

Abstract
Transient hepatomegaly often accompanies acute bacterial infections. Reversible, dose-dependent hepatomegaly also occurs when animals are given intravenous infusions of bacterial lipopolysaccharide (LPS). We found that recovery from LPS-induced hepatomegaly requires a host enzyme, acyloxyacyl hydrolase (AOAH), that inactivates LPS. When we challenged Aoah−/−mice with low doses of LPS or Gram-negative bacteria, their livers remained enlarged (as much as 80%above normal) many weekslonger than did the livers of Aoah+/+ animals. When compared with livers from LPS-primed Aoah+/+ mice, LPS-primed Aoah−/− livers had (a) more numerous and larger Kupffer cells, (b) intrasinusoidal leukocyte aggregates and activated sinusoidal endothelial cells, and (c) sustained production of IL-10 and mRNAs for TNF, IL-10, and IRAK-M. Depleting Kupffer cells decreased the liver enlargement by approximately 40%, whereas depletion of neutrophils, dendritic cells, NK cells, NK-T cells or B cells had no effect. Pre-treatment with dexamethasone almost completely prevented prolonged hepatomegaly in Aoah−/− mice, whereas neutralizing TNF or interleukin-1β was only partially effective. In contrast, an antagonistic antibody to the IL-10 receptor increased LPS-induced hepatomegaly by as much as 50%. Conclusions: our findings suggest that persistently active LPS induces Kupffer cells to elaborate mediators that promote the accumulation of leukocytes within enlarged sinusoids. Large increases in IL-10 and several other modulatory molecules are unable to prevent prolonged hepatomegalyin mice that cannot inactivate LPS. The striking findings in this mouse model should encourage studies to find out how AOAH contributes to human liver physiology and disease. 242 words
Keywords: lipopolysaccharide, Kupffer cell, interleukin-10, acyloxyacyl hydrolase, detoxification
INTRODUCTION
Although the liver’s ability to remove and inactivate bloodborne bacterial endotoxin (lipopolysaccharide, LPS) has been appreciated for many years, the uptake and detoxification mechanisms remain controversial and poorly understood. Many studies have concluded that Kupffer cells are largely responsible for LPS clearance (1, 2) although there is also evidence that hepatocytes can take up LPS (3). How the liver detoxifies endotoxin has also been debated, with some authors supporting inactivation by binding to lipoproteins (4) while others have favored enzymatic dephosphorylation (5) or deacylation by Kupffer cells, hepatocytes, or other cells (6). Despite these differences, there is general agreement that LPS uptake and detoxification contribute to normal liver physiology and may influence the course of some of the inflammatory and metabolic diseases of the liver (7–10).
Our laboratory has sought to define the role of a host enzyme, acyloxyacyl hydrolase (AOAH), in hepatic LPS degradation and inactivation. AOAH is a highly conserved lipase that inactivates LPS by removing fatty acyl chains from the lipid A moiety (11). We found previously that AOAH is produced in the liver by Kupffer cells (KCs) and dendritic cells, and that depleting phagocytic cells with clodronate-liposomes greatly reduced the liver’s ability to deacylate LPS (6). Although AOAH-deficient mice recovered normally from the usual acute reactions to intravenous LPS, they developed dose-related hepatomegaly that lasted for at least 21 days (6). Microscopic examination revealed enlarged sinusoids that contained leukocyte aggregates; the appearance of the hepatocytes was normal and there was no change in hepatic triglyceride content or serum transaminase levels.
Hepatomegaly has been observed during the course of experimental Pseudomonas aeruginosa (12) and Propionibacterium acnes (13) infections, in animals with subacute intraabdominal abscesses (14) and in response to LPS (15) or TNF infusion (16). The liver enlargement experienced by LPS-challenged Aoah−/− mice is more pronounced (as much as 80% increase in liver weight) and persists much longer than was noted in these reports. We have now explored the basis for this unexpected phenotype, asking “How does LPS induce prolonged hepatomegaly in animals that cannot deacylate [inactivate] it?” After characterizing the phenotype in greater detail, we present here the results of several interventions that, by depleting cells or neutralizing potential mediators, help define its pathophysiology.
EXPERIMENTAL PROCEDURES
Reagents
LPS from E. coli O14 was prepared by phenol-chloroform-petroleum ether extraction. [3H/14C]LPS was prepared using S. typhimurium PR122 as previously described (17). Clodronate-liposomes and PBS-liposomes were prepared by J. Niederkorn (UT Southwestern Medical Center) using clodronate provided by Roche. Nω-Nitro-L-arginine methyl ester hydrochloride (L-NAME) and Nω-Nitro-D-arginine methyl ester hydrochloride (D-NAME) were from Sigma-Aldrich, Inc. (St. Louis, MO). 5-Bromo-2′-deoxy-uridine (Brdu) was from Roche Diagnostics. PEGsTNF-R1, a pegylated form of the TNF neutralizing domain of Etanercept, was provided by Amgen. Anakinra and Actemra were purchased from Amgen and Genentech, respectively.
Animals
Aoah−/− C57Bl/6 mice and μMT, Aoah−/− (double knockout) mice were produced as described (6, 18). The mice were maintained in specific pathogen-free conditions in the UT Southwestern Animal Resources Center and used for experiments when they were 5 – 12 weeks of age. All protocols were approved by the UT Southwestern Institutional Animal Care and Use Committee.
Antibodies
PE-or Alexa Fluor® 647-labeled rat-anti mouse F4/80 (BM8), Alexa Fluor® 555 conjugated goat anti-rat IgG and Qdot® 565 conjugated goat anti-FITC antibody were from Invitrogen. Biotin-conjugated rat anti-mouse CD11b (M1/70), rat anti-mouse CD144 (11D4.1) and FITC labeled anti-Brdu antibody were from BD Biosciences. An agonistic monoclonal antibody (UT12) to the Toll-like receptor 4 (TLR4)—MD-2 complex was produced by S. Ohta (19) and prepared as described (20). Rat anti-mouse IL-10R antibody (YL03.1b1.3a-34ABS) and isotype control Ab (MB819.7D7.180) were generously provided by Schering-Plough Biopharma, Palo Alta, CA. Antibodies for flow cytometry were from BD Biosciences.
UT12 dose response and time course experiments
Groups of 3 Aoah−/− and Aoah+/+ mice were injected i.v. with UT12 IgG (20) (0.0125, 0.05, 0.1 or 0.25 μg/g body weight). Livers were harvested 7 days postinjection. In another experiment, Aoah−/− and Aoah+/+ mice were injected i.v. with 0.1 μg/g body weight and studied on days 7, 14 or 21 postinjection.
Liver fixation for electron microscopy
Aoah−/− and Aoah+/+ mice were injected i.v. with 0.5 μg E.coli O14 LPS/g body weight or an equal volume of PBS. Seven days later, animals were deeply anesthetized with isoflurane and perfused with 15 ml of PBS followed by 20 ml of fixative (4% paraformaldehyde and 1% glutaraldehyde in 0.1M cacodylate buffer) through the left ventricle. The liver was then removed, cut into small pieces, and immersed in fixative for 1 hour at room temperature.
Scanning and transmission electron microscopy
SEM and TEM liver samples were prepared by Tom Januszewski (Molecular and Celluar Imaging Facility, UT Southwestern). The samples were examined with a XL30 ESEM scanning electron microscope and a JEOL 1200 EX transmission electron microscope at voltages of 30 and 120. At least 15 images of each specimen were taken.
Confocal microscopy
FITC-LPS was made by conjugating fluorescein isothiocyanate (FITC) with E.coli O14 LPS. The FITC/LPS molar ratio in the conjugate was 0.53. Conjugation did not alter the ability of the LPS to stimulate IL-6 production by mouse peritoneal macrophages (not shown). Aoah−/− and Aoah+/+ mice were injected via the lateral tail vein with 200 μl PBS containing 0.5 μg FITC-LPS, 0.5 μg FITC-BSAor 0.03 μg FITC (matching the amount of FITC in FITC-LPS) per gram body weight. Livers (3 mice/group) were harvested at selected time points, embedded in OCT compound (Sakura Finetek USA, CA) and frozen in liquid nitrogen. Six μm sections were stained with rat anti-mouse CD144 antibody followed by Alexa Fluor® 555 conjugated goat anti-rat IgG to locate sinusoidal endothelial cells. The sections were then blocked with normal rat IgG before Alexa Fluor® 647 -conjugated rat anti-mouse F4/80 antibody was added to identify Kupffer cells. Qdot® 565 conjugated goat anti-FITC antibody was then used to amplify the FITC signal. Nuclei were stained with DAPI. Z-stack Images were taken using a Leica TCS SP5 confocal microscope (Leica Microsystems) and 3 dimensional rendering of images was performed using Bitplane Imaris software.
Detection of liver cell proliferation by 5-bromo-2-deoxyuridine (Brdu) incorporation
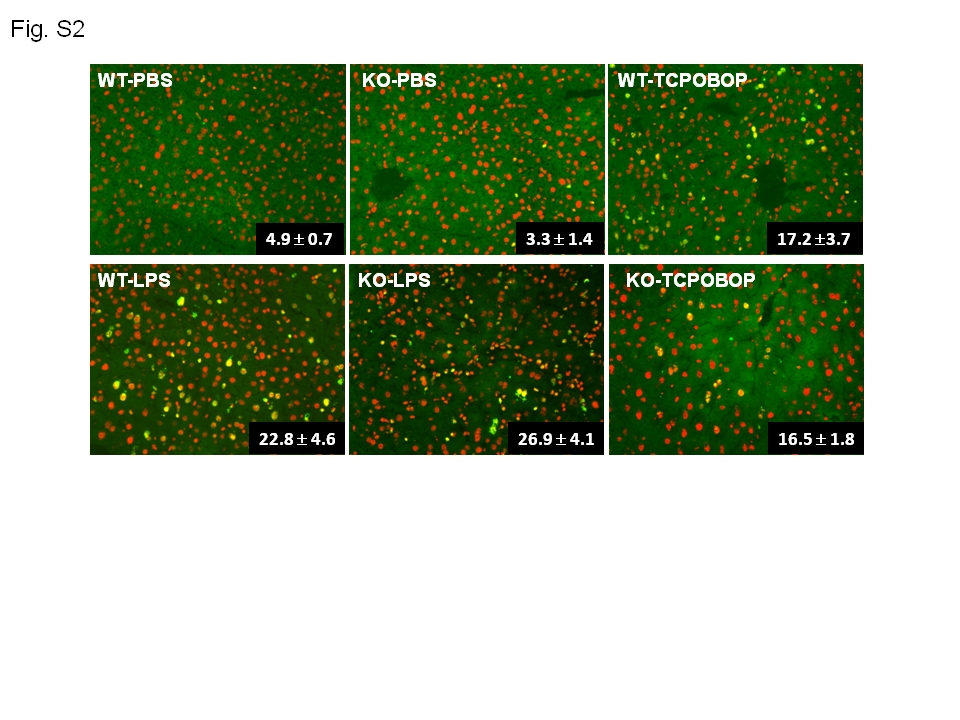
Aoah−/−; and Aoah+/+ mice were injected i.v. with 0.5 μg LPS per g body weight, PBS, or 0.4 mg/kg TCPOBOP (1,4-bis[2-(3,5-dichloropyridyloxy)] benzene; Sigma). TCPOBOP induces hepatocyte proliferation without causing liver injury (21). Brdu (1 mg / mouse) was given i.p.2 hours after LPS injection and repeated daily for 6 days. On day 7 after LPS challenge, livers were harvested, embedded in OCT compound and frozen in liquid nitrogen. Cryostat liver sections were fixed with 70% methanol/30% acetone, DNA was denatured with 2N HCl (0.5% Triton X-100) and neutralized with 0.1 M borate sodium, then the sections were stained with FITC-conjugated anti-Brdu antibody (BD Biosciences, San Jose, CA) and propidium iodide (PI). Images were taken using a Zeiss Axioplan 2 fluorescence microscope. The labeling index (percentage of Brdu-positive cells observed in five different 20X images) was calculated to measure liver cell proliferation.
Plasma Levels of cytokines/chemokines
Blood samples were collected into EDTA-containing tubes. Plasma levels of TNF, IFN-γ, IL-6, IL-10 and MCP-1 were measured using mouse OptEIA ELISA sets (BD Biosciences) according to the manufacturer’s instructions. Plasma RANTES was measured using an ELISA kit from Santa Cruz Biotechnology, Inc. Plasma IL-1β was measured using the IL-1β ELISA kit from eBiosciences.
Real-time PCR
Quantification of gene expression was performed using the ABI Prism 7000 Sequence Detection System (Applied Biosystems). Primers were designed using Primer express software (Applied Biosystems). The primer sequences are listed in Table S3. Liver samples for PCR were quickly snap-frozen in liquid nitrogen and stored at −70ºC. RNA was isolated using RNeasy Mini kit (Qiagen), contaminated genomic DNA was removed with RNase-free DNase I (Roche), and cDNA was prepared using the iScript cDNA synthesis kit (Bio-Rad). All procedures were performed according to the manufacturer’s instructions. Real-time PCR was performed using SYBR green Master mix reagent (Applied Biosystems) under standard conditions (10 minutes at 95ºC, 40 cycles involving denaturation at 95ºC for 15 seconds, annealing/extension at 60ºC for 1 minute). 36B4 was the internal control gene. Relative mRNA levels were quantitated as the fold-change relative to PBS-treated wildtype mice. All assays were performed in duplicate.
Kupffer cell depletion
Kupffer cells were depleted by injecting 200 μl clodronate-liposomes i.v. Two days later, we injected 8 μg LPS in PBS or an equal volume of PBS i.v. Kupffer cell depletion was documented as a decrease in the number of F4/80+ cells in cryostat liver sections obtained 6 days after LPS or PBS treatment. To minimize the impact of photobleaching, digital photographs were taken (5 different fields/liver section, 20X magnification) using a Zeiss Axioplan 2 fluorescence microscope, and cells were counted from these images.
Other interventions (Table S2)
To analyze the role of IL-10 in the development of hepatomegaly in LPS-treated Aoah−/− mice, Aoah−/− mice were given0.5 mg/mouse of rat anti-mouse IL-10R antibody or isotype control Ab (generously provided by Schering-Plough/Merck) intraperitoneally on the first day after i.v. LPS injection (0.1 μg/g body weight). Livers were harvested 7 days after LPS treatment. To inhibit circulating TNF, we gave Aoah−/− mice an intraperitoneal injection of 100 μg of PEGsTNF-R1 (Amgen) 1.5 hour before administering LPS i.v. (0.2 μg/g body weight) and then every other day until the end of the experiment (5 days after LPS challenge). Plasma was obtained one hour after LPS administration to measure TNF by ELISA (BD Biosciences). To inhibit IL-1β, Aoah−/− mice were given Anakinra (IL-1R blocker, 25μg/mouse) 1 hour before LPS administration and then twice every day until the end of the experiment. In some experiments, mice were given both PEGsTNF-R1 and Anakinra. PEGsTNF-R1 was given i.p. every other day, and Anakinra was given i.p. twice daily until the end of experiment (5 to 7 days after LPS administration). To test if LPS-induced hepatomegaly in Aoah−/− mice is influenced by the sympathetic nervous system, we delivered epinephrine (beta agonist, 2mg/kg/day), norepinephrine (alpha agonist, 2.5mg/kg/day), metoprolol (beta-antagonist, 20mg/kg/day) and Prazosin (alpha-antagonist, 3mg/kg/day) via implantable osmotic pumps (100 ul, Alzet, Cupertino, CA) that were placed in the peritoneal cavity 6 days before intravenous LPS administration. Dexamethasone was given intraperitoneally (1mg/kg/day) daily from 3 days before LPS challenge until the end of the experiment 7 days after challenge. In another protocol, dexamethasone was given daily starting one day after LPS administration. Ibuprofen (600 ug, Sigma) was given i.p. from 2 days before LPS challenge until the end of the experiment 5 days after challenge. In all experiments, control mice received an equal volume of PBS.
To test the role of nitric oxide synthase (NOS) in inducing hepatomegaly, Aoah−/− mice were provided L-NAME or D-NAME (Sigma) in their drinking water (1000 mg/l) from 7 days before LPS or PBS administration to the end of the experiment on day 7. One day after i.v. LPS injection, blood was obtained from tail vein and anticoagulated with EDTA to measure nitrate/nitrite concentration by colorimetric assay (Cayman Chemical, Ann Arbor, Michigan). In other experiments, sodium nitrite (Sigma, 500 mg/l) was added to the drinking water of Aoah−/− mice from 7 days before to 7 days after intravenous LPS administration.
RESULTS
An agonistic monoclonal antibody to MD-2—TLR4 does not induce prolonged hepatomegaly in AOAH-deficient mice
To confirm that the hepatic enlargement observed in Aoah+/+ and Aoah−/− mice requires exposure to LPS, we challenged the mice with an agonistic monoclonal antibody to MD-2—TLR4, the LPS receptor complex. This antibody, UT2 (19), elicited equivalent dose-related increases in liver size in both mouse genotypes (Supplemental Fig. S1A), and liver weight had returned to normal in both Aoah−/− and Aoah+/+ mice by day 21 after injection (Supplemental Fig. S1B). Activation of MD-2—TLR4 in Aoah−/− mice using a non-LPS agonist thus does not produce the persistent hepatomegaly observed following LPS exposure (Supplemental Fig. S1C and D), confirming that the prolonged hepatomegaly response is LPS-dependent.
LPS remains in hepatic sinusoids for many days after i.v. injection
We have previously shown that approximately 80% of an intravenous dose of LPS is taken up by the liver, where it remains at least two weeks (6). To track the cellular localization of injected FITC-LPS within the liver, we used confocal microscopy to detect its association with Kupffer cells (F4/80+), sinusoidal endothelial cells (VE-cadherin [CD144]+) (22), and hepatocytes. One day after i.v. injection, the FITC-LPS was largely found within, or attached to, Kupffer cells (Fig. 1A), although some of the FITC was also “free” within sinusoids (Fig. 1A and 1B, arrows). Even 7 days after injection, almost all of the detectable FITC-LPS was within sinusoids; most of it was again associated with Kupffer cells and very little co-localized with hepatocytes (Fig. 1C and 1D). The cellular localization of FITC-LPS was qualitatively similar in Aoah+/+ and Aoah−/− mice 7 days after FITC-LPS injection (Fig. 1, C and E and D and F), suggesting that deacylation does not substantially influence the retention of LPS by Kupffer cells.
Figure 1. LPS remains within sinusoids for at least one week after i.v. injection.

Aoah−/− and Aoah+/+ mice were injected i.v. with 0.5μg FITC-LPS per g body weight. Liver sections were immunolabeled as in Methods to detect Kupffer cells (F4/80+, red), the junctions of sinusoidal endothelial cells (CD144 or VE-cadherin, white), and nuclei (blue). A. Aoah−/− liver, one day after injection (63x, zoom 2.4). B. 3-dimensional rendering of A. C and D. Images (x63, zoom 1) of Aoah+/+ (C) and Aoah−/− (D) livers on day 7 after injection. E and F. Higher magnification (x63, zoom 2.4) views of C and D (areas indicated by the rectangles). Seven days after injection, the LPS is seen largely within sinusoidal spaces and a large fraction of it is within, or closely associated with, Kupffer cells. Examples of extrasinusoidal LPS are indicated by the arrows in A and B.
Morphological evidence for Kupffer cell and sinusoidal endothelial cell activation
Since liver sections stained with hematoxylin-eosin revealed prominent, blood-filled sinusoids in LPS-primed Aoah−/− mice (6), we defined these changes further using transmission (TEM) and scanning (SEM) electron microscopy on sections of perfused livers obtained 7 days after i.v. LPS injection. Both TEM and SEM revealed evidence of cell thickening (Fig. 2A, B), Kupffer cell activation (prominent cytoplasmic extensions and adhesion and/or phagocytosis of erythrocytes and leukocytes) [Fig. 2C, D], and changes consistent with sinusoidal injury (i.e. a remarkable loss of fenestrae [Fig. 2E, F]). These morphological changes resemble those reported by Sarphie and colleagues 24 hrs after LPS administration to rats (23).
Figure 2. Morphological evidence for Kupffer cell activation and sinusoidal injury.

Aoah−/− and Aoah+/+ mice were injected i.v. with 0.5μg E.coli O14 LPS /g body weight. Seven days later, perfused and fixed livers from these mice were prepared for scanning electron microscopy (SEM) and transmission electron microscopy (TEM) as in Methods. A and B. TEM (Original magnification x10,000) of livers from Aoah+/+ (A) and Aoah−/− mice (B). In Aoah−/− livers, Kupffer cells often were enlarged and contained other cells. C and D. SEM (Original magnification 2,500x) of livers from Aoah+/+ (C) and Aoah−/− mice (D). In Aoah−/− livers, Kupffer cells had numerous cytoplasmic extensions and adherent or phagocytosed cells. E and F. SEM (original magnification, 10,000) showing normal sinusoidal fenestrae in Aoah+/+ livers (E) and a decrease in their number and size in Aoah−/− livers (F).
In addition, immunohistochemistry performed on sections obtained on day 7 after LPS injection showed that the LPS-primed, Aoah−/− livers contained many more large, F4/80-positive cells (Kupffer cellsor recruited monocytes) than did LPS-primed, Aoah+/+ livers (Fig. 3A and B), and that many of these macrophages appeared to contain phagocytosed, CD11b positive neutrophils (Fig. 3C and D).
Figure 3. Intrahepatic macrophage (Kupffer cell) activation.

Mice were injected i.v. with 0.5 μg E.coli O14 LPS/g body weight and studied 7 days later. Cryostat liver sections were immunolabled with PE-conjugated anti-mouse F480 (red) and biotin-conjugated anti-mouse CD11b followed by FITC-streptavidin (green) as in Methods. A and C, Aoah+/+, B and D, Aoah−/−. C and D. Enlargement views of areas indicated by the rectangles in A and B. F480 positive cells were much more prominent in Aoah−/− liver sections, and they often contained CD11b+ neutrophils. No difference in Kupffer cell size or appearance was apparent in liver sections from LPS-unexposed Aoah−/− and Aoah+/+ mice (not shown).
The morphological changes seen in the livers of LPS-treated Aoah−/− mice are thus consistent with activation of Kupffer cells (and possibly recruited monocyte-macrophages) and sinusoidal endothelial cell injury in livers that retain fully acylated LPS.
Many cell types accumulate in the liver
We used flow cytometry to identify individual nonparenchymal cell types within the liver. As shown in Figure 4, LPS-challenged Aoah−/− mice experienced significantly greater intrahepatic accumulation of B cells, monocyte-macrophages, neutrophils, dendritic cells, CD3+ T cells and NK1.1+ natural killer cells than did LPS-treated Aoah+/+ mice. The hepatic content of these cell types had returned almost to baseline within 3 weeks after LPS exposure in Aoah−/− mice (Fig. 4) yet liver size did not decrease (Fig. S1D and (6)).
Figure 4. Manycell types accumulate in the liver.

Aoah−/− and Aoah+/+ mice were injected i.v. with 10 μg LPS per mouse as in Methods. On days 3, 7, 14 and 21, livers were perfused and harvested to isolate non-parenchymal cells (NPCs), B cells (B220+CD3−), Kupffer cells and monocyte-macrophages (F4/80+), neutrophils (CD11b+Ly-6G+), dendritic cells (CD11c+), T cells (CD3+), NK cells (CD3-NK1.1+). Cell densities were calculated as follows: (Percentage of cell type X total NPCs number)/liver weight (g)). Open squares = Aoah−/−, closed circles = Aoah+/+. n = 3 mice/time point. Significance of differences between Aoah+/+ and Aoah−/− mice: * = p < 0.05, ** = p <0.01, *** = p < 0.001.
Hepatocyte proliferation does not contribute to prolonged hepatomegaly
To test the hypothesis that hepatocyte proliferation contributes to LPS-induced hepatomegaly (21), we used BRdU to quantitate cell division. Beginning 2 hrs after i.v. LPS challenge, mice received BRdU daily until they were studied on day 7. As shown in Fig. S2, LPS-induced cell proliferation was similar in LPS-primed WT and KO mice. Treatment with the mitogen TCPOBOP, used as a control, also induced equivalent liver cell proliferation in Aoah+/+ and Aoah−/− mice.
Prolonged hepatomegaly is associated with persistently high liver and plasma cytokine levels
LPS induced similar acute plasma cytokine responses in Aoah+/+ and Aoah−/− mice (Fig 5A). Plasma levels of certain cytokines (e.g., IL-10) persisted much longer in LPS-treated Aoah−/− mice than they did in LPS-treated wildtype mice, whereas other cytokine levels followed a similar time-course in the two groups (RANTES, IL-6, TNF, MCP-1). Quantitation of hepatic mRNA abundance using real-time PCR showed striking elevations in IL-10 and TNF mRNAs in Aoah−/− mice over a 7-day period after LPS injection (Fig. 5B); mRNAs for several anti-inflammatory proteins (IRAK-M, SHIP, SOCS1, A-20) were also elevated in these mice 5 to 7 days after LPS injection (Fig. 5C), as were the mRNAs for IL-1β, inducible nitric oxide synthase (NOS2) and CCL2 (MCP-1). Although liver TNF and IL-1β mRNA levels remained elevated for many days, we were unable to detect TNF or IL-1β protein in either liver lysates or plasma beyond 24 hrs after LPS injection. Plasma MCP-1 levels were similar in Aoah−/− and Aoah+/+ mice. These data suggest that the prolonged hepatic response to LPS in Aoah−/− mice involves sustained elevations in the mRNAs for several anti-inflammatory mediators; in contrast, many pro-inflammatory cytokine mRNAs or proteins were not produced in excess.
Fig. 5. Prolonged hepatomegaly is associated with persistently high liver and plasma cytokine levels.

Aoah+/+ and Aoah−/− mice were given 0.5 μg LPS/ g body weight i.v. on day 0. Plasma cytokines and chemokines were measured by ELISA on blood samples obtained on the indicated days (A). Liver cytokine mRNA was quantitated by Real-time PCR (B, C). The normal values (day 0) were from wildtype mice that received PBS. Open squares = Aoah−/−, closed squares = Aoah+/+. mRNA abundance for some modulatory protein genes was also quantitated in livers studied 5–7 days after LPS injection (C). n = 3 mice/group. Significance of differences between Aoah+/+ and Aoah−/− mice: * = p <0.05, ** = p <0.01, *** = p<.001.
Kupffer cells play an important role
In order to better understand the contributions of individual cell types to the hepatomegaly phenotype, we performed several cell depletion experiments. In each of these experiments, we defined “hepatomegaly” as a significant (p <0.05, Student’s t test) increase above normal in the (liver weight/body weight) ratio, expressed as a percentage. In normal 6 – 10 week-old mice, the liver weight is approximately 5% of body weight. “Prolonged hepatomegaly” was defined as hepatomegaly that persisted six or more days after LPS infusion. As detailed in Table S1, depleting neutrophils, NK and NK-T cells, or dendritic cells did not prevent LPS-induced hepatomegaly in Aoah−/− animals. LPS also induced prolonged hepatomegaly in mice that lacked both AOAH and B cells (Aoah−/−, μMT). We concluded that none of these cell types was required to produce the phenotype. In contrast, clodronate-liposome treatment to deplete KCs reduced both LPS uptake by the liver (Fig. 6C) and the hepatomegaly response to LPS (Fig. 6D) by approximately 40%, with similar reductions in mRNA abundance for TNF, IL-10 (Fig. 6E and 6F) and IRAK-M (not shown). Kupffer cells thus play an important role in producing prolonged hepatomegaly in Aoah−/− mice.
Figure 6. Kupffer cells contribute to the development of LPS-induced hepatomegaly in Aoah−/−mice.

Aoah−/− mice were injected i.v. with 200 μl clodronate-liposomes or PBS-liposomes on day 0. On day 2, they were injected i.v. with 0.4 ug/g body weight of 3H/14C-labeled LPS or an equal volume of PBS. A. On day 8, cryostat liver sections from mice which had been given PBS-liposomes [1] or clodronate liposomes [2] were stained with PE-labeled anti-mouse F4/80 antibody (red) and biotin-conjugated anti-mouse CD11b followed by FITC-conjugated streptavidin (green). Picture [3] shows a section from the liver of a mouse that received clodronate-liposomes then LPS (harvested 6 days after LPS). B. Kupffer cell quantitation. The number of F4/80-positive cells per 20X field (mean +/− range, 5 fields/mouse, 2 mice). X axis labels indicate the pre-treatment (PBS or clodronate) and challenge (PBS or LPS) agents. C. LPS recovery from liver: per cent of injected 14C dpm found in liver 6 days after injection of 3H/14C LPS i.v. n = 7–8 mice/group. Mean +/− 1 SE. D. Liver weight/body weight ratio (%) on day 8. n = 7 or 8/group. The levels of TNFα (E) and IL-10 (F) mRNA were quantitated by real-time PCR on samples snap-frozen on day 8. Their levels are expressed as fold-change relative to the value in livers from animals that received PBS-liposomes and no LPS. n = 3–4 mice/group. Mean +/− 1 SE.
We found that FITC-LPS was associated with Kupffer cells for many days in vivo as well as morphological evidence for KC activation following LPS infusion (see above). When we depleted KCs using clodronate-liposomes and studied the animals 8 days later, we found an 85% reduction in hepatic F4/80-positive macrophages (Fig. 6A and 6B). In contrast, the livers of mice that received clodronate-liposomes on day 0 and LPS on day 2 had ~50% of the control numbers of hepatic macrophages when they were studied on day 8. These results suggest that clodronate treatment effectively reduced the resident macrophage (Kupffer cell) population yet did not prevent the recruitment of monocyte-macrophages to the liver during the 6 day period following LPS administration (24). The FITC-LPS remaining in the liver did not associate with these macrophages (not shown) and their role in perpetuating the hepatomegaly phenotype is uncertain.
Preventing the prolonged hepatomegaly phenotype: inhibiting TNF, IL-1β, IL-6, interleukin-10, or nitric oxide synthase
Pre-treating mice with dexamethasone almost completely prevented the prolonged hepatomegaly phenotype (Table S2), confirming the inflammatory nature of the process. To explore TNF’s role, we infused a TNF-neutralizing form of the pegylated, soluble human TNF receptor 1 (PEGsTNF-R1; Amgen) before injecting i.v. LPS. There was a 27% reduction in prolonged hepatomegaly (Table S2). In parallel experiments we found that interleukin-1 receptor antagonist (Anakinra) also inhibited LPS-induced hepatomegaly by 23%. Simultaneous pre-treatment with both antagonists did not enhance the inhibitory effect. TNF and IL-1 thus seem to play minor roles in inducing or maintaining the hepatomegaly phenotype. Inhibiting IL-6 with Actemra did not prevent or enhance LPS-induced hepatomegaly (Table S2).
Finding increased hepatic levels of mRNA for NOS2, the inducible isoform of nitric oxide synthase, raised the possibility that inducible nitric oxide production might contribute to the sinusoidal enlargement noted in LPS-treated Aoah−/− mice (25). This hypothesis was tested by adding L-NAME to the drinking water for 7 days prior to, and for 7 days following, the LPS challenge. Although a reduction in LPS-induced plasma nitrite/nitrate levels documented the L-NAME effect (Fig. 7A), this treatment did not prevent prolonged hepatomegaly in Aoah−/− mice (Fig. 7B). In other experiments, we provided nitrite in the drinking water for 7 days prior to the LPS challenge and for 7 days thereafter (26); there was no effect on LPS-induced hepatomegaly (not shown). Other ineffective interventions included anticoagulation with low molecular weight heparin, inhibition of prostanoid synthesis with ibuprofen, and infusions of adrenergic receptor agonists and antagonists (Table S2).
Figure 7. Effect of NOS inhibitor and IL-10 receptor antagonist on LPS-induced hepatomegaly in Aoah−/− mice.

Treatment with L-NAME decreased basal and LPS-induced plasma nitrate/nitrate levels (A) but did not prevent LPS-induced hepatomegaly (B). Aoah−/− mice were given rat anti-mouse IL-10R antibody or an isotype matched Ab 1 day after intravenous LPS injection (2 μg/20g body weight). Animals were studied on day 7. The liver weight/body weight ratio (C) and plasma IL-10 levels (D) were measured.
The strikingly elevated levels of plasma IL-10 and hepatic IL-10 mRNA raised the possibility that persistently elevated IL-10 might also contribute to the hepatomegaly phenotype. To test this possibility, we gave Aoah−/− mice a monoclonal antibody that blocks the IL-10 receptor. The mice that received the antibody one day after LPS administration developed significantly greater hepatomegaly than did mice that received an isotype control antibody (Fig. 7C); additionally, their plasma IL-10 levels (Fig. 7D) were significantly elevated. IL-10 thus appeared to be very important for controlling and/or preventing the response. Liver size almost doubled in Aoah−/− mice that received LPS before IL-10 receptor blockade.
DISCUSSION
Hepatomegaly is a dose-dependent response to TLR4 agonists. After enlarging for a few days, the liver returns to its previous size. This transient phenomenon has attracted little interest and its physiology has evidently not been studied. Here we explored the basis for the much longer-lasting hepatomegaly that occurs in mice that cannot inactivate LPS because they lack the LPS-deacylating enzyme, AOAH. These animals exhibit wildtype acute cytokine responses to intravenous doses of LPS (Fig. 5A,B), yet they develop impressive hepatic enlargement that lasts for many weeks. Importantly, TLR4 activation by a non-LPS agonist, the monoclonal antibody UT12, induced hepatomegaly of similar degree and duration in wildtype and Aoah−/− animals (Fig. S1), indicating that the AOAH-dependent phenotype is also LPS-dependent.
For these studies we used E. coli Ra (O14) LPS. A complete “rough-form” LPS, it offered several advantages over smooth (long polysaccharide-containing) LPS preparations: a more uniform size and structure, CD14-and LBP-independent activation of TLR4 (27), and an aggregation state that promotes rapid uptake from the blood, largely by Kupffer cells. We found previously that Kupffer cells produce AOAH (6). Whereas Kupffer cell depletion reduced hepatic LPS deacylation by approximately 90%, LPS uptake by the liver fell only 60%, indicating that other hepatic cells can also remove LPS from the blood (6). Here we found that some FITC-LPS remained associated with Kupffer cells for at least 7 days, long after the liver has returned to baseline size in wildtype animals; although dispersion of the FITC-LPS may have limited its detection, there were no evident differences in LPS location in the livers of wildtype and Aoah−/− mice. LPS induced Aoah−/− Kupffer cells to become larger and more phagocytic than Aoah+/+ Kupffer cells for at least one week. Kupffer cell depletion reduced LPS-induced hepatomegaly in Aoah−/− mice by ~40%, suggesting that molecules produced by Kupffer cells contribute importantly to the prolonged hepatomegaly phenotype.
The most striking morphological feature of the phenotype was found in the sinusoids, where there were aggregates of cells (KCs, PMN, platelets, erythrocytes) with occasional hemophagocytosis. Scanning electron microscopy suggested subtle changes in the appearance of sinusoidal endothelial cell fenestrae (Fig. 2), and intravital flow studies (D. Rockey, not shown) found slower, less consistent flow through the sinusoids of LPS-treated Aoah−/− livers. Leukocyte recruitment or trapping in the liver peaked during the second week after LPS exposure, then returned to baseline by 21 days (Fig. 4). Importantly, spleen size increased transiently then returned to baseline whereas liver size remained large (6), suggesting that portal hypertension does not develop during prolonged hepatomegaly. Hepatomegaly persisted for at least 3 weeks, with no evident increase in hepatocyte proliferation (Fig. S2) or accumulation of triglyceride (6). Serum transaminase and alkaline phosphatase levels also did not increase (6).
We used several interventions to neutralize potential mediators in vivo (Table S2). We prevented coagulation using low-molecular weight heparin (28), blocked prostanoid synthesis using ibuprofen, and, to look for a role for the sympathetic nervous system (29–31), we provided adrenoreceptor stimulation (epinephrine and norepinephrine) or antagonism (metroprolol and prazosin) using indwelling osmotic pumps. None of these interventions prevented LPS-induced prolonged hepatomegaly in Aoah−/− animals. Damping nitric oxide/nitrite synthesis using L-NAME or providing nitrite in the drinking water (26) also had no effect on the phenotype.
Acute plasma TNF levels were somewhat lower in LPS-infused Aoah−/− mice than in the wildtype controls and we were unable to detect TNF in plasma or liver lysates more than 2 hours after LPS injection. Liver TNF mRNA levels remained elevated for at least one week, however, and a role for TNF was found when pre-treatment with the TNF-binding protein, PEGsTNF-R1, reduced prolonged hepatomegaly by 27% (Table S2). IL-1β was similarly implicated using an IL-1 receptor antagonist (Anakinra). Increases in several anti-inflammatory mediators, most notably IL-10, were unable to check the response.
In summary, AOAH is required to prevent prolonged hepatomegaly after intravenous challenge with low doses of LPS. LPS is largely taken up from the bloodstream by Kupffer cells, which retain at least some of it for a week or more. In animals that lack AOAH, the Kupffer cells enlarge and phagocytose blood cells; many leukocyte cell types are either recruited to the liver or retarded there as blood flow slows through congested sinusoids. Dexamethasone pre-treatment largely prevents LPS-induced prolonged hepatomegaly, neutralizing TNF or IL-1β has a partial inhibitory effect, and blocking the IL-10 receptor greatly enhances the phenotype. Future studies will address the ability of persistently-active (fully acylated) LPS to stimulate Kupffer cells for prolonged periods in vivo.
AOAH’s key role in recovery from endotoxin exposure in mice should encourage efforts to identify the enzyme’s role in human liver physiology and disease. The enzyme may be particularly important in those conditions, such as alcoholic liver disease (8), in which gut-derived endotoxin is thought to play a contributory role.
Supplementary Material
A. UT12-induced hepatomegaly is dose-dependent. Aoah−/− (open squares) and Aoah+/+ (closed squares) mice were injected i.v. with different amounts of UT12 antibody. Livers were excised and weighed 7 days later. B. UT12 induces transient hepatomegaly in both Aoah−/− and Aoah+/+ mice. Aoah−/− and Aoah+/+ mice were injected i.v. with UT12 (0.1 μg/g body weight) and groups of 3 mice were euthanized on days 7, 14 and 21. The baseline values (day 0) were derived from mice that received PBS instead of UT12 or LPS. C. Dose response for LPS-induced hepatomegaly in Aoah+/+ and Aoah−/− mice. D. Time course of LPS-induced, hepatomegaly in Aoah+/+ and Aoah−/− mice. Mice (three per group) were injected with 10 ug LPS/mouse i.v., and the liver to body weight ratio determined at 7, 14 and 21 days. Note that this dose is not sufficient to cause significant hepatomegaly in Aoah+/+ mice.
{kind=link}
Aoah−/− and Aoah+/+ mice were injected i.v. with 0.5 μg of LPS per g body weight, PBS or 0.4 mg/kg of TCPOBOP. Two hours later, Brdu (1 mg / mouse) was given i.p. and once daily to the end of experiment at day 7. Liver sections were stained with anti-Brdu antibody and propidium iodide (PI). Images were taken using a Zeiss Axioplan 2 fluorescence microscope. Brdu (green) and PI (red) positive cells were quantitated from 6 images of each group. The labeling index is expressed as number of Brdu-positive cells/100 nuclei. The labeling index indicates the level of liver cell proliferative response.
{kind=link}
Acknowledgments
We thank Shearing-Plough Biopharma/Merck for providing the antibody to IL-10R, Amgen for providing the pegylated soluble TNF receptor 1 (PEGsTNF-R1), and Dr. S. Ohta for providing agonistic antibody UT-12 to TLR4.
Financial support: extramural grant AI18188 from the National Institute for Allergy and Infectious Diseases and, in part, by the Division of Intramural Research, NIAID, NIH.
Abbreviations
- FITC
Fluorescein isothiocyanate
- KC
Kupffer cell
- L-NAME
Nω-Nitro-L-arginine methyl ester hydrochloride
- LPS
lipopolysaccharide
- NOS
nitric oxide synthase
- TNF
tumor necrosis factor
- SEM
scanning electron microscopy
- TEM
transmission electron microscopy
- BRdU
bromodeoxyuridine
- TCPOBOP
1,4-bis[2-(3,5-dichloropyridyloxy)]benzene
REFERENCESReference List
- 1.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006 Dec;26(10):1175–1186. doi: 10.1111/j.1478-3231.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- 2.Nolan JP. Endotoxin, reticuloendothelial function, and liver injury. Hepatology. 1981 Sep;1(5):458–465. doi: 10.1002/hep.1840010516. [DOI] [PubMed] [Google Scholar]
- 3.Scott MJ, Billiar TR. beta 2-integrin induced p38 MAPK activation is a key mediator in the CD14/TLR4/MD2-dependent uptake of LPS by hepatocytes. J Biol Chem. 2008 Aug 13; doi: 10.1074/jbc.M803905200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szabo G, Romics L, Jr, Frendl G. Liver in sepsis and systemic inflammatory response syndrome. Clin Liver Dis. 2002 Nov;6(4):1045–66. x. doi: 10.1016/s1089-3261(02)00058-2. [DOI] [PubMed] [Google Scholar]
- 5.Poelstra K, Bakker WW, Klok PA, Hardonk MJ, Meijer DKF. A physiologic function for alkaline phosphatase: Endotoxin detoxification. Lab Invest. 1997;76:319–327. [PubMed] [Google Scholar]
- 6.Shao B, Lu M, Katz SC, Varley AW, Hardwick J, Rogers TE, et al. A Host Lipase Detoxifies Bacterial Lipopolysaccharides in the Liver and Spleen. J Biol Chem. 2007 May 4;282:13726–13735. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 7.Su GL. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am J Physiol Gastrointest Liver Physiol. 2002 Aug;283(2):G256–G265. doi: 10.1152/ajpgi.00550.2001. [DOI] [PubMed] [Google Scholar]
- 8.Szabo G, Bala S. Alcoholic liver disease and the gut-liver axis. World J Gastroenterol. 2010 Mar 21;16(11):1321–1329. doi: 10.3748/wjg.v16.i11.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004 Jul;114(2):147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pastor CM, Billiar TR, Losser MR, Payen DM. Liver injury during sepsis. J Crit Care. 1995 Dec;10(4):183–197. doi: 10.1016/0883-9441(95)90010-1. [DOI] [PubMed] [Google Scholar]
- 11.Munford R, Lu M, Varley A. Chapter 2: Kill the bacteria…and also their messengers? Adv Immunol. 2009;103:29–48. doi: 10.1016/S0065-2776(09)03002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minei JP, Fong Y, Marano MA, Moldawer LL, Jones WG, Wei H, et al. Hepatocellular membrane function during chronic burn injury. J Surg Res. 1989 Apr;46:311–316. doi: 10.1016/0022-4804(89)90193-5. [DOI] [PubMed] [Google Scholar]
- 13.Margenthaler JA, Landeros K, Kataoka M, Eilers M, Ku G, Flye MW. Effects of endotoxin tolerance on Propionibacterium acnes-primed lipopolysaccharide hepatic injury. J Surg Res. 2003 Jun 1;112(1):102–110. doi: 10.1016/s0022-4804(03)00133-1. [DOI] [PubMed] [Google Scholar]
- 14.Vary TC, Kimball SR. Regulation of hepatic protein synthesis in chronic inflammation and sepsis. Am J Physiol. 1992 Feb;262(2 Pt 1):C445–C452. doi: 10.1152/ajpcell.1992.262.2.C445. [DOI] [PubMed] [Google Scholar]
- 15.Brosnan JT, Qian D. Endotoxin-induced increase in liver mass and hepatocyte volume. Biochem Soc Trans. 1994 May;22(2):529–532. doi: 10.1042/bst0220529. [DOI] [PubMed] [Google Scholar]
- 16.Feingold KR, Grunfeld C. Tumor necrosis factor-alpha stimulates hepatic lipogenesis in the rat in vivo. J Clin Invest. 1987 Jul;80(1):184–190. doi: 10.1172/JCI113046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall CL, Munford RS. Enzymatic deacylation of the lipid A moiety of Salmonella typhimurium lipopolysaccharides by human neutrophils. Proc Natl Acad Sci USA. 1983;80:6671–6675. doi: 10.1073/pnas.80.21.6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu M, Zhang M, Kitchens RL, Fosmire S, Takashima A, Munford RS. Stimulus-dependent deacylation of bacterial lipopolysaccharide by dendritic cells. J Exp Med. 2003;197:1745–1754. doi: 10.1084/jem.20030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohta S, Bahrun U, Shimazu R, Matsushita H, Fukudome K, Kimoto M. Induction of Long-Term Lipopolysaccharide Tolerance by an Agonistic Monoclonal Antibody to the Toll-Like Receptor 4/MD-2 Complex. Clin Vaccine Immunol. 2006 Oct 1;13:1131–1136. doi: 10.1128/CVI.00173-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu M, Varley AW, Ohta S, Hardwick J, Munford RS. Host inactivation of bacterial lipopolysaccharide prevents prolonged tolerance following gram-negative bacterial infection. Cell Host Microbe. 2008 Sep 11;4(3):293–302. doi: 10.1016/j.chom.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ledda-Columbano GM, Curto M, Piga R, Zedda AI, Menegazzi M, Sartori C, et al. In vivo hepatocyte proliferation is inducible through a TNF and IL-6-independent pathway. Oncogene. 1998 Aug 27;17(8):1039–1044. doi: 10.1038/sj.onc.1202018. [DOI] [PubMed] [Google Scholar]
- 22.Vestweber D. VE-Cadherin: The Major Endothelial Adhesion Molecule Controlling Cellular Junctions and Blood Vessel Formation. Arterioscler Thromb Vasc Biol. 2008 Feb 1;28:223–232. doi: 10.1161/ATVBAHA.107.158014. [DOI] [PubMed] [Google Scholar]
- 23.Sarphie TG, Deaciuc IV, Spitzer JJ, D’Souza NB. Liver sinusoid during chronic alcohol consumption in the rat: an electron microscopic study. Alcohol Clin Exp Res. 1995 Apr;19(2):291–298. doi: 10.1111/j.1530-0277.1995.tb01505.x. [DOI] [PubMed] [Google Scholar]
- 24.Freudenberg MA, Freudenberg N, Galanos C. Time course of cellular distribution of endotoxin in liver, lungs and kidneys of rats. Br J Exp Pathol. 1982 Feb;63(1):56–65. [PMC free article] [PubMed] [Google Scholar]
- 25.Chen T, Zamora R, Zuckerbraun B, Billiar TR. Role of nitric oxide in liver injury. Curr Mol Med. 2003 Sep;3(6):519–526. doi: 10.2174/1566524033479582. [DOI] [PubMed] [Google Scholar]
- 26.Cauwels A, Buys ES, Thoonen R, Geary L, Delanghe J, Shiva S, et al. Nitrite protects against morbidity and mortality associated with TNF- or LPS-induced shock in a soluble guanylate cyclase-dependent manner. J Exp Med. 2009 Dec 21;206(13):2915–2924. doi: 10.1084/jem.20091236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huber M, Kalis C, Keck S, Jiang Z, Georgel P, Du X, et al. R-form LPS, the master key to the activation ofTLR4/MD-2-positive cells. Eur J Immunol. 2006 Mar;36(3):701–711. doi: 10.1002/eji.200535593. [DOI] [PubMed] [Google Scholar]
- 28.Miyazawa Y, Tsutsui H, Mizuhara H, Fujiwara H, Kaneda K. Involvement of intrasinusoidal hemostasis in the development of concanavalin A-induced hepatic injury in mice. Hepatology. 1998 Feb;27(2):497–506. doi: 10.1002/hep.510270225. [DOI] [PubMed] [Google Scholar]
- 29.Izeboud CA, Hoebe KH, Grootendorst AF, Nijmeijer SM, van Miert AS, Witkamp RR, et al. Endotoxin-induced liver damage in rats is minimized by beta 2-adrenoceptor stimulation. Inflamm Res. 2004 Mar;53(3):93–99. doi: 10.1007/s00011-003-1228-y. [DOI] [PubMed] [Google Scholar]
- 30.Abraham E, Kaneko DJ, Shenkar R. Effects of endogenous and exogenous catecholamines on LPS-induced neutrophil trafficking and activation. Am J Physiol Lung Cell Mol Physiol. 1999 Jan;276:L1–L8. doi: 10.1152/ajplung.1999.276.1.L1. [DOI] [PubMed] [Google Scholar]
- 31.Oben JA, Diehl AM. Sympathetic nervous system regulation of liver repair. Anat Rec A Discov Mol Cell Evol Biol. 2004 Sep;280(1):874–883. doi: 10.1002/ar.a.20081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. UT12-induced hepatomegaly is dose-dependent. Aoah−/− (open squares) and Aoah+/+ (closed squares) mice were injected i.v. with different amounts of UT12 antibody. Livers were excised and weighed 7 days later. B. UT12 induces transient hepatomegaly in both Aoah−/− and Aoah+/+ mice. Aoah−/− and Aoah+/+ mice were injected i.v. with UT12 (0.1 μg/g body weight) and groups of 3 mice were euthanized on days 7, 14 and 21. The baseline values (day 0) were derived from mice that received PBS instead of UT12 or LPS. C. Dose response for LPS-induced hepatomegaly in Aoah+/+ and Aoah−/− mice. D. Time course of LPS-induced, hepatomegaly in Aoah+/+ and Aoah−/− mice. Mice (three per group) were injected with 10 ug LPS/mouse i.v., and the liver to body weight ratio determined at 7, 14 and 21 days. Note that this dose is not sufficient to cause significant hepatomegaly in Aoah+/+ mice.
Aoah−/− and Aoah+/+ mice were injected i.v. with 0.5 μg of LPS per g body weight, PBS or 0.4 mg/kg of TCPOBOP. Two hours later, Brdu (1 mg / mouse) was given i.p. and once daily to the end of experiment at day 7. Liver sections were stained with anti-Brdu antibody and propidium iodide (PI). Images were taken using a Zeiss Axioplan 2 fluorescence microscope. Brdu (green) and PI (red) positive cells were quantitated from 6 images of each group. The labeling index is expressed as number of Brdu-positive cells/100 nuclei. The labeling index indicates the level of liver cell proliferative response.