [Google Scholar]
- 12.Prestera T, Holtzclaw W D, Zhang Y, Talalay P. Proc Natl Acad Sci USA. 1993;90:2965–2969. doi: 10.1073/pnas.90.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie T, Belinsky M, Xu Y, Jaiswal A K. J Biol Chem. 1995;270:6894–6900. doi: 10.1074/jbc.270.12.6894. [DOI] [PubMed] [Google Scholar]
- 14.Wasserman W W, Fahl W E. Proc Natl Acad Sci USA. 1997;94:5361–5366. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Venugopal R, Jaiswal A K. Proc Natl Acad Sci USA. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venugopal R, Jaiswal A K. Oncogene. 1998;17:3145–3156. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- 17.Kwong M, Kan Y W, Chan J Y. J Biol Chem. 1999;274:37491–37498. doi: 10.1074/jbc.274.52.37491. [DOI] [PubMed] [Google Scholar]
- 18.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et al. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 19.Chan K, Kan Y W. Proc Natl Acad Sci USA. 1999;96:12731–12736. doi: 10.1073/pnas.96.22.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alam J, Stewart D, Touchard C, Boinapally S, Choi A M, Cook J L. J Biol Chem. 1999;274:26071–26078. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 21.Chan J Y, Kwong M. Biochim Biophys Acta. 2000;1517:19–26. doi: 10.1016/s0167-4781(00)00238-4. [DOI] [PubMed] [Google Scholar]
- 22.Moinova H R, Mulcahy R T. Biochem Biophys Res Commun. 1999;261:661–668. doi: 10.1006/bbrc.1999.1109. [DOI] [PubMed] [Google Scholar]
- 23.Wild A C, Moinova H R, Mulcahy R T. J Biol Chem. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- 24.Cohen S D, Hoivik D J, Khairallah E A. In: Toxicology of the Liver. Plaa G L, Hewitt W R, editors. Vol. 1. Bristol, PA: Taylor & Francis; 1998. pp. 159–186. [Google Scholar]
- 25.Lu S C. FASEB J. 1999;13:1169–1183. [PubMed] [Google Scholar]
- 26.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel J D, Yamamoto M. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]