

ABSTRACT
Cyclic-di-GMP is a near-ubiquitous bacterial second messenger that is important in localized signal transmission during the control of various processes, including virulence and switching between planktonic and biofilm-based lifestyles. Cyclic-di-GMP is synthesized by GGDEF diguanylate cyclases and hydrolyzed by EAL or HD-GYP phosphodiesterases, with each functional domain often appended to distinct sensory modules. HD-GYP domain proteins have resisted structural analysis, but here we present the first structural representative of this family (1.28 Å), obtained using the unusual Bd1817 HD-GYP protein from the predatory bacterium Bdellovibrio bacteriovorus. Bd1817 lacks the active-site tyrosine present in most HD-GYP family members yet remains an excellent model of their features, sharing 48% sequence similarity with the archetype RpfG. The protein structure is highly modular and thus provides a basis for delineating domain boundaries in other stimulus-dependent homologues. Conserved residues in the HD-GYP family cluster around a binuclear metal center, which is observed complexed to a molecule of phosphate, providing information on the mode of hydroxide ion attack on substrate. The fold and active site of the HD-GYP domain are different from those of EAL proteins, and restricted access to the active-site cleft is indicative of a different mode of activity regulation. The region encompassing the GYP motif has a novel conformation and is surface exposed and available for complexation with binding partners, including GGDEF proteins.
IMPORTANCE
It is becoming apparent that many bacteria use the signaling molecule cyclic-di-GMP to regulate a variety of processes, most notably, transitions between motility and sessility. Importantly, this regulation is central to several traits implicated in chronic disease (adhesion, biofilm formation, and virulence gene expression). The mechanisms of cyclic-di-GMP synthesis via GGDEF enzymes and hydrolysis via EAL enzymes have been suggested by the analysis of several crystal structures, but no information has been available to date for the unrelated HD-GYP class of hydrolases. Here we present the multidomain structure of an unusual member of the HD-GYP family from the predatory bacterium Bdellovibrio bacteriovorus and detail the features that distinguish it from the wider structural family of general HD fold hydrolases. The structure reveals how a binuclear iron center is formed from several conserved residues and provides a basis for understanding HD-GYP family sequence requirements for c-di-GMP hydrolysis.
Introduction
Cyclic-di-GMP (c-di-GMP) is a near-ubiquitous bacterial signaling molecule implicated in the regulation of diverse processes such as virulence, motility, and biofilm formation (1, 2). Discovered initially as a regulator of cellulose biosynthesis (3), c-di-GMP has risen to prominence in recent times because whole-genome analysis has indicated that many bacteria are likely to utilize c-di-GMP signaling (1, 4). c-di-GMP is synthesized from GTP by enzymes containing GGDEF diguanylate cyclase domains and is hydrolyzed by phosphodiesterase enzymes containing either EAL or HD-GYP domains, each domain nomenclature corresponding to a subset of conserved amino acids responsible for enzymatic activity (2). Structures of isolated GGDEF and EAL domains have become available, as have those of GGDEF/EAL fusion proteins (reviewed by Schirmer and Jenal [2]). In contrast, HD-GYP domains are usually found separate from GGDEF domains (although they are often appended to a variety of other sensory domains [5]). HD-GYP proteins have so far resisted high-resolution crystallography, with the only structural information available arising from distantly related members of the HD superfamily that catalyze disparate, non-c-di-GMP-related functions.
Bdellovibrio bacteriovorus is a predatory deltaproteobacterium which follows two different life cycles—invasive predation of other Gram-negative bacteria with rapid intracellular replication within its prey and slow, more conventional axenic growth on complex microbiological media (for a population with mutational changes, becoming host independent, so-called HI strains [6, 7]). The B. bacteriovorus HD100 genome is predicted to encode one EAL protein and six HD-GYP proteins (8). One of the six HD-GYP candidates, Bd1817, has a nonconsensus sequence in the GYP motif, G-P, lacking the conserved tyrosine (Fig. 1). Realizing that an unconventional Bd1817 structure might inform the cdi-GMP field where structures of conventional HD-GYP domains have not been forthcoming, we proceeded to crystallize and characterize the Bd1817 HD-GYP protein and thus reveal the features shared by Bd1817 and other characterized HD-GYP proteins (Fig. 1).
FIG 1 .

Sequence alignment of B. bacteriovorus Bd1817 and other HD-GYP proteins. Bacterial strains and associated UNIPROT accession codes are as follows: Bd1817 (B. bacteriovorus, Q6MM30, aa 131 to 308), RpfG (Xanthomonas campestris, Q4UU85, aa 180 to 349), PA4108 (Pseudomonas aeruginosa, Q9HWS0, aa 141 to 308), PA4781 (P. aeruginosa, Q9HV27, aa 161 to 344), VCA_0681 (Vibrio cholerae, Q9KLR1, aa 237 to 405), 3,508 bp (Bordetella pertussis, Q7VTL7, aa 14 to 183), KPK_3322 (Klebsiella pneumoniae, B5XSE7, aa 204 to 372), SCO5218 (Streptomyces coelicolor, Q9K4A3, aa 226 to 393), BAS0914 (Bacillus anthracis, Q81UA6, aa 187 to 355). Conserved residues are boxed in white font on a red background, and partially conserved residues are boxed in red font on a white background. Metal-liganding and phosphate-liganded residues are indicated by blue triangles and magenta ovals, respectively. Secondary structure elements for Bd1817 are given above the alignment, corresponding to the HD-GYP domain only. Residue numbering refers to Bd1817, which terminates at L308; the other sequences have been cropped to match the region from A131 to the common DP element preceding α7. This alignment was prepared using T-Coffee (33) and ESPript (34).
RESULTS AND DISCUSSION
Bd1817 expression and function.
We verified that Bdellovibrio expresses Bd1817 by transcript profiling (9) (Fig. 2), finding that its expression was highest at the late 3- and 4-h stages of predatory Bdellovibrio growth inside a dead prey Escherichia coli cell and also during axenic (host/prey-independent, HI) growth. Fluorescence from a Bd1817-mCherry fusion protein was detectable at modest levels, distributed throughout the Bdellovibrio cytoplasm (see Fig. S1 in the supplemental material) in axenically growing HI cells, especially those elongating or dividing. Deletion of the Bd1817 gene did not significantly affect predatory growth or the rate of axenic (HI) growth of the knockout mutant, which was within the range of growth rates of diverse wild-type strains. Thus, we established that Bd1817 protein is expressed but have not yet detected the phenotype it produces.
FIG 2 .

Comparative transcriptional profiling of Bd1817 expression. Lanes AP to 4h contain total RNA taken from predatory B. bacteriovorus HD100 attacking E. coli prey cells, i.e., wild-type attack phase cells outside prey (AP) and B. bacteriovorus HD100 wild-type cells that invaded and were replicating inside E. coli prey cells for 15, 30, or 45 min or 1, 2, 3, 4 h. Lanes 1 to 4 contain Bd1817 RT-PCR products showing expression levels from matched 10-ng total RNA samples from attack phase Bdellovibrio (lane 1) and axenically grown, independently isolated wild-type HI strains of B. bacteriovorus HD100. Lane 2, strain HID2; lane 3, strain HID13; lane 4, strain HID26. Control lanes are as follows: S17-1, E. coli S17-1 RNA alone; –, no-template negative control; +, B. bacteriovorus genomic DNA positive control; L, 100-bp DNA ladder. This figure shows that Bd1817 is most highly expressed in the growth and septation phases of the Bdellovibrio axenic and predatory life cycles.
Bd1817 retains most of the conserved residues of a HD-GYP protein (e.g., showing 24% identity and 48% similarity with the family archetype RpfG, Fig. 1), and despite, or possibly because of, the lack of a strong c-di-GMP phosphodiesterase phenotype, it crystallized readily—and so, structural studies were instigated to gain insight into the function of HD-GYP proteins for which no high-resolution crystal structures exist to date.
Structure determination and protein fold.
We report here the structure of Bd1817 (from four different crystal forms, the highest-resolution P21A form extending to 1.28 Å, examples of active-site and GYP motif electron density are provided in Fig. 3); the data were phased using a single-wavelength anomalous dispersion (SAD) protocol that took advantage of the intrinsic transition metals (Table 1). Our expression construct contains an N-terminal polyhistidine/thrombin tag, the uncharacterized N-terminal domain (NTD), and the C-terminal HD-GYP phosphodiesterase domain. The structure of Bd1817 confirms the modular nature of HD-GYP proteins (Fig. 4a), with the NTD (amino acids [aa] 1 to 78) separated from the HD-GYP domain by a compact linker region composed of 3 α-helices (aa 79 to 146); the two regions are physically distinct from one another, and it is intuitive that different sensory modules could be easily appended to the HD-GYP fold for distinct signaling purposes, such as is the case for RpfG of Xanthomonas (10). The ~40-aa region preceding the catalytic domain of RpfG is predicted to be α-helical, and so the linker conformation we observe in Bd1817 may also be relevant to other multidomain HD-GYP proteins. The juxtaposition of the NTD, linker helices, and HD-GYP domain results in an arrangement where a conserved hydrophobic residue, following the HD motif (Y185), packs against residues at the C-terminal end of one of the linker helices (F104, P107). The NTD of Bd1817 has no strong homology to other protein folds but does possess weak structural similarity to pleckstrin homology domains (e.g., that of the Exo84 RalA-binding domain [11]; root mean square [RMS] deviation of 3.4 Å for common Cα; Fig. 5a). Coupled with the observation that in one of our crystal forms the affinity tag has bound in an ordered fashion into a cleft in the NTD (binding in an identical region to RalA in the Exo84-RalA complex [11] but in the opposite orientation; Fig. 5b), this raises the possibility that this domain could function in a protein-protein interaction. Interestingly, the NTD has an RxxD motif with the arginine and aspartate side chains “stacked” in a conformation identical to that of the c-di-GMP binding inhibitory site of several GGDEF proteins (e.g., residues R34/D37 of Bd1817 matching R359/D362 of PleD [12]). The HD-GYP domain (aa 147 to 308) is an all-alpha fold composed of 7 helices and superficially resembles that of other HD superfamily proteins, with several important differences that cluster around the active site (Fig. 6). The closest structural match to a characterized HD superfamily member is the E. coli 5′-deoxynucleotidase YfbR (13) (RMS deviation of 3.8 Å for common Cα). Analysis of protein-protein contacts in the four different crystal forms indicates that Bd1817 is likely monomeric (at least in this unliganded form).
FIG 3 .

Electron density of chain A P21A crystal form, 1.28-Å resolution data, final refined map at 1.2σ. (a) Binuclear metal-binding site, with bridging hydroxide ion (W1) and tetrahedral ion, modeled as phosphate. (b) Region of GYP motif (glycine 245 and proline 246, “missing” the conserved tyrosine), electron density of aa 237 to 249 demonstrating the ordered nature of the fold.
TABLE 1 .
Data collection and refinement statisticsa
| Parameter | P21A | P21B | P1 | P3121 |
|---|---|---|---|---|
| Data collectionb | ||||
| Space group | P21 | P21 | P1 | P3121 |
| Cell dimensions | ||||
| a, b, c (Å) | 47.2, 79.8, 84.8 | 83.2, 45.5, 98 | 47.1, 49.4, 78.4 | 101, 101, 94.5 |
| α, β, γ (°) | 90, 96, 90 | 90, 113.3, 90 | 73.2, 75.9, 67.4 | 90, 90, 120 |
| Resolution (Å) | 1.28 (1.35–1.28) | 1.7 (1.79–1.7) | 1.55 (1.63–1.55) | 2.6 (2.78–2.6) |
| Rsym | 11.0 (65.4) | 5.3 (64.4) | 8.0 (17.2) | 7.9 (46.7) |
| Rpim | 6.9 (41.6) | 1.7 (29.3) | 4.7 (13.8) | 6.7 (42.2) |
| I/σI | 9.8 (3.4) | 25.0 (2.4) | 13.4 (3.3) | 5.9 (1.6) |
| Completeness (%) | 99.6 (98.6) | 97.7 (85.4) | 90.0 (62.9) | 97.2 (97.5) |
| Redundancy | 6.9 (6.6) | 10.1 (4.8) | 5.5 (2.0) | 1.9 (1.9) |
| Refinement | ||||
| Resolution (Å) | 1.28 | 1.7 | 1.55 | 2.6 |
| No. of reflections | 161,271 | 72,760 | 80,548 | 4,568 |
| Rwork/Rfree | 16.5/19.6 | 19.4/21.9 | 21.0/23.6 | 21.2/25.1 |
| No. of atoms | ||||
| Protein | 5,024 | 4,997 | 4,887 | 2,424 |
| Ligand/ion | 34 | 14 | 22 | 7 |
| Water | 574 | 316 | 352 | 11 |
| B factors | ||||
| Protein | 13.1 | 31.8 | 10.9 | 86.5 |
| Ligand/ion | 21.0 | 22.2 | 8.5 | 74.0 |
| Water | 25.0 | 32.6 | 16.2 | 64.6 |
| RMS deviation | ||||
| Bond length (Å) | 0.005 | 0.006 | 0.006 | 0.008 |
| Bond angle (°) | 1.0 | 1.0 | 1.0 | 1.1 |
Values in parentheses are for highest-resolution shell.
Rsym, ∑|(Ihkl) − <I>|/∑(Ihkl); Rpim, multiplicity-weighted merging R-factor.
FIG 4 .

Modular nature of the Bd1817 HD-GYP protein and active-site coordination of metal ions. Selected side chains and bound phosphate are shown in stick form, and metal ions (tan, M1 leftmost and M2 rightmost) and bound hydroxide (red, W1) are shown in sphere form. (a) Ribbon diagram of Bd1817 with individual domains colored separately as follows: NTD (aa 1 to 78), blue; linker helices (aa 79 to 146), yellow; HD-GYP domain (aa 147 to 308), white; lid region subdomain (aa 188 to 211), green; GYP motif subdomain (aa 239 to 255), orange. (b) Detail of binuclear metal active site (HD-GYP domain). Fe-protein interactions, purple dashed lines; hydroxide-protein interaction, green dashed line. (c) Schematic of binuclear metal site with coordination distances shown (taken from chain A of the 1.28-Å P21A crystal form).
FIG 5 .

Features of the NTD. (a) The Bd1817 NTD (blue) possesses weak structural homology with the Ral-binding domain of Exo84 (cyan; PDB code 1ZC3 [11]), ending at the point where the Bd1817 linker region (yellow) starts. In the P21B crystal form, residues −11 to 3 of chain B (the polyhistidine tag with the thrombin site left uncleaved [red]) are highly ordered and bind into a cleft formed between the NTD and linker region of chain A. This interaction occurs in a region identical to that of the Exo84 binding partner RalA (magenta), albeit running in different chain directions. (b) Detail of affinity tag-NTD interaction. Selected side chains are shown in stick form.
FIG 6 .
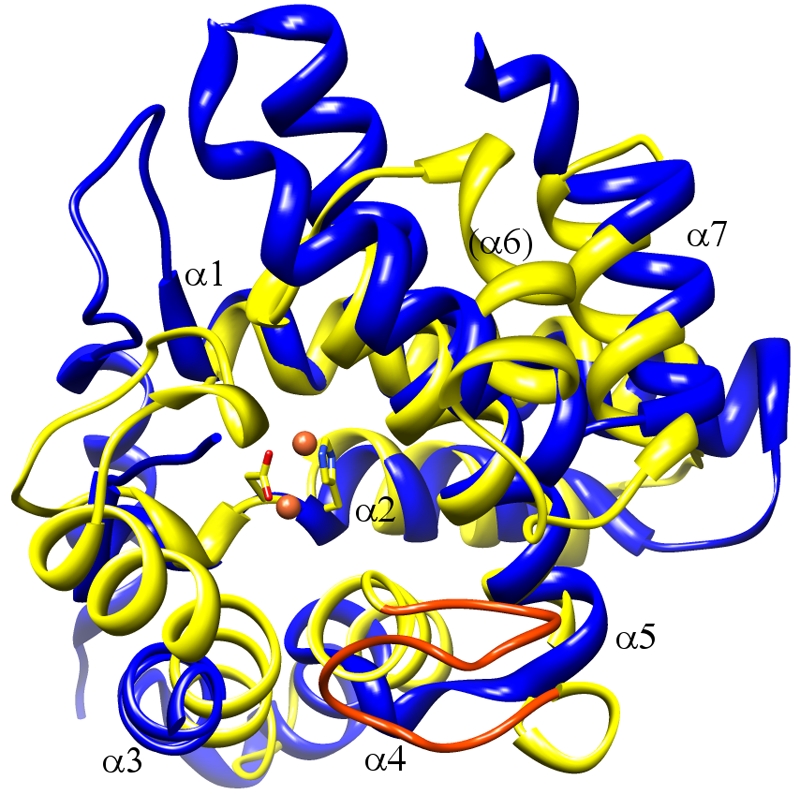
Comparison of the HD-GYP fold with a representative of the wider HD family. Bd1817 (yellow; GYP motif orange) and the HD protein YfbR (PDB code 2PAQ [13]; blue) are shown superimposed, with the HD residues of Bd1817 in stick form and the active-site binuclear metal center in sphere form. It is apparent that all of the α-helices of the HD-GYP fold have counterparts in the generalized HD fold, although α6 sits at an angle different from that of its counterpart in YfbR. The lid region of Bd1817 corresponds to a flexible loop of YfbR (lower left-hand area of image, left unmodeled). Comparatively, the GYP motif of Bd1817 replaces the short interhelical loop of HD proteins (between α4 and α5 equivalents, the 20 residues between Bd1817 aa 236 and 255 contrasting with the 5 residues between YfbR aa 122 and 126).
Binuclear metal-binding pocket.
The active site of the Bd1817 HD-GYP domain contains a binuclear metal arrangement (Fig. 4b, 4c) held in place by conserved residues that were previously highlighted as important in a large-scale alignment of HD-GYP sequences (14). This binuclear arrangement is seen in a subset of HD superfamily members that place liganding residues on the third and fourth helices to create a second site after the classical HD site on helix two (15). Each metal assumes an approximate octahedral geometry (Fig. 4b), with both sites bidentately “bridged” by three separate entities—D184 of the HD motif, a bound solvent molecule (W1), and a tetrahedral ion (with excellent electron density [Fig. 3a], modeled as phosphate, presumably copurified with the protein, as no phosphate was present in buffers or the crystallization conditions). Metal one (M1) is further coordinated by H183 of the HD motif, H150 (from the first helix of the HD domain), and N265. Metal two (M2) is further coordinated by H212 and conserved residues situated immediately prior to the GYP motif (H237 and E238). The nature of the liganding residues, anomalous diffraction experiments at various wavelengths (1, 1.6, and 1.8 Å, used to discriminate among Fe, Zn, and Mn; A. L. Lovering, data not shown), and precedent with homologous HD domain proteins (15) suggest that these two metal sites are Fe ions. Metal preference could, however, be protein/motif specific—a Borrelia burgdorferi phosphodiesterase with a nonconsensus HK-GYP motif was found to have enhanced activity with the addition of Mn2+ (16). The liganding histidines at positions H183, H212, and H237 are oriented via hydrogen bonding of their Nδ1 atoms to helical backbone carbonyl groups, whereas H150 is oriented via interaction with D268, providing an explanation for the conservation of this residue on helix α5.
Structure of the GYP motif.
The region of the fold containing the GYP motif is located near the solvent-exposed end of the active-site pocket (Fig. 7a). The GYP motif is better described as an extended conserved region with the consensus sequence HHExxDGxGYP, as highlighted by Ryan and Dow (17). The Bd1817 protein has the somewhat nonstandard sequence H237EExxNGxG-P, where the protein is “missing” the tyrosine of the classic GYP signature and has a glutamate in place of the second histidine. Nevertheless, this section of the fold is completely ordered in our structures (respective electron density is shown in Fig. 3b) and is located in a larger region (aa 237 to 255) that can be described as a structural insertion into the usual HD superfamily topology. Remarkably, this region (to the best of our knowledge, checked by a ψ/φ angle search using generous tolerance in PDBeMotif [18]) appears to have a novel conformation, forming a pair of U-shaped turns whose planes are approximately 90° apart. Despite resembling a β-meander, the backbone residues in this region make very few classical β-sheet hydrogen bonds, and the carbonyl groups of T240 and L249 point toward each other (2.9 Å apart). A solvent molecule (W2) sits at the core of the two turns, hydrogen bonding to E239, T240, G245, and L249. The first two residues of the extended motif (H237 and E238) complex M2, whereas conserved E239 acts to stabilize the backbone conformation of the second turn by interacting with the amide nitrogens of K247 and L249. Residues N242GTGP (equivalent to the consensus DGxGYP) form the common strand between the two turns and thus are exposed (Fig. 7a). The first conserved glycine, G243, allows for a tight kink in the backbone, and the second, G245, appears to be necessary in order to prevent steric clashes with E238 and E239. The lack of a tyrosine in the Bd1817 GYP sequence precludes a thorough analysis of its role—we speculate that the concurrent alteration of E238 in our structure (replacing the consensus histidine) may hint at coevolution of these residues, and in other HD-GYP proteins, the tyrosine may orient this histidine (at Nδ1) for coordination with M2 (via Nε2). The proximity of this tyrosine location to the active site (G245 is ~12 Å from the bound phosphate) may mean that it also has a role in substrate binding. Mutagenesis of the HD-GYP protein RpfG (17, 19) implicated the GYP residues in binding to GGDEF domains, an event important in fine-tuning levels of c-di-GMP. The presence and exposure of the representative N242GTG-P region in Bd1817 on the extremity of the protein fold are consistent with binding by a partner protein (Fig. 4a). Despite the absence of the conserved tyrosine in our structure, we speculate that backbone atoms will play a large role in such an interaction—all of the carbonyl groups of residues 241 to 248 (across the region which includes the DGxGYP consensus sequence of canonical HD-GYP proteins) are available in our structure.
FIG 7 .

Structure of the GYP region of the fold and possible site for c-di-GMP binding. (a) Detail of the GYP motif (residues 239 to 255; orange), illustrating fold stabilization by the conserved E239 side chain and tightly bound solvent molecule W2 (hydrogen bond interactions represented by dashed green line, backbone amide and carbonyl groups represented by blue and red spheres, respectively). Proline 246 of the GYP motif (G-P for Bd1817, this structure) sits at the edge of the second of two perpendicular U-shaped turns (with glycine 245, N atom shown as a blue sphere, preceding it). The GYP motif is located in a region close to that of the binuclear metal active site (liganding residues are yellow). (b) Possible binding site modeled for a c-di-GMP substrate in HD-GYP proteins. The bound phosphate in the Bd1817 structure was used as a guide to place c-di-GMP (stick form, C atoms magenta), with the 3′ end located proximal to H187 (postulated to protonate this group upon bond cleavage; see text). One of the guanine bases is positioned toward the GYP motif and a region formed by the S262 and H293 residues (consensus A and D, respectively), while the other base clashes with the lid region (green). Regardless of how c-di-GMP is positioned next to the binuclear center, the restricted space available in this region suggests that a small conformational rearrangement of the lid region would be required to accommodate the substrate if this structure of Bd1817 is representative of other HD-GYP active-site geometry. (c) Detail of several nonconsensus residues of Bd1817 (single-letter code, C atoms blue, HD-GYP family consensus in parentheses; derived from Fig. 1), illustrating the position around the predicted c-di-GMP-binding site (in surface representation, transparent to show side chain detail, metal sites and bound phosphate ion). In particular, the consensus arginine at Bd1817 E274 would be well placed to contact the second c-di-GMP phosphate group (oriented toward the top in this view). The nonconsensus GYP motif at aa 245 and 246 has been left unnumbered for clarity.
Mechanistic implications for HD-GYP family proteins.
The metal-bound phosphate ion makes further contacts with residues H187, W209, and R269 and is likely to represent the position of the phosphodiester group of the c-di-GMP substrate in HD-GYP proteins (a model of which is shown in Fig. 7b). The nonconsensus residues of Bd1817 appear to cluster around the predicted site of c-di-GMP binding (Fig. 7c), and conservation at these positions (Fig. 1) is presumably related to interaction with the substrate, as H187, W209, D242, R267, E274, or H293 cannot be said to play an essential structural role, as ascertained from our well-ordered, different crystal forms. In keeping with this observation of deviation from the consensus, purified Bd1817 had no detectable phosphodiesterase activity or c-di-GMP binding in vitro (J. Dahbi and M. Gomelsky, personal communication), a finding that may tally with the nonconsensus nature of several active-site residues apparent from our structure herein (or, less likely, may reflect a lack of appropriate stimulus at the appended NTD). Identification of the role of the NTD of Bd1817 (and also the homologous Bd2325, Bd1762, Bd1822, and Bd3880 Bdellovibrio HD-GYP proteins) would potentially allow mutagenesis experiments to assess the function of the above conserved residues.
Access to the phosphate-binding pocket is restricted by a smaller domain formed by residues 188 to 211 which resembles a “lid” over the active site (Fig. 4a and 7b). The bridging solvent molecule (W1) is located 2 Å from M1 and 1.9 Å from M2 and is therefore likely to be a hydroxide ion and not H2O or a μ-oxo species (15). The EAL c-di-GMP phosphodiesterases possess a binuclear metal center with softer metal ions (e.g., the Mn2+ of BlrP1; hence, acidic residues dominate the metal-binding pocket, in contrast to the histidines of Bd1817) and utilize an active-site lysine residue to assist in the generation of hydroxide species (20, 21). The Bd1817 structure reveals that HD-GYP proteins probably utilize one of the M1 coordinating residues for hydroxide activation, as the Nd2 group of N265 is 2.9 Å from W1 (Fig. 4b). Most HD-GYP proteins possess an aspartate residue at the position equivalent to N265 which would be able to perform acid-base catalysis and be functionally homologous to similarly placed residues in other binuclear phosphodiesterases (e.g., D392 of PDE4 [22]). Like the proposed EAL enzymatic mechanism (20, 21), the bridging hydroxide is positioned to perform an in-line nucleophilic attack on the phosphorus atom (of the bound c-di-GMP substrate; W1 is 3.1 Å from the bound phosphate ion). The direct coordination of the phosphate oxygens by M1 (2.1 Å) and M2 (2.1 Å) will also polarize the substrate and assist in bond breakage and transition state stabilization (formation of a pentacoordinate intermediate). With the leaving group (O3′) situated opposite the attacking species (hydroxide), the Bd1817 structure predicts that the O3′ end of the c-di-GMP substrate would face toward L194 and F273, with the O5A end oriented toward E238 and the GYP motif (Fig. 7b). A consensus arginine (corresponding to Bd1817 E274, at the end of α5) is located opposite the edge of the lid, and its conservation and position suggest possible complexation with the second phosphodiester group of c-di-GMP. Leaving group protonation in EAL enzymes has been postulated to occur via a water-acidic residue relay (20, 21), but the precedent in HD superfamily proteins is to use a residue one helical turn on from the HD motif (e.g., HDxxE72 in YfbR, correlating with no observable activity for the E72A mutant [13]). The equivalent residue in Bd1817 is H187, which hydrogen bonds to the M1-interacting phosphate oxygen atom, not the atom corresponding to the c-di-GMP O3′ group. This situation may be altered in other members of the HD-GYP family, where the consensus residue at this position is lysine, with its longer side chain; indeed, the HDxxK127 region in the HD superfamily protein myo-inositol oxygenase is in such a “productive” orientation (15). The lack of space around the non-metal-complexed oxygens of the bound phosphate ion suggests that a conformational change would be necessary to accommodate the c-di-GMP substrate, commensurate with the proposed regulation of HD-GYP activity by appended sensory domains (2). A small rearrangement of the lid region would allow an expansion of the active-site cleft, and the analogous region in other HD superfamily proteins has been shown to envelop the substrate in a lid-like manner (15) and also to be subject to mobility/disorder (e.g., the unstructured nature of this loop in YfbR [13]). Residues W209 and R269 have smaller counterparts in consensus sequences (Fig. 1 and 7c), which may also aid substrate binding and may be the reason why no c-di-GMP binding was observed—although again, the possibility of a lack of an appropriate stimulus at the NTD cannot be ruled out. Comparatively, the HD-GYP domain active site is likely to “enclose” the substrate (as judged by the relative burial of the binuclear metal center), in contrast to EAL α8β8 barrels, which bind the substrate largely via one face (20). It is tempting to speculate that this enclosure, in tandem with an architecture that results in greater contact with the “proximal,” as opposed to “distal,” phosphate of the cyclic substrate, would endow HD-GYP proteins with relatively greater activity than EAL proteins in hydrolyzing the linear pGpG intermediate occurring during the conversion of c-di-GMP to GMP. Further studies with substrate or substrate analogues are necessary to determine the precise mechanism of recognition and enzymatic regulation, but the general features of the HD-GYP family outlined by the structure of Bd1817 (modularity, active-site architecture, a GYP domain, and a binuclear metal center) will provide a model for investigating other members of this diverse class of signaling proteins.
MATERIALS AND METHODS
Cloning of the Bd1817 gene.
Nucleotide primers 5′ GCAGCGGCCTGGTGCCGCGCGGCAGCCATATGGATTATGTTTCTATACGAGTAAGC 3′ and 5′ CTCAGTGGTGGTGGTGGTGGTGCTCGAGCTAGAGCCCCTTCAGGATGTCGTTCAG 3′ were used to amplify the region encoding Bd1817 (the full-length protein, aa 1 to 308) from B. bacteriovorus HD100 genomic DNA. The product, inserted into expression plasmid pET28b (Novagen) in a restriction-free process (23), was utilized in a second-round PCR. The construct was confirmed by sequencing and introduced into E. coli expression strain BL21 λDE3.
In vivo studies of Bd1817.
The Bd1817 gene was deleted silently to leave the first two and the last three codons in frame, using reciprocal recombination of a construct conjugated into B. bacteriovorus HD100 from suicide vector pK18mobsacB, and exconjugants were screened by Southern blotting and reverse transcription (RT)-PCR to verify the gene deletion. The mobilizable vector pSUP404.2 (24) was used to express Bd1817 with 200 bp of up- and downstream DNA for complementation studies. RT-PCR was used to confirm gene expression during complementation by this plasmid construct, and RT-PCR by standard methods (25) was used to detect the expression of Bd1817 during predatory and axenic growth. The predicted open reading frame of Bd1817 without the stop codon was fused to the mCherry gene with a 5-aa linker, and this construct was cloned into pK18mobsacB. This construct was conjugated into B. bacteriovorus HD100 to recombine with the genomic copy of the gene, resulting in a tagged gene with the nascent promoter and a promoterless second copy of the gene. Kanamycin selection maintained this construct.
Protein expression and purification.
Cells were grown at 37°C until they reached an optical density at 600 nm of ~0.8. Gene expression was then induced with 0.2 mM isopropyl-β-d-thiogalactopyranoside for 21 h at 20°C. Harvested cells (approximately 10 g from a 1.5-liter cell culture in 2× yeast extract-tryptone medium) were resuspended by tumbling in 30 ml buffer A (20 mM HEPES [pH 7.2], 0.3 M NaCl, 20 mM imidazole, 5% [vol/vol] glycerol) and lysed using sonication. Unbroken cells were pelleted by centrifugation at 6,000 × g for 20 min, the supernatant was clarified by a second centrifugation at 200,000 × g for 1.5 h, and the final supernatant was applied to a 1-ml Hi-Trap His column preequilibrated in buffer A. Fractions were eluted in a stepwise manner using buffer A containing 40 and 300 mM imidazole. Approximately pure fractions of Bd1817 were dialyzed overnight in buffer B (10 mM Tris [pH 7.2], 150 mM NaCl) and concentrated to a protein concentration of ~40 mg/ml. The final yield from 1.5 liters of cell culture was ~80 mg of purified protein. Bd1817 protein (and crystals) was noticeably yellow in color.
Crystallization and cryoprotection.
Crystals were grown by the hanging-drop method at 18°C using 1 μl of protein solution mixed with an equal volume of reservoir solution. Initial crystallization conditions were identified, giving several crystal forms—P21A (0.1 M bis-Tris [pH 5.5], 0.2 M ammonium acetate, 25% [wt/vol] PEG 3350, 20% [vol/vol] dimethyl sulfoxide [DMSO]), P21B (identical to the P21A condition except that no DMSO was used), P1 (identical to the P21B condition except for a doubling to 0.4 M ammonium acetate), and P3121 (2.1 M dl-malic acid [pH 7.0]). Cryoprotection was attained by the sequential addition of increments of mother liquor supplemented with 20% (vol/vol) ethylene glycol, followed by subsequent flash cooling in liquid nitrogen.
Data collection and structure determination.
Diffraction data (Table 1) were collected on a home source (P21B and P1 crystal forms) and at beamline ID 23-1 of the European Synchrotron Radiation Facility (ESRF), Grenoble, France (P21A and P3121 crystal forms). Data were processed using XDS (26) and scala (27), and data file manipulations were performed using the CCP4 suite of programs (28). Initial phasing was accomplished using the P21B crystal form home source data (Cu-Kα, Rigaku Micromax). Despite a relatively low anomalous multiplicity of 5.3, excellent SAD phases were obtained using PHENIX autosolve (29), which located 24 sites at 2.5-Å resolution (4 Fe, 2 PO4, 16 Met sulfur, 2 Cys sulfur). The overall figure of merit for phasing was 0.49. Density modification and 2-fold noncrystallographic symmetry averaging with PHENIX yielded a readily interpretable map, and the molecule was built manually using COOT (30). Chain A from the P21B crystal form was used as a search model to determine the other crystal form structures by molecular replacement with PHASER (31). All crystal forms were refined using PHENIX (29) and possess essentially identical structures. The final models are of excellent stereochemical quality (Table 1). Structural figures were prepared using Chimera (32).
Protein structure accession number.
The atomic coordinates and structure factors determined in this study have been deposited in the Protein Data Bank (PDB; http://www.rcsb.org) under the following PDB ID codes: 3TM8, P21A; 3TMB, P21B; 3TMC, P1; 3TMD, P3121.
SUPPLEMENTAL MATERIAL
Fluorescence phase-contrast imaging of Bd1817 with a C-terminal mCherry fluorescent tag. Host-independent axenic cultures were grown overnight to an optical density at 600 nm of ~1, harvested by centrifugation, resuspended in fresh peptone-yeast extract broth, and incubated for a further 2 h with shaking at 200 rpm at 29°C before being imaged with a Nikon Eclipse E600 with 1,000× phase-contrast and epifluorescence optics using the Simple PCI program. Cells of different morphology (typical of host-independent cells) demonstrated a small but significant fluorescence signal, which was brighter in elongated cells, demonstrating that the tagged Bd1817 protein was expressed in the cells. B, bright-field image; F, fluorescence image; M, merged image. Download Figure S1, DOC file, 3.3 MB.
ACKNOWLEDGMENTS
We acknowledge the use of beamline ID 23-1 at the ESRF, Grenoble, France, as part of a Block Allocation Group. We thank Mark Gomelsky (University of Wyoming) for personal communication of c-di-GMP activity studies.
This work was supported in part by Royal Society research grant RRCI14995 to A.L.L. and BBSRC grant GO13632/1 to R.E.S.
Footnotes
Citation Lovering AL, Capeness MJ, Lambert C, Hobley L, Sockett RE. 2011. The structure of an unconventional HD-GYP protein from Bdellovibrio reveals the roles of conserved residues in this class of cyclic-di-GMP phosphodiesterases. mBio 2(5):e00163-11. doi:10.1128/mBio.00163-11.
REFERENCES
- 1. Römling U, Gomelsky M, Galperin MY. 2005. C-di-GMP: the dawning of a novel bacterial signalling system. Mol. Microbiol. 57:629–639 [DOI] [PubMed] [Google Scholar]
- 2. Schirmer T, Jenal U. 2009. Structural and mechanistic determinants of c-di-GMP signalling. Nat. Rev. Microbiol. 7:724–735 [DOI] [PubMed] [Google Scholar]
- 3. Ross P, et al. 1987. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature 325:279–281 [DOI] [PubMed] [Google Scholar]
- 4. Amikam D, Galperin MY. 2006. PilZ domain is part of the bacterial c-di-GMP binding protein. Bioinformatics 22:3–6 [DOI] [PubMed] [Google Scholar]
- 5. Ryan RP, et al. 2006. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc. Natl. Acad. Sci. U. S. A. 103:6712–6717 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6. Stolp H, Starr MP. 1963. Bdellovibrio bacteriovorus gen. et sp. n., a predatory, ectoparasitic, and bacteriolytic microorganism. Antonie Van Leeuwenhoek 29:217–248 [DOI] [PubMed] [Google Scholar]
- 7. Thomashow MF, Cotter TW. 1992. Bdellovibrio host dependence: the search for signal molecules and genes that regulate the intraperiplasmic growth cycle. J. Bacteriol. 174:5767–5771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rendulic S, et al. 2004. A predator unmasked: life cycle of Bdellovibrio bacteriovorus from a genomic perspective. Science 303:689–692 [DOI] [PubMed] [Google Scholar]
- 9. Lambert C, et al. 2006. Characterizing the flagellar filament and the role of motility in bacterial prey-penetration by Bdellovibrio bacteriovorus. Mol. Microbiol. 60:274–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ryan RP, et al. 2010. Cell-cell signal-dependent dynamic interactions between HD-GYP and GGDEF domain proteins mediate virulence in Xanthomonas campestris. Proc. Natl. Acad. Sci. U. S. A. 107:5989–5994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jin R, et al. 2005. Exo84 and Sec5 are competitive regulatory Sec6/8 effectors to the RalA GTPase. EMBO J. 24:2064–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wassmann P, et al. 2007. Structure of BeF3-modified response regulator PleD: implications for diguanylate cyclase activation, catalysis, and feedback inhibition. Structure 15:915–927 [DOI] [PubMed] [Google Scholar]
- 13. Zimmerman MD, Proudfoot M, Yakunin A, Minor W. 2008. Structural insight into the mechanism of substrate specificity and catalytic activity of an HD-domain phosphohydrolase: the 5′-deoxyribonucleotidase YfbR from Escherichia coli. J. Mol. Biol. 378:215–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Galperin MY, Natale DA, Aravind L, Koonin EV. 1999. A specialized version of the HD hydrolase domain implicated in signal transduction. J. Mol. Microbiol. Biotechnol. 1:303–305 [PMC free article] [PubMed] [Google Scholar]
- 15. Brown PM, et al. 2006. Crystal structure of a substrate complex of myo-inositol oxygenase, a di-iron oxygenase with a key role in inositol metabolism. Proc. Natl. Acad. Sci. U. S. A. 103:15032–15037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sultan SZ, et al. 2011. Analysis of the HD-GYP domain cyclic dimeric GMP phosphodiesterase reveals a role in motility and the enzootic life cycle of Borrelia burgdorferi. Infect. Immun. 79:3273–3283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ryan RP, DOW JM, Intermolecular interactions between HD-GYP and GGDEF domain proteins mediate virulence-related signal transduction in Xanthomonas campestris. 2010. Virulence. 1:404–408 [DOI] [PubMed] [Google Scholar]
- 18. Golovin A, Henrick K. 2008. MSDmotif: exploring protein sites and motifs. BMC Bioinform. 9:312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andrade MO, et al. 2006. The HD-GYP domain of RpfG mediates a direct linkage between the Rpf quorum-sensing pathway and a subset of diguanylate cyclase proteins in the phytopathogen Xanthomonas axonopodis pv citri. Mol. Microbiol. 62:537–551 [DOI] [PubMed] [Google Scholar]
- 20. Barends TR, et al. 2009. Structure and mechanism of a bacterial light-regulated cyclic nucleotide phosphodiesterase. Nature 459:1015–1018 [DOI] [PubMed] [Google Scholar]
- 21. Tchigvintsev A, et al. 2010. Structural insight into the mechanism of c-di-GMP hydrolysis by EAL domain phosphodiesterases. J. Mol. Biol. 402:524–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huai Q, Colicelli J, Ke H. 2003. The crystal structure of AMP-bound PDE4 suggests a mechanism for phosphodiesterase catalysis. Biochemistry 42:13220–13226 [DOI] [PubMed] [Google Scholar]
- 23. van den Ent F, Löwe J. 2006. RF cloning: a restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 67:67–74 [DOI] [PubMed] [Google Scholar]
- 24. Roschanski N, Strauch E. 2011. Assessment of the mobilizable vector plasmids pSUP202 and pSUP404.2 as genetic tools for the predatory bacterium Bdellovibrio bacteriovorus. Curr. Microbiol. 62:589–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Evans KJ, Lambert C, Sockett RE. 2007. Predation by Bdellovibrio bacteriovorus HD100 requires type IV pili. J. Bacteriol. 189:4850–4859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kabsch W. 2010. XDS . Acta Crystallogr. D Biol. Crystallogr. 66:125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Evans P. 2006. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62:72–82 [DOI] [PubMed] [Google Scholar]
- 28. Collaborative Computational Project , Number 4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed]
- 29. Zwart PH, et al. 2008. Automated structure solution with the PHENIX suite. Methods Mol. Biol. 426:419–435 [DOI] [PubMed] [Google Scholar]
- 30. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 31. McCoy AJ, et al. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pettersen EF, et al. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 33. Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217 [DOI] [PubMed] [Google Scholar]
- 34. Gouet P, Courcelle E, Stuart DI, Métoz F. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fluorescence phase-contrast imaging of Bd1817 with a C-terminal mCherry fluorescent tag. Host-independent axenic cultures were grown overnight to an optical density at 600 nm of ~1, harvested by centrifugation, resuspended in fresh peptone-yeast extract broth, and incubated for a further 2 h with shaking at 200 rpm at 29°C before being imaged with a Nikon Eclipse E600 with 1,000× phase-contrast and epifluorescence optics using the Simple PCI program. Cells of different morphology (typical of host-independent cells) demonstrated a small but significant fluorescence signal, which was brighter in elongated cells, demonstrating that the tagged Bd1817 protein was expressed in the cells. B, bright-field image; F, fluorescence image; M, merged image. Download Figure S1, DOC file, 3.3 MB.