

Abstract
MRI contrast agents providing very high relaxivity values can be obtained through the attachment of multiple gadolinium(III) complexes to the interior surfaces of genome-free viral capsids. In previous studies, the contrast enhancement was predicted to depend on the rigidity of the linker attaching the MRI agents to the protein surface. To test this hypothesis, a new set of Gd-hydroxypyridonate based MRI agents was prepared and attached to genetically introduced cysteine residues through flexible and rigid linkers. Greater contrast enhancements were seen for MRI agents that were attached via rigid linkers, validating the design concept and outlining a path for future improvements of nanoscale MRI contrast agents.
Keywords: Virus, capsid, MRI contrast, gadolinium, HOPO, relaxivity, linker rigidity
Introduction
Magnetic resonance imaging (MRI) is a powerful medical diagnostic technique due to its ability to provide images of the human body with great anatomical detail.1,2 However, its ability to distinguish between two adjacent regions is often limited by the inherently narrow range of relaxation rates for water protons. As a result, a great deal of effort has been directed toward increasing tissue contrast through the administration of contrast enhancement agents prior to scanning. The most effective small molecules for this purpose have contained paramagnetic metal ions, which are capable of improving contrast by shortening the relaxation time of nearby protons.1-6
Gadolinium(III) is most commonly used in MRI contrast agents due to its large magnetic moment and long electronic relaxation time.2 However, due to the high toxicity of its free form, the metal must be complexed tightly by a coordinating agent prior to injection into humans. Current commercially available Gd(III)-based contrast agents are based on poly(aminocarboxylate) chelators (e.g. DOTA and DTPA), which have fairly low relaxivity values (4-5 mM-1s-1 at 60 MHz and 25 °C). As a result, gram quantities are typically injected to obtain the desired level of contrast enhancement.7-9 A further limitation of these agents is their low tissue retention and blood circulation times (less than 30 min), which restrict their use in MRI applications with larger data collection times.10 As a result, these agents have only been successfully used to target sites where they can accumulate in large concentrations. For the selective imaging of biological targets bearing low (e.g. micromolar to nanomolar) concentrations of specific markers, contrast agents with much greater relaxivity values must be combined with signal amplification strategies to generate a sufficient amount of signal difference to allow their detection.3,4
According to the Solomon-Bloembergen-Morgan (SBM) theory of paramagnetic relaxation,11-15 the relaxivity of a Gd-based contrast agent can be enhanced1 in three ways: by increasing the number of water molecules (represented by q) coordinated to the Gd center, by reducing the tumbling rate (1/τR) of the contrast agent, and by keeping the exchange rate of the inner sphere water molecules on Gd (1/τM) at an optimum value (e.g. τM ∼10 ns). Previous studies in our labs have found that Gd(III)-hydroxypyridonate (Gd-HOPO) based contrast agents have a higher number of inner-sphere water molecules (q = 2 or even 3) compared with other commercial agents.9,16,17 Furthermore, HOPO complexes also have optimum water exchange rates, with τM values ranging between 10 and 20 ns.18 By reducing the tumbling rates of these contrast agents, it should be possible to obtain even higher relaxivity values.
The tumbling rates of contrast agents can be reduced via their covalent or non-covalent attachment to macromolecules such as modified proteins,19 dendrimers,20,21 liposomes,22 carbohydrates,23 or synthetic and micellar nanoparticles.4,24 In each case, enhancements in the relaxivity values have been observed. These approaches have the added benefit that they can allow the attachment of multiple contrast agents to each carrier, further increasing the total relaxivity.4
As an alternative strategy, the construction of nanoscale MRI contrast agents from viral capsids has been explored by various laboratories in recent years because of the inherent multivalency of these particles and the slow solution tumbling rates resulting from their large size. In the first example, very high relaxivity values (T1 = 202 mM-1s-1 at 61 MHz) were obtained upon the chelation of gadolinium ions to the protein coat of the cowpea chlorotic mottle virus (CCMV, 28.5 nm diameter).25 However, the dissociation constant for gadolinium was only 31 μM, and clinical applications require much stronger chelation to prevent the release of toxic Gd3+ ions. In another example, the attachment of Magnevist (Gd-DTPA) to the exterior surface of MS2 viral capsids via lysine residue modification26 has resulted in a three-fold increase in relaxivity (14-16.9 mM-1s-1 at 64 MHz) compared to free Magnevist. Copper-catalyzed click chemistry has been used for the covalent attachment of Gd(DOTA) to the exterior lysine residues of cowpea mosaic virus(CPMV) while simultaneously exploiting the natural affinity of the genomic RNA for Gd3+ ions in order to increase the total metal loading. This resulted in relaxivities of 11-15 mM-1s-1 per complex at 64 MHz.27 All these examples demonstrate the exceptional potential of viral capsids for building highly efficient nanoscale contrast agents.
Additional studies by our labs have explored the use of bacteriophage MS2 to construct high-relaxivity MRI agents. The capsid of this virus is icosahedral in symmetry and is comprised of 180 sequence identical protein subunits.28 The resulting spherical shells are 27 nm in diameter and can be prepared in genome-free assembled form through direct protein expression in E. coli.29 Access to the interior surface of the capsid is afforded by thirty-two pores that are 2 nm in diameter, permitting modification of the internal surface residues, Figure 1a,b.30 The interior and exterior surfaces of the MS2 capsid can therefore be modified differentially by targeting a variety of side chain groups, including lysine, tyrosine, and artificial amino acids.29,30,31
Figure 1.
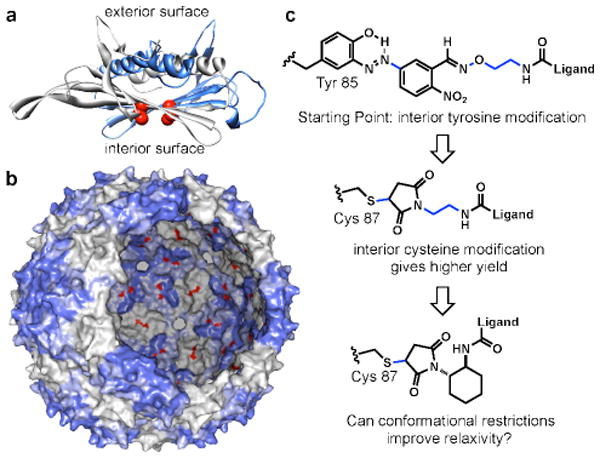
Design of highly efficient MRI contrast enhancement agents. (a) A crystal structure rendering of an MS2 coat protein dimer indicates the positions of the introduced cysteine residues (Cys 87) in red. (b) A rendering of the full 27 nm MS2 capsid shows the interior and exterior surfaces, with the 180 internal N87C positions highlighted in red. (c) The plan for increasing the overall relaxivity involved the use of more efficient cysteine modification chemistry and the rigidification of several rotable bonds (indicated in blue).
We have previously reported the site-specific covalent modification of the exterior and the interior surfaces of MS2 viral capsids with Gd-HOPO-based complexes.32,33 The Gd-complexes were attached to tyrosine residues on the internal surface and to lysine residues on the external surface. Using this approach, we demonstrated the advantage of housing the Gd-HOPO complexes within MS2 capsids to improve their solubility at high levels of modification. We also found that water transport through the capsid shell was fast on the NMR timescale and that the internally modified capsids yielded high relaxivity values (31 mM-1s-1, 25 °C, pH 7, at an external field of 60 MHz). The externally modified capsids gave lower relaxivity values, which was attributed to the increased flexibility of the lysine modification products relative to those formed with the internal tyrosine residues.
In order to convert the MS2 capsids to imaging agents for targeted imaging, we sought to improve the relaxivity further by both increasing the bioconjugation efficiency of the Gd complexes (resulting in the attachment of more contrast agents to each capsid) and by rigidifying the linker that attaches them to the protein scaffold. We recently reported the use of a mutagenically introduced cysteine at position 87 (the N87C mutant shown in Figure 1a,b) and targeted this residue with maleimide reagents to introduce drug molecules34,35 or imaging agents36 to the interior surface of the capsids with high efficiency (approaching 100%). Based on this success, this site was also chosen for modification with Gd-HOPO complexes through the use of linking groups that possessed a minimal number of rotatable bonds. It was found that these design concepts were indeed successful, providing capsids with effective relaxivities as high as 7416 mM-1s-1 at 60 MHz while maintaining the high affinity for the gadolinium ions that is conferred by the HOPO ligands.
Results and Discussion
To test the hypothesis that increasing the linker rigidity would increase the relaxivity, we designed HOPO-based contrast agents bearing maleimide groups through either a flexible linear linker or a rigid cyclohexyl linker. The synthesis of the newly designed cysteine reactive HOPO-contrast agents involved the attachment of a linker containing a terminal maleimide group to the heteropodal trisaminoethylamine(TREN)-bis(HOPO)-terephthalimide(TAM) moiety (Scheme 1). The TAM starting material was activated as the pentafluorophenyl (PFP) ester (2). This molecule was then treated with linear maleimide-amine 3a37 to give TAM-PFP-ethyl-maleimide 4a. This compound was next coupled to TREN-bis-HOPO-(OBn)2 (5) to yield the benzyl protected ligand. Benzyl deprotection with concentrated hydrochloric and acetic acid was not possible, as it led to chloride addition to the maleimide double bond. Optimization of this reaction using a mixture of 50% trifluoroacetic acid in dichloromethane with 10% thioanisole as a scavenger afforded fully deprotected ligand 6a with minimal amounts of side products. The material was purified via ether precipitation and used without further purification.
Scheme 1.
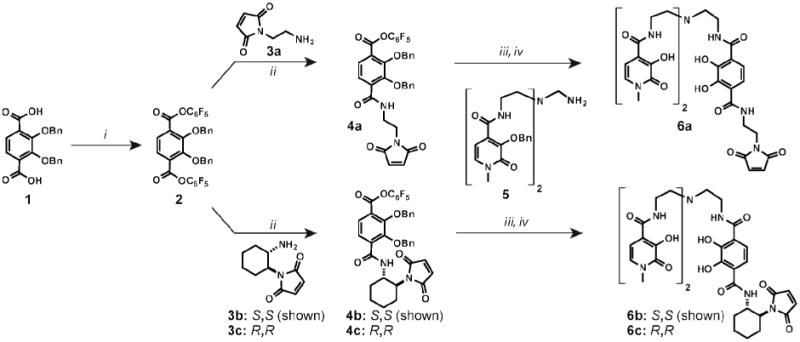
Synthesis of cysteine reactive HOPO-based contrast agents. Reagents and conditions: (i) PFP-trifluoroacetate, DIPEA, dry THF, 77 %; (ii) 3a, 3b, or 3c, DIPEA, DCM, slow addition, rt, 24 h. 4a=69%, 4b=66%, 4c=67%; (iii) 5, DIPEA, DCM, rt, 3 h; (iv) TFA:DCM (1:1), 10% thioanisole, rt, 3 h, followed by ether precipitation. 6a=50%, 6b=55%, 6c=56% (after two steps). DIPEA = N,N-diisopropylethyl amine.
For the synthesis of the rigid linker, the two enantiomers of trans-1,2-cyclohexyldiamine were converted to corresponding maleimide-amines 3b and 3c in three steps (see the Supporting Information for details). Both enantiomers were used in order to determine if there would be a difference in relaxivity due to their interaction with the chiral capsid protein. Activated PFP ester 2 was reacted with 3b and 3c and then deprotected to yield rigid ligands 6b(S,S) and 6c(R,R) in a sequence similar to that used to access the linear linker.
Exposure of N87C MS2 capsids to 20 equiv of ligands 6a-6c (based on protein monomer concentration) for 2-3 h led to virtually quantitative modification, resulting in 180 copies of the HOPO-ligand attached to the inside surface of each capsid. The extent of attachment was determined using LC/ESI-MS following disassembly of the capsids (see Figure 2 and Supporting Information). This level of modification therefore provided a two-fold increase in bioconjugation efficiency over the previous tyrosine-based strategy. There was no detectable protein precipitation and the capsids remained soluble despite the high loading of the hydrophobic ligand. Capsid assembly (both before and after the introduction of Gd(III) ions) was verified using TEM, size-exclusion chromatography (SEC), and dynamic light scattering (DLS) measurements (see Supporting Information).
Figure 2.
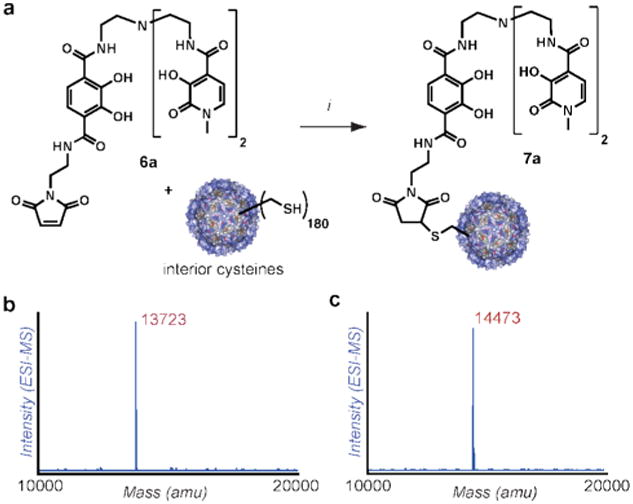
Bioconjugation of HOPO-maleimide 6a to N87C MS2 and mass spectral characterization of the MS2-conjugates (See Supporting Information for additional spectra). (a) Conditions: 10 mM phosphate, TRIS buffer pH 8, 6a (20 eq), 3h. (b) ESI-MS reconstruction of MS2 N87C monomers after capsid dissociation. Expected mass: [M+H+] = 13719 m/z. (c) ESI-reconstruct of linear linker protein conjugate 7a, showing virtually complete conversion (corresponding to 180 copies per capsid). Expected mass of HOPO-linear-MS2: [M+H+] = 14469 m/z. All mass values agreed to within 0.03% of those expected. Full charge ladders appear in Figure S1 of the Supporting Information.
Next, the conjugates were metallated with a solution containing 0.9-1.0 eq of GdCl3 to afford the corresponding Gd complexes. Initial control experiments in which the targeted cysteine residues were capped with N-ethyl maleimide before exposure to GdCl3 resulted in a significant amount of background metal binding, Table 1. Dialysis against a citrate solution and size exclusion chromatography did not remove this non-specifically bound Gd, with ∼49% of the amount added remaining. This indicated that the N87C capsids have a non-negligible affinity for gadolinium ions without a strong affinity ligand like HOPO being present in solution.
Table 1.
Quantitation of Gd(III) binding to HOPO-modified viral capsids.
| Entry | Sample | [Protein] (μM)a | [Gd] (uM)b | %Gd bound |
|---|---|---|---|---|
| 1 | MS2-NEMc | 413 | 201 | 49 |
| 2 | MS2 + HOPO-EAd | 413 | 20 | 5 |
| 3 | MS2-HOPO-Lin (7a) | 41 | 41.1±0.4 | >95% |
| 4 | MS2-HOPO-S,S (7b) | 33 | 33.0±0.2 | >95% |
| 5 | MS2-HOPO-R,R (7c) | 23 | 20.6±0.4 | 89 |
Protein concentrations were determined using UV/vis, and are taken to be within 10% of the actual values.
Gadolinium concentrations were determined using ICP-AES. Control reactions were conducted to check for non-specific binding.
These involved N-ethyl maleimide capped MS2 treated with 0.95-1 equiv. of GdCl3 (entry 1)
N-ethyl maleimide capped MS2 treated with 0.95-1 equiv. of GdCl3 and 1 equiv. of free HOPO-ethyl amine (Entry 2).
However, it was found that background binding did not occur in the presence of one equivalent of free bis-HOPO-TREN-TAM-ethyl amine ligand17—conditions that more accurately represented the metallation reactions with conjugates 7a-7c. This ligand is similar to the HOPO-maleimide ligands that were synthesized, but lacked the bioconjugation handle. In this case, we observed that only a minimal amount (<5%) of the Gd3+ added remained non-specifically bound to MS2.
To prepare the MS2-HOPO-Gd(III) conjugates under analogous conditions, capsids bearing internal HOPO ligands (MS2-HOPO-Lin, MS2-HOPO-R,R and MS2-HOPO-S,S) were prepared and concentrated using ultrafiltration. The complexes were next metallated using 0.95 eqivalents of GdCl3 relative to the estimated concentration of protein after the ultrafiltration step (which could vary by about 10%). Following complex formation, any remaining Gd3+ was removed by gel filtration and dialysis against a solution of ammonium citrate (as a low-affinity competitive ligand), followed by overnight dialysis against 12.5 mM HEPES buffer at pH 7. To ensure accurate relaxivity measurements, the Gd-content of the resulting capsid samples was measured using inductively coupled plasma-atomic emission spectroscopy (ICP-AES), and the protein concentrations were independently determined using UV-vis spectroscopy, Table 1. The comparison of these values indicated a high degree of Gd incorporation (>95% for MS2-HOPO-Lin (7a) and MS2-HOPO-S,S (7b) and 89% for MS2-HOPO-R,R (7c)). DLS and SEC analyses confirmed that the capsids remained stable and intact after metallation (see the Supporting Information for full characterization of the capsid conjugates).
The relaxivity values of the bioconjugates were measured to quantify the effect of linker rigidity on contrast enhancement. T1 relaxation times of the protein conjugate solutions (approximately 200 μM in [Gd3+]) were measured at 60 MHz field strengths using an inversion recovery pulse sequence. The relaxivity values for the capsids conjugated to the linear and rigid linkers were calculated using the following equation:
| Equation 1 |
where [Gd] represents the metal concentration measured by ICP-AES and T1d is the intrinsic diamagnetic solvent relaxation time. The relaxivity values were measured in triplicate at pH 7 in 12.5 mM HEPES solution at 25 °C as well as the more physiologically relevant temperature of 37 °C (Table 2).
Table 2.
Comparison of relaxivity values at 60 MHz.a
| Sample | Relaxivity at 60 MHz (mM-1s-1)b | Relaxivity/capsid at 60 MHz (mM-1s-1)c | ||
|---|---|---|---|---|
| 25 °C | 37 °C | 25 °C | 37 °C | |
| Lin (Gd-7a) | 32.6 ± 0.1 | 29.7 ± 0.1 | 5868 | 5346 |
| SS (Gd-7b) | 41.2 ± 0.2 | 38.2 ± 0.6 | 7416 | 6876 |
| RR (Gd-7c) | 29.6 ± 0.1 | 25.4 ± 0.3 | 4736 | 4064 |
Relaxivity values were measured in 12.5 mM HEPES (pH 7) at 25 °C and 37 °C for the MS2-Gd-HOPO conjugates.
Reported per Gd(III) complex.
The total relaxivities of the full capsids were calculated assuming 180 Gd(III) complexes for 7a, 180 Gd(III) complexes for 7b, and 160 Gd(III) complexes for 7c. These estimates are taken to be within 5-10% of the actual values, based on the errors associated with protein concentration determination.
The measured relaxivity values indicated that increasing the linker rigidity had a positive effect on contrast enhancement in some cases. The (S,S)-linked rigid conjugate gave a higher relaxivity value (41 mM-1s-1 at 60 MHz) compared with the (R,R) linker (30 mM-1s-1, 60 MHz) and the linear linker (33 mM-1s-1, 60 MHz). Also, due to the nearly quantitative conversion achieved in the maleimide coupling reaction, the total relaxivity per capsid was as high as 7416 mM-1s-1 for MS2-HOPO-S,S (7c). The lysine and tyrosine bioconjugates from our previous work gave relaxivity values of 23.2 and 30.9 mM-1s-1, respectively, at 60 MHz.32,33 Since each capsid contained only ∼95 complexes, total relaxivities of up to 2900 mM-1s-1 were observed. The current MS2-Gd-HOPO conjugates therefore possess up to 2.5-fold higher overall relaxivity through the combination of rigidification effects and higher loading.
A marked difference in relaxivity was observed for the two enantiomeric ligands when attached to the protein. This discrepancy is reasonable considering the intrinsic chirality of the protein, which will experience complex and different interactions with the two enantiomeric linkers. Previous studies38-42 have reported differences in water exchange rates for diastereomeric small molecule contrast agents that include Gd3+. In particular, Burai et al.38 have observed differential water-exchange rates for the diastereomers of [Gd(EPTPA-bz-NH2)H2O)]2- and [Gd(DTPA-bz-NH2)H2O)]2-. They predicted that upon slowing the tumbling rates of these diastereomers, the differences in water-exchange rates would be reflected in the relaxivity values. Caravan et al.39 have studied the diastereomers of the contrast agent MS-325, which can bind non-covalently to human serum albumin (hSA) and give different relaxivity enhancements for the two diastereomers. The authors presented detailed NMRD studies on the complexes and predicted that the relaxivity differences were also possibly due to differences in water-exchange rates between the two diastereomers.
To clarify the way in which the chiral linkers alter the interactions of the complexes with the protein surface, molecular models were generated starting with the crystal structure of the MS2 coat protein.28 The Gd(HOPO) complexes were based on a reported crystal structure43 and altered to display the linkers corresponding to 7b and 7c. Each complex was generated as a set of three possible coordination isomers (defined in Supporting Information Figure S5). For the modeling studies, two copies of each complex were attached to an adjacent pair of C2-symmetric Cys 87 residues (green in Figure 3 c-f). The structures were minimized using MacroModel, using no constraints on the metal complexes or the amino acids within a 20 Å radius.
Figure 3.
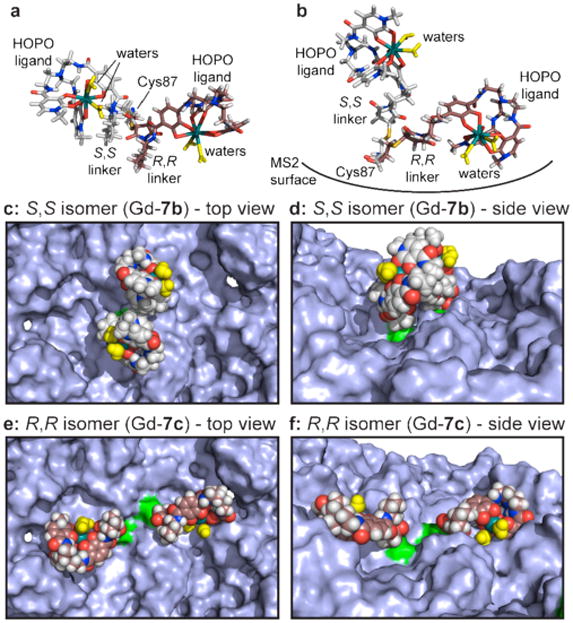
Models of sterically hindered Gd-HOPO complexes attached to the interior MS2 surface. Structures attached to two adjacent Cys87 groups were minimized simultaneously using MacroModel, and three different coordination isomers were considered for each. The Gd coordination spheres were based on the crystal structure reported in reference 43. Representative structures are shown here, with the full set appearing in Supporting Information Figures S5 and S6. Overlays of S,S (Gd-7b, gray) and R,R (Gd-7c, brown) complexes are shown for (a) a top view and (b) a side view. The rigid linker of S,S-complex Gd-7b (c and e) forces an upright conformation that leaves the water molecules (yellow) unobstructed. R,R-complex Gd-7c (d and f) places the water molecules much closer to the capsid surface, likely restricting their exchange. The Cys 87 attachment points are shown in green in c-f.
Representative results for Gd-7b and Gd-7c complexes are shown in Figure 3, with the full set appearing in Supporting Information Figure S8. Little change was observed for the protein backbone or the gadolinium coordination geometries, but the orientations of the cysteine 87 side chains were altered somewhat in response to the linker chirality, Figure 3a,b. Interestingly, the chiral 1,2-diaminocyclohexane groups exhibited striking effects on the display of the ligands attached to the sulfur atoms, with the S,S-linker (Gd-7b) orienting the complexes above the protein surface, Figure 3c,d. This likely leads to unencumbered exchange of the bound water molecules with the bulk solvent. In contrast, the R,R-linker (Gd-7c) flips the complex toward the protein surface (Figure 3b). This geometry places significant constraints on the water molecules and may allow the protein side chains to compete with their binding. Either of these effects could explain the lower overall relaxivity of the R,R-complexes. The S,S-linkages also generated markedly increased interactions between the complexes, which could further restrict their conformational flexibilities. These observations were consistent throughout the full series of complexes that were modeled. In ongoing studies we are analyzing the temperature dependence of relaxivity and the NMRD profiles for these capsid-attached complexes, with the goal of gaining further insight into the nature of these differences.
Conclusion
These studies demonstrate that increasing the rigidity of the linker between Gd-containing contrast agents and a viral capsid scaffold can lead to significant increases in relaxivity properties. The obtained relaxivity values are some of the highest reported for high-affinity Gd3+ complexes, paving the way for their future use in applications that target non-abundant biological markers. Current efforts are focused on the addition of cell binding ligands on the external surfaces of the assemblies35 and the determination of the detection limits associated with these complexes under physiological conditions.
Supplementary Material
Acknowledgments
The work by P.G., D.W.R., and M.B.F. was supported by the Director, Office of Science, Materials Sciences and Engineering Division, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. A.D. and K.N.R. were supported by the NIH (HL069832). We also thank Prof. Christopher J. Chang and his laboratory for advice and instrumentation access. Prof. Mauro Botta and Dr. Kathy Durkin are also acknowledged for helpful discussion.
Footnotes
Supporting Information Available: Full synthetic procedures are provided, as are descriptions of the analysis techniques. A summary of the molecular modeling techniques is also included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Caravan P. Chem Soc Rev. 2006;35:512–523. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- 2.Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Chem Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 3.Major JL, Meade TJ. Acc Chem Res. 2009;42:893–903. doi: 10.1021/ar800245h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.L Villaraza AJ, Bumb A, Brechbiel MW. Chem Rev. 2010;110:2921–2959. doi: 10.1021/cr900232t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Werner E, Datta A, Jocher C, Raymond K. Angew Chem Int Ed. 2008;47:8568–8580. doi: 10.1002/anie.200800212. [DOI] [PubMed] [Google Scholar]
- 6.Chan KWY, Wong WT. Coord Chem Rev. 2007;251:2428–2451. [Google Scholar]
- 7.Bottrill M, Nicholas LK, Long NJ. Chem Soc Rev. 2006;35:557–571. doi: 10.1039/b516376p. [DOI] [PubMed] [Google Scholar]
- 8.Lowe MP. Aust J Chem. 2002;55:551–556. [Google Scholar]
- 9.Datta A, Raymond KN. Acc Chem Res. 2009;42:938–947. doi: 10.1021/ar800250h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinmann HJ, Press WR, Gries H. Inves Radiol. 1990;25:S49–S50. doi: 10.1097/00004424-199009001-00023. [DOI] [PubMed] [Google Scholar]
- 11.Bloembergen N. J Chem Phys. 1956;27:572–573. [Google Scholar]
- 12.Bloembergen N. Phys Rev. 1956;104:1542–1547. [Google Scholar]
- 13.Bloembergen N, Morgan LO. J Chem Phys. 1961;34:842–850. [Google Scholar]
- 14.Solomon I. Phys Rev. 1955;99:559–565. [Google Scholar]
- 15.Solomon I, Bloembergen N. J Chem Phys. 1956;25:261–266. [Google Scholar]
- 16.Xu J, Franklin SJ, Whisenhunt DW, Raymond KN. J Am Chem Soc. 1995;117:7245–7246. [Google Scholar]
- 17.Pierre VC, Botta M, Aime S, Raymond KN. Inorg Chem. 2006;45:8355–8364. doi: 10.1021/ic061262q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson MK, Botta M, Nicolle G, Helm L, Aime S, Merbach AE, Raymond KN. J Am Chem Soc. 2003;125:14274–14275. doi: 10.1021/ja037441d. [DOI] [PubMed] [Google Scholar]
- 19.Yang JJ, Yang JH, Wei LX, Zurkiya O, Yang W, Li SY, Zou J, Zhou YB, Maniccia ALW, Mao H, Zhao FQ, Malchow R, Zhao SM, Johnson J, Hu XP, Krogstad E, Liu ZR. J Am Chem Soc. 2008;130:9260–9267. doi: 10.1021/ja800736h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pierre VC, Botta M, Raymond KN. J Am Chem Soc. 2005;127:504–505. doi: 10.1021/ja045263y. [DOI] [PubMed] [Google Scholar]
- 21.Langereis S, de Lussanet QG, van Genderen MHP, Meijer EW, Beets-Tan RGH, Griffioen AW, van Engelshoven JMA, Backes WH. NMR Biomed. 2006;19:133–141. doi: 10.1002/nbm.1015. [DOI] [PubMed] [Google Scholar]
- 22.Strijkers GJ, Mulder WJM, van Heeswijk RB, Frederik PM, Bomans P, Magusin P, Nicolay K. Magn Reson Mat Phys Biol Med. 2005;18:186–192. doi: 10.1007/s10334-005-0111-y. [DOI] [PubMed] [Google Scholar]
- 23.Claude BSirlin, David RVera, Jacqueline ACorbeil, Marlon BCaballero, Richard BBuxton, Robert FMattrey. Acad Radiol. 2004;11:1361–1369. [Google Scholar]
- 24.Manus LM, Mastarone DJ, Waters EA, Zhang X, Schultz-Sikma EA, MacRenaris KW, Ho D, Meade TJ. Nano Lett. 2010;10:484–489. doi: 10.1021/nl903264h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allen M, Bulte JWM, Liepold L, Basu G, Zywicke HA, Frank JA, Young M, Douglas T. Magn Reson Med. 2005;54:807–812. doi: 10.1002/mrm.20614. [DOI] [PubMed] [Google Scholar]
- 26.Anderson EA, Isaacman S, Peabody DS, Wang EY, Canary JW, Kirshenbaum K. Nano Lett. 2006;6:1160–1164. doi: 10.1021/nl060378g. [DOI] [PubMed] [Google Scholar]
- 27.Prasuhn DE, Yeh RM, Obenaus A, Manchester M, Finn MG. Chem Commun. 2007:1269–1271. doi: 10.1039/b615084e. [DOI] [PubMed] [Google Scholar]
- 28.Valegard K, Liljas L, Fridborg K, Unge T. Nature. 1990;345:36–41. doi: 10.1038/345036a0. [DOI] [PubMed] [Google Scholar]
- 29.Carrico ZM, Romanini DW, Mehl RA, Francis MB. Chem Commun. 2008:1205–1207. doi: 10.1039/b717826c. [DOI] [PubMed] [Google Scholar]
- 30.Hooker JM, Kovacs EW, Francis MB. J Am Chem Soc. 2004;126:3718–3719. doi: 10.1021/ja031790q. [DOI] [PubMed] [Google Scholar]
- 31.Kovacs EW, Hooker JM, Romanini DW, Holder PG, Berry KE, Francis MB. Bioconjugate Chem. 2007;18:1140–1147. doi: 10.1021/bc070006e. [DOI] [PubMed] [Google Scholar]
- 32.Hooker JM, Datta A, Botta M, Raymond KN, Francis MB. Nano Lett. 2007;7:2207–2210. doi: 10.1021/nl070512c. [DOI] [PubMed] [Google Scholar]
- 33.Datta A, Hooker JM, Botta M, Francis MB, Aime S, Raymond KN. J Am Chem Soc. 2008;130:2546–2552. doi: 10.1021/ja0765363. [DOI] [PubMed] [Google Scholar]
- 34.Wu W, Hsiao SC, Carrico Z, Francis M. Angew Chem Int Ed. 2009;48:9493–9497. doi: 10.1002/anie.200902426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stephanopoulos N, Tong GJ, Hsiao SC, Francis MB. ACS Nano. 2010;4:6014–6020. doi: 10.1021/nn1014769. [DOI] [PubMed] [Google Scholar]
- 36.Meldrum T, Seim KL, Bajaj VS, Palaniappan KK, Wu W, Francis MB, Wemmer DE, Pines A. J Am Chem Soc. 2010;132:5936–5937. doi: 10.1021/ja100319f. [DOI] [PubMed] [Google Scholar]
- 37.Adapted from Mehta NB, Philips AP, Lui FF, Brooks RE. J Org Chem. 1960;25:1012–1015. [Google Scholar]
- 38.Burai U, Toth E, Sour A, Merbach AE. Inorg Chem. 2005;44:3561–3568. doi: 10.1021/ic048645d. [DOI] [PubMed] [Google Scholar]
- 39.Caravan P, Parigi G, Chasse JM, Cloutier NJ, Ellison JJ, Lauffer RB, Luchinat C, McDermid SA, Spiller M, McMurry TJ. Inorg Chem. 2007;46:6632–6639. doi: 10.1021/ic700686k. [DOI] [PubMed] [Google Scholar]
- 40.Dunand FA, Aime S, Merbach AE. J Am Chem Soc. 2000;122:1506–1512. [Google Scholar]
- 41.Aime S, Barge A, Bruce JI, Botta M, Howard JAK, Moloney JM, Parker D, de Sousa AS, Woods M. J Am Chem Soc. 1999;121:5762–5771. [Google Scholar]
- 42.Woods M, Aime S, Botta M, Howard JAK, Moloney JM, Navet M, Parker D, Port M, Rousseaux O. J Am Chem Soc. 2000;122:9781–9792. [Google Scholar]
- 43.Xu J, Churchill DG, Botta M, Raymond KN. Inorg Chem. 2004;43:5492–5494. doi: 10.1021/ic049028s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.