

Abstract
Advancing our understanding of neuroplasticity and the development of novel therapeutics based upon this knowledge is critical in order to improve the treatment and prevention of a myriad of nervous system disorders. Epigenetic mechanisms of neuroplasticity involve the post-translational modification of chromatin and the recruitment or loss of macromolecular complexes that control neuronal activity-dependent gene expression. While over a century after Ramón y Cajal first described nuclear subcompartments and foci that we now know correspond to sites of active transcription with acetylated histones that are under epigenetic control, the rate and extent to which epigenetic processes act in a dynamic and combinatorial fashion to shape experience-dependent phenotypic and behavioral plasticity in response to various types of neuronal stimuli over a range of time scales is only now coming into focus. With growing recognition that a subset of human diseases involving cognitive dysfunction can be classified as ‘chromatinopathies’, in which aberrant chromatin-mediated neuroplasticity plays a causal role in the underlying disease pathophysiology, understanding the molecular nature of epigenetic mechanisms in the nervous system may provide important new avenues for the development of novel therapeutics. In this review, we discuss the chemistry and neurobiology of the histone deacetylase (HDAC) family of chromatin-modifying enzymes, outline the role of HDACs in the epigenetic control of neuronal function, and discuss the potential relevance of these epigenetic mechanisms to the development of therapeutics aiming to enhance memory and neuroplasticity. Finally, open questions, challenges, and critical needs for the field of ‘neuroepigenetics’ in the years to come will be summarized.
Keywords: epigenetics, chromatin, neuroplasticity, memory, HDAC, therapeutics
1. INTRODUCTION
1.1 Chromatin as a Substrate for Epigenetic Control
Recent molecular, cellular, and behavioral findings have revealed the importance of epigenetic mechanisms that alter chromatin structure in maintaining stable patterns of gene expression and altering neuroplasticity associated with memory formation (reviewed in Levenson and Sweatt 2005; Barret and Wood, 2008), mood (Berton and Nestler 2006; Tsankova et al. 2007), drug addiction (reviewed in Renthal, Nestler 2007), neuroprotection (Kazantsev and Thompson 2008), and other forms of experience-dependent input into the nervous system. Collectively, these findings have provided new insight into the cellular and molecular mechanisms through which gene expression affects neurotransmission and behavioral plasticity over long time periods. This in turn has led to a growing desire to understand the nature of epigenetic regulation in the nervous system in greater detail.
At the heart of epigenetic regulatory mechanisms is the fundamental unit of chromatin in all eukaryotic cells, the nucleosome, composed of 147 base pairs of DNA wrapped around two copies of specific variants of each of the core histones H2A, H2B, H3, and H4, along with one copy of the liker histone H1. By packaging DNA and controlling the access of other factors, epigenetic mechanisms provide an important level of control of gene expression throughout development and in post-mitotic cells, such as neurons.
With its repeating nucleosomal units, chromatin as a polymer is well designed to be a ‘plastic’ substrate that can respond to both fast and short-term changes in neuronal signaling and cell states within the nervous system. To alter gene expression states in neurons, as in all other cell types, epigenetic regulatory processes involves the dynamic interplay of two major classes of multiprotein, macromolecular complexes: 1) ATP-dependent remodeling complexes, which alter the position of nucleosomes to either increase or decrease transcription (reviewed in Racki, Narlikar 2008); and 2) histone-modifying complexes, which post-translationally modify the N-terminal tails of histone proteins through acetylation, methylation, phosphorylation, ubiquitinylation, sumoylation, glycosylation, and ribosylation (Grozinger, Schreiber 2002; Ruthenburg et al. 2007; Borrelli et al. 2008).
1.2 Histone Deacetylase: HDACs
Of the various histone modifications, the reversible acetylation and deacetylation of the ε-amino group of lysine side chains within the N-terminal tails of histones has emerged as a central regulator of transcriptional programming and brain plasticity (Levenson and Sweatt 2005; Borrelli et al. 2008). The enzymes responsible for the acetylation of histones are known as histone acetyltransferases (HATs), which have been shown to have a critical role in memory formation (Guan et al. 2002; Korzus et al. 2004; Levenson et al. 2004; Alarcon et al. 2004; Wood et al. 2005; Wood et al. 2006; Vecsey et al. 2007) and are discussed in detail in other reviews (Berndsen et al. 2008; Selvi et al. 2010; Anamika et al. 2010), and the complementary family of histone deacetylases (HDACs), which are the focus here.
1.2.1 HDAC Family
HDACs remove the acetyl group from the ε-amino group of lysine side chains with the N-terminal tails of histones (and other non-histone substrates). In doing so, HDACs favor the closed, repressive state of chromatin through ‘cis’ regulatory mechanisms involving interaction of positively charged histone tails with negatively charged phosphodiester backbone, and the further recruitment of other transcriptional co-repressors through ‘trans’ regulatory mechanisms involving changes in bromodomain-mediated recruitment of proteins (Grozinger, Schreiber 2002; Ruthenburg et al. 2007). There are a total of 18 HDAC enzymes in the mammalian genome (reviewed in de Ruijter et al. 2003; Smith et al. 2008). These enzymes generally divided into four classes including class I, II, III and IV, based on sequence homology to their yeast counterparts. Among the HDACs, class I, II and IV HDACs are the zinc-dependent hydrolases. Class I HDACs include 1, 2, 3, and 8, which have been well documented to exert deacetylase activity on histone substrates as well as non-histone substrates. Class II HDACs can be divided into class IIa members, which include HDAC 4, 5, 7 and 9, and class IIb members, which include HDAC6 and 10.
In the case of HDAC5, a role in the brain has been identified in response to both antidepressant action (Tsankova et al. 2006), to chronic emotional stimuli (Renthal et al. 2005), as well as regulation of long-term potentiation (Guan et al. 2002). HDAC4 and HDAC5 have also been shown to undergo nucleocytoplasmic trafficking in response to neural activity (Chawla et al. 2003). Similarly, an additional class IIa family member, HDAC9, has recently been shown to regulate activity-dependent gene expression and dendritic growth in developing cortical neurons (Sugo et al. 2010).
Class IIb family members, HDAC6 and 10 are mainly localized in the cytoplasm. HDAC6 is unique in the family in its possession of two deacetylase domains. HDAC6 has been shown to function as both an α-tubulin (K40) deacetylase (Haggarty et al. 2003), and to regulate neurotrophic factor trafficking (Dompierre et al. 2007). Through its actives as a tubulin deacetylases, HDAC6 has been identified as having an important role in the modulation of mitochondrial transport in hippocampal neurons in response to serotonergic neurotransmission in a manner that is dependent upon GSK3β activity (Chen et al. 2010).
HDAC11 is classified as a class IV HDAC, and despite its high levels of expression in the mouse brain very little is know about its biological role and inhibitor sensitivity.
In contrast to the class I/II/IV HDACs, class III HDACs (sirtuins; SIRT1-7) are NAD(+)-dependent enzymes, which exhibit a non-overlapping sensitivity to most structural classes of inhibitors (reviewed in Smith et al. 2008). For reasons of limited space, and the fact that most HDAC inhibitors that have been shown to enhance memory formation do not target this class of HDACs (see below), we will not consider these family members in more detail here.
1.2.2 Brain Expression of HDACs
Analysis of the expression levels and distribution of HDAC1-11 in the mouse brain using the Allen Brain Atlas indicates that all isoforms are expressed at varying levels throughout the brain. Expression studies in rat have shown that most HDACs are expressed in the adult brain predominantly in neurons (Broide et al. 2007). Recent studies by MacDonald and Roskams (2008) and Guan et al. (2009) have shown that HDAC1 is predominantly expressed in glia and neural progenitor cells. In contrast, HDAC2 is more highly expressed in mature neurons and to a lesser extent in differentiated glial cells. These findings suggest important roles for class I HDACs in the development of the nervous system. Indeed, loss of both HDAC1 and HDAC2 leads to severely aberrant brain development through disruption of neural precursor differentiation (Montgomery et al. 2009).
1.2.3 HDAC Complexes
Numerous studies have shown that HDACs function as part of large multiprotein complexes that are targeted to chromatin by DNA binding proteins. A number of biochemically purified HDAC-containing complexes have been characterized, including Sin3 complexes, CoREST complexes, and NuRD complexes (Grozinger, Schreiber 2002; Ruthenburg et al. 2007; Bantscheff et al. 2011). However, the exact composition and mechanisms of regulation of these chromatin-modifying complexes in the brain and in different cell types remains poorly understood but a fascinating area for future investigation. Given the differences and cofactors and complex components, there are likely a number of allosteric regulatory mechanisms that govern the function of HDACs.
1.3 Targeting HDACs with Small-Molecule Probes
Efforts are underway in the field of neuroepigenetics to develop selective, brain-penetrant, small-molecule probes of chromatin-modifying and chromatin-remodeling complexes that affect neural activity-regulated gene transcription and other epigenetic mechanisms of regulation. Most advanced in this area are efforts to selectively target the enzymatic activity of members of the HDAC family. As discussed below in more detail, it has been demonstrated that it is possible with HDAC inhibitors to manipulate the acetylation state of histones in the promoters of certain genes thereby affecting neural activity-regulated gene transcription and neuroplasticity leading under certain conditions to enhanced memory formation. These findings have important implications to the fundamental mechanisms of memory and may potentially provide new avenues for therapeutic development for a range of disorders involving altered neuroplasticity.
1.3.1 Classes of HDAC Inhibitors
Four major classes of small-molecule probes of HDAC function presently exist, including inhibitors presently in clinical trials or already approved by the F.D.A.: 1) carboxylic acids (e.g, butyrate, valproate), 2) hydroxamic acids (e.g., trichostatin A and SAHA (suberoylanilide hydroxamic acid), 3) ortho-aminoanilines (e.g., MS-275), and 4) natural products (e.g., trapoxin, FK228) (Fig. 1A). Only the first thee of these classes have to date been explored in the context of animal models of learning and memory (Table 1). Most, if not all, HDAC inhibitors function through chelating the active site zinc ion, and differences in class I and class II HDAC isoform selectivity can be obtained by varying the chelator moiety. On the basis of the structures of these inhibitors, a general model for HDAC inhibition has been put forth consisting of “cap-linker-chelator” functionalities, which is supported by structural models of HDACs and bound inhibitors and numerous structure-activity-relationship studies (Fig. 1B) (Finnin et al. 1999; Yoshida 2003; Bieliauskas, Pflum 2008). Considering the natural substrate of HDACs and the underlying reaction mechanism for deacetylation reveals the similarity of the known inhibitors that enhance memory to acetyl-lysine, which is consistent with the fact that these inhibitors are all competitive with the acetyl-lysine substrate (Fig. 1C).
Figure 1. HDAC inhibitors as probes of chromatin-mediated neuroplasticity.

(A) Structural classes of HDAC inhibitors known to enhance memory formation. (B) Cap-linker-chelator model for HDAC inhibitors bound in an HDAC2 homology model based upon the x-ray crystal structure of trichostatin A with an HDAC-like protein (Finnin et al. 1999). Trichostatin chelates the active site zinc (grey ball; white arrow) and extends its capping element to interact with surface residues. (C) Opposing enzymatic activities of histone deacetylases (HDAC) and histone acetyltransferases (HAT).
Table 1.
In vivo behavioral studies of the effects of different classes of HDAC inhibitors on memory formation.
| CARBOXYLIC ACID | HYDROXAMIC ACID | ORTHO- AMINOANILINE | |||||
|---|---|---|---|---|---|---|---|
| Butyric acid | Butyric acid | 4-Phenyl-butyric acid | Valproic acid | Pentyl-4n-valproic acid | TSA | SAHA | RGFP136 |
|
Ref:Levenson et al. 2004 Behavior: FC (cont) Strain: rat Route: IHC Dose: 1.2 g/kg Time: acute (+ 60 min) |
Ref:Yeh et al. 2004 Behavior: FC (cont) Strain: rat Route: IAM Dose: Time: acute (+ 60 min) |
Ref:Ricobaraza et al. 2009 Behavior: Strain: Tg2576 AD Route: IP Dose: 200 mg/kg Time: chronic (5 w) |
Ref:Li et al. 2006 Behavior: FE (cont) Strain: WT Route: IP Dose: 200 mg/kg Time: chronic (27 d) |
Ref:Li et al. 2006 Behavior: WM; SIA Strain: rat Route: IP Dose: 84 mg/kg Time: acute (+ 3 hr); chronic |
Ref:Korzus et al. 2004 Behavior: NOR Strain: CBPdn Route: IP Dose: 2 mg/kg Time: acute (+ 2 hr) |
Ref:Alcaron et al. 2004 Behavior: FC (cont; cue) Strain: CBP+/−Route: ICV Dose: Time: acute (+ 3 hr) |
Ref:McQuown et al. 2011 Behavior: NOR Strain: WT Route: SC Dose: 30 mg/kg Time: acute |
|
Ref:Fischer et al. 2007 Behavior: FC (cont); WM Strain: p25 Route: IP Dose: (1.2 g/kg) Time: chronic (28 d) |
Ref:Bredy et al. 2007 Behavior: FE (cue) Strain: WT Route: IP Dose: 1 g/kg Time: acute (+ 2 hr) |
Ref:Ito et al. 2009 Behavior: NOR, WM Strain: PQBP1-KD Route: IP Dose: 100 mg/kg Time: chronic (4 w) |
Ref:Bredy et al. 2007 Behavior: FE (cue) Strain: WT Route: IP Dose: 300 mg/kg Time: acute (+ 2 hr) |
Ref:O’Loinsigh et al. 2004 Behavior: WM Strain: rat Route: IP Dose: 84 mg/kg Time: acute (+ 20 min); chronic (4 d) |
Ref:Lattal et al. 2007 Behavior: FE (cont) Strain: WT Route: IHC Dose: Time: acute (+ 0 min) |
Ref:Guan et al. 2009 Behavior: FC (cont) Strain: WT; HDAC2 OE Route: systemic (i.p.) Dose: 25 mg/kg Time: chronic (10–21 d) |
|
|
Ref:Lattal et al. 2007 Behavior: FE (cont) Strain: WT Route: IP Dose: 1.2 g/kg Time: acute (+ 0 min) |
Ref:Fontan-Lozano et al. 2008 Behavior: NOR Strain: WT; KA; SAMP-8 Route: IP Dose: 250 mg/kg Time: acute (+ 30 min) |
Ref:Bredy et al. 2008 Behavior: FC (cue); RFC (cue); FE (cue) Strain: WT Route: IP Dose: 100 mg/kg Time: acute (+ 2 hr) |
Ref:Foley et al. 2004 Behavior: WM Strain: rat (aged) Route: IP Dose: 84 mg/kg Time: acute (+ 20 min); chronic (4 d) |
Ref:Vecsey et al. 2007 Behavior: FC (cont) Strain: WT Route: IHC Dose: Time: acute (+ 0 min) |
Ref:Kilgore et al. 2010 Behavior: FC (cont) Strain: APPswe/PS1dE9 Route: IP Dose: 50 mg/kg Time: chronic (19 d) |
||
|
Ref:Fontan-Lozano et al. 2008 Behavior: EBCC Strain: WT Route: IP Dose: 250 mg/kg Time: acute (+ 30 min); chronic (8 d) |
Ref:Guan et al. 2009 Behavior: FC (cont) Strain: WT Route: IP Dose: 1.2 g/kg Time: chronic (21 d) |
Ref:Kilgore et al. 2010 Behavior: FC (cont) Strain: WT Route: IP Dose: 100 mg/kg Time: chronic (14 d) |
Ref:Fontan-Lozano et al. 2008 Behavior: EBCC Strain: WT Route: IP Dose: 1 mg/kg Time: acute (+ 30 min); chronic (8 d) |
||||
|
Ref:Stefanko et al. 2009 Behavior: NOR Strain: WT; CBPKIX Route: IP Dose: (1.2 g/kg) Time: acute (+ 0 min) |
Ref: Dash et al. 2009 Behavior: WM Strain: WT + TBI Route: IP Dose: (1.2 g/kg) Time: chronic (8 d) |
Ref: Fontan-Lozano et al. 2008 Behavior: NOR Strain: WT; KA; SAMP-8 Route: IP Dose: 1 mg/kg Time: acute (+ 30 min) |
|||||
|
Ref: Kilgore et al. 2010 Behavior: FC (cont) Strain: APPswe/PS1dE9 Route: IP Dose: 1.2 g/kg Time: chronic (21 d) |
Ref: Francis et al. 2009 Behavior: FC Strain: APPswe/PS1m Route: IP Dose: 2 mg/kg Time: acute (+ 2 hr) |
Abbreviations: APPswe/PS1dE9 double-transgenic mice containing the Swedish double mutations (Lys595Asn/Met596Leu) as well as a mutant human PS1 transgene carrying the deleted exon 9 variant under control of mouse prion promoter elements; APPswe/PS1m double transgenic mice expressing mutant human APP (Lys670Asn/Met671Leu) and mutant human PS1 (Met146Val); cont = contextual; cue = cued; CBP = CREB binding protein; dn = dominant negative; EBCC = eyeblink classical conditioning; FC = fear conditioning memory; FE = fear extinction; hr = hour; HDAC2 = transgenic histone deacetylase 2 overexpression mice; IAM = intraamygdala; ICV = intracerebroventricular; IHC = intrahippocampal; IP = intraperitoneal; KA= kainic acid-induced hippocampal degeneration; KIX = CBP KIX domain mutant; min = minutes; p25 = CDK5 activator; pm = point mutation; PQBP1-KD = polyglutamine tract-binding protein 1 knockdown; RFC = reconsolidation of fear conditioning memory; SAMP-8 = senescence accelerated mouse model of aging; SIA = scopolamine-induced amnesia; TBI = traumatic brain injury; Tg2576 AD = transgenic mice expressing human APP containing the Swedish double mutation (Lys670Asn/Met671Leu); WM = water maze WT= wildtype mice; +/− = heterozygous; w = weeks.
1.3.2 Effects of HDAC Inhibitors on Neurons
Treatment of neurons with inhibitors of HDACs results in the hyperacetylation of histones, creating a more open, accessible conformation of chromatin, which in turn leads to the recruitment of additional chromatin-remodeling complexes that bind to acetylated histone tails via specialized protein-binding domains (Ruthenburg et al. 2007; Borrelli et al. 2008). As discussed in other chapters in more detail, models for the effectiveness of HDAC inhibitors toward memory enhancement suggest a key role for CBP-CREB dependent transcription of genes (Alarcon et al. 2004; Korzus et al. 2004; Vecsey et al. 2007; Barrett, Wood 2008). Notably, although HDACs play a fundamental role in the regulation of gene expression, in contrary to the common assumption that the effect of administering HDAC inhibitors would lead to the global increases in most if not all genes within neurons, numerous studies have demonstrated the restricted expression of a subset of genes, usually in a bidirectional manner. For example, Vecesy et al. (2007) observed a CBP-dependent expression of the orphan nuclear receptors Nr4a1 (Nur77 and NGFI-B) and Nr42a after trichostatin A induced memory enhancement, but not a number of other CREB target genes, as was observed also in cultured cell lines (Fass et al. 2003; Lamb et al. 2006).
1.3.3 Epigenetic Regulation of Immediate-Early Genes Involved in Memory
One of the key signaling pathways under epigenetic control is that involving brain-derived neurotrophic factor (BDNF)-TrkB signaling (Martinowich et al. 2003; Bredy et al. 2007; Tsankova et al. 2007; Yasuda et al. 2009). In turn, one of the transcriptional targets of BDNF-TrkB signaling that has also been shown to be under epigenetic control is the regulatory immediate-early gene (IEG) Egr1 (Zif268) (Bozon et al. 2002; Knapska, Kaczmarek 2004; Nott et al. 2008; Guan et al. 2009). Egr1 encodes a zinc finger transcription factor and its mRNA expression is known to be upregulated in the hippocampus by associative learning (Knapska, Kaczmarek 2004). Egr1 heterozygous mice show intact short-term memory but have impaired long-term memory (Jones et al. 2001). The 5′ cis-regulatory elements in the promoter of Egr1 contains binding sites for several regulatory factors, including two cAMP response elements (CRE) sites that can be bound by CREB, six serum response element (SRE) sites that can be bound by ELK1, activating protein-1/2 (AP-1/2) sites that can be bound Fos/Jun dimers, an SP1 site, an CCAAT/enhancer binding protein (C/EBP) site, and GSG box sites that bind EGR-family members. BDNF has been shown to regulate the transcription of Egr1 through the nitrosylatation of cysteine residues (Cys262 and Cys274) on HDAC2, which results in its dissociation from chromatin and increases acetylation of H3 and H4 in the Egr1 promoter (Nott et al. 2008). Furthermore, using chromatin immunoprecipitation, HDAC2, but not HDAC1, was enriched in binding to the Egr1 promoter (Guan et al., 2009). Consequently, the transcription of genes such as Egr1 may provide a useful surrogate assay for measuring epigenetic regulation of memory processes.
Another example of an important IEG under epigenetic control that has been shown to be subject to activity-dependent regulation and to be critically involved in the process of memory formation is nuclear receptor subfamily 4 group A member 2 (Nr4a2; Nurr1) (Peña de Ortiz et al., 2000; Fass et al. 2003; Colón-Cesario et al., 2006; Vecsey et al., 2007, McQuown et al., 2011). Nr4a2, which is a member of an evolutionarily conserved family of highly homologous nuclear receptors, including Nr4a1 (Nur77; NGFI-B) and Nr4a3 (NOR1), was found along with Nr4a1 to increased upon trichostatin A treatment in vivo in the hippocampus after contextual fear conditioning (Vecsey et al., 2007) whereas a number of other CRE motif containing genes did not increase their expression levels matching what had had been observed in vitro in culture neuronal cells subject to forskolin treatment to mimic activity-dependent increases in cAMP (Fass et al. 2003). Trichostatin A treatment was also shown to enhance the expression of Nr4a2 and Nr4a1 in wild-type control mice but not in mutant mice with deletions of α and Δ isoforms of Creb indicating that the regulation of Nr4a2 and Nr4a1 was CREB-dependent. Taken together with the in vitro cell culture studies, these findings demonstrated that HDAC inhibitor treatment has a selective effect on gene expression with only certain CRE motif containing genes being regulated. Remarkably, the silencing of Nr4a2, through delivery of small interfering RNA to the hippocampus, has been shown to attenuate the memory enhancing effects of HDAC3 loss-of-function in test of novel object memory (McQuown et al. 2011). This suggests a key role for Nr4a2 target genes in the regulation of novel object memory. Of note, Nr4a2 has been shown to exist in co-repressor complexes that repress genes with TCF/LEF binding sites through interaction with Lef-1 that upon accumulation of sufficient levels of β-catenin become remodeled to CBP-containing co-activator complexes that can activate genes (Saijo et al. 2009). Given previous data suggesting a critical role for β-catenin in memory consolidation (Maguschak, Ressler 2008), it is tempting to speculate that β-catenin, separate from its validated synaptic functions, may also play a role in memory consolidation through an Nr4a2-dependent transcriptional mechanism. This mode of regulating Nr4a2-mediated gene expression may possibly underlie, at least in part, the beneficial effects of lithium and other GSK3 inhibitors that increase β-catenin levels on memory in mouse models of Alzheimer’s disease (Fiorentini et al. 2010; Toledo et al. 2010).
1.4 Enhancement of Memory Formation by HDAC Inhibitors
Collectively, the ability of HDAC inhibitors to enhance memory formation has implications for the treatment of cognitive deficits, such as Alzheimer’s disease, and learned fear disorders (e.g., posttraumatic stress disorder, panic, and phobias) and related anxiety disorders (reviewed in Levenson and Sweatt 2005; Barret and Wood, 2008). Although it can be inferred from the biochemical and behavioral effects of HDAC inhibitors, there is currently only limited knowledge of their pharmacokinetics and brain penetration, which is an area of investigation absent from most studies that needs to be addressed in order to make headway in future probe development (see below in section 1.4.2 for more discussion).
Studies performed to date with the HDAC inhibitors in various rodent models of learning and memory are summarized in Table 1. Key differences when comparing the results in the literature include: 1) the structural class of the inhibitor, 2) the rodent species (rat vs. mouse), 3) the genotype (wildtype vs. genetic model), 4) the animal age, 5) the route of compound administration, 6) the dose of compound administered, and 7) the time-frame of compound administration relative to the behavior (acute vs. chronic).
1.4.1 Isoform Selectivity of HDAC Inhibitors That Enhance Memory Formation
Harnessing the therapeutic potential of HDAC inhibitors linked to cognitive enhancement, and understanding the mechanistic basis through which HDACs regulate the molecular mechanisms of memory, requires knowledge of the specific HDAC family member(s) involved. Standard approaches to measuring HDAC activity involve the use of a radioactive [3H]acetyl-histones (or peptide) substrates that require organic extraction prior to scintillation counting and thus are not readily adaptable to a high-throughput screening format to drive medicinal chemistry. To overcome this limitation, fluorometric deacetylase assays making use of acetylated tripeptide substrates based upon the N-terminal tail of histone H4 Lys12 that are amide-coupled to 7-amino-4-methylcoumarin (AMC) have been developed (Lahm et al. 2007; Bradner et al. 2010). With these substrates, it is now possible to measure a robust deacetylase activity for both class I and class IIa/ HDACs affording the ability for the first time to accurately determine the selectivity of HDAC inhibitors (Lahm et al. 2007; Kilgore et al. 2010; Bradner et al. 2010). Important considerations in these experiments include: 1) running the assays under conditions of enzyme linearity, 2) use of the acetylated substrate at its Km, 3) proper normalization to DMSO controls since DMSO is known to inhibit HDAC activity, 4) the proper use of a trypsin counter screen to rule out inhibition of trypsin, and 5) most importantly to determine the purity and quality of the enzymatic preparation being used.
Using recombinant human HDACs expressed and purified from Sf9 insect cells, the half-maximal inhibitory concentrations (IC50s) for several known HDAC inhibitors that enhance memory formation have been determined using HDAC1-9 with HDAC10 and HDAC11 remaining challenging for routine assaying using currently available synthetic substrates (Kilgore et al. 2010; Brander et al. 2010). For example, as shown in Table 2, the carboxylic acids, valproic acid and butyric acid both have IC50s in the micromolar range against the class I HDAC1-3 and HDAC8, with effectively no inhibition against class IIa HDAC family members or the class IIb HDAC6. In contrast, hydroxamates, such as SAHA, demonstrates nanomolar potency toward HDAC1-3 and 6 and HDAC8, while having IC50s > 25 μM for class IIa enzymes. Here the use of the MAZ1675 substrate for measuring class IIa isoform activity reveals that previously published data showing inhibition of SAHA (and other hydroxamates such as trichostatin A) of class IIa isoforms is most likely due to a contaminating class I HDAC activity (e.g., HDAC3 bound to HDAC4/5) being measured (Lahm et al. 2007; Bradner et al. 2010). An additional class of memory enhancers recently described by McQuown et al. (2011) is that represented by the ortho-aminoaniline RGFP136. This class of compounds is known to show slow-on/slow-off inhibition kinetics and to provide selective inhibition of class I HDACs. In the case of RGFP136, the greatest potency reported is toward HDAC3. However, given the slow-binding kinetics for this class of compounds, the relationship between in vitro selectivity and in vivo selectivity given their pharmacokinetic profile when administered acutely requires further clarification (see below in section 1.4.2 for more discussion).
Table 2.
In vitro HDAC IC50 (μM) with isoform-specific substrates.
| Class I | Class IIa | Class IIb | |||||
|---|---|---|---|---|---|---|---|
| Type | Compound | HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC5 | HDAC6 |
| carboxylic acid | butyric acid1 | 8.3 | 7.0 | 4.8 | 10.4 | >2000 | >2000 |
| carboxylic acid | valproate1 | 35.5 | 59.3 | 218.5 | 97.1 | >2000 | >2000 |
| hydroxamic acid | SAHA1 | 0.002 | 0.003 | 0.006 | 0.7 | 19.3 | 0.004 |
| ortho-aminoaniline | RGFP1362 | 5.2 | 3.0 | 0.4 | nd | nd | nd |
Overall, that valproic acid, butyric acid, SAHA, and RGFP136 share in common the inhibition of the class I HDACs suggests that in vivo the activity of all, or a subset, of these HDAC family isoforms may be responsible for inhibiting memory formation at least for the types of memory tested to date. Consequently, inhibitors that can target class I HDAC isoforms are an active area of investigation. Efforts are under way, in research groups both in academia and industry, to optimize such compounds in terms of their selectivity and pharmacological properties.
1.4.2 Brain Penetration and Pharmacokinetic Assessment of Epigenetic Probes
One of the major issues associated with the use of small-molecule probes that, in the context of targeting epigenetic mechanisms involved in memory processes and behaviors, to date has received little attention but will increasingly need to be addressed in order to advance the field toward a precise understanding of molecular mechanisms and efforts toward novel therapeutic developments, is the study of their absorption, distribution, metabolism, excretion and pharmacokinetics (ADME/PK).
Overall, the extent and rate of brain penetration by small-molecule probes is affected by a number of parameters, including its physiochemical properties, plasma exposure, intrinsic blood-brain barrier permeability, and plasma and brain protein binding (Hammarlund-Udenaes et al. 2008; Lacombe et al. 2011). Therefore in order to properly interpret efficacy results and infer aspects of the underlying mechanism and selectivity in vivo, key ADME/PK parameters that should be measured include, but are not limited to: 1) Cmax, the peak concentration of the probe in the tissue of interest (e.g. brain and blood plasma); 2) T1/2, the biological half-like or the time required for the concentration of probe toe become half its original amount; and 3) AUC, the area under the curve or the integral of the drug concentration over time that provides a metric of the exposure to the probe. However, while determining total brain concentrations measured during in vivo experiments as a function of time and dose provides a useful starting point to relate to exposure achieved to known in vitro potency and selectivity properties, only the fraction of the small-molecule probe that is free and unbound to plasma and brain proteins is able to interact with its intended target. For these reasons, brain concentration levels should ideally be corrected by an estimate of the fraction unbound measured through in vitro experiments using brain tissue homogenate or brain tissue slices as this has been shown to correlate better with a variety of target occupancy and pharmacodynamic readouts (Liu, Chen, & Smith, 2008; Read & Braggio, 2010; Summerfield et al., 2007). Furthermore, since the ADME/PK properties of small-molecule probes can be affected by the dose administered, the site of administration, and the vehicle through alteration of the absorption rate and other parameters, as well as be variable between species, significant care needs to be taken when comparing the results between different studies and with different small-molecule probes, particularly in the absence of analysis of pharmacodynamic markers and when moving between different model organisms.
In the specific case of interpreting the in vivo results from administering HDAC inhibitors, these ADME/PK considerations are of paramount importance because of the fact that detailed enzymatic characterization and mechanistic pharmacology studies have begun to reveal that different structural classes of HDAC inhibitors have different rates of association and dissociation from each HDAC isoforms. For example, hydroxamic acids have been observed to be ‘fast-on/fast-off’ inhibitors and certain benzamides ‘slow-on/slow-off’ inhibitors with consequently different mechanisms of inhibition (Cho et al. 2008). While these differences in kinetic properties can be controlled for in closed in vitro systems to enable equilibrium affinity measurements, in an in vitro setting the extent and duration of responses to small molecule-probe/receptor interactions depends upon the ‘residence time’ during which the small-molecule probe is actually engaged with its receptor (Tummino, Copeland 2008). As a consequence, without detailed knowledge of the PK profile of HDAC inhibitors in plasma and brain tissues it is problematic to interpret the extent and rate at which different HDAC isoforms are inhibited.
While administering probes directly into the brain has the advantage of bypassing a number of these ADME/PK issues, and has been used effectively to demonstrate a critical role for the hippocampus in HDAC inhibitor induced memory formation and for fear memory extinction (Vecsey et al. 2007; Lattal et al. 2007), a major limitation with this approach is the inability to accurately determine the actual concentration that the target is being exposed to, which limits the accuracy through which one can infer aspects of selectivity between different targets. These questions are becoming of increased importance for the interpretation of the effects of selective HDAC inhibitors and lead optimization efforts aiming to develop drug candidates. A second limitation, more from the perspective of target validation for therapeutic development, is the inability to discern the consequence of exposing other brain regions or tissues in the body. For example, if a target of interest is expressed in other tissues where it plays a critical role, for example the liver, then it is possible that the effects of inhibiting the target of interest in that tissue may limit the safety window or obscure the desired effects.
Finally, while the measurement and optimization of these ADME/PK properties of probe compounds and candidate therapeutics will undoubtedly remain a bottleneck for future probing epigenetic mechanisms in the brain with pharmacological probes, advances in two areas are likely to impact the extent and rate at which these problems are addressed. The first is the development of improved human and animal cell-based blood-brain barrier models that will aid in probe design particularly in the early stages of optimization of novel probes and the transition in to in vivo studies (Lacombe et al. 2011). Second, there are nascent efforts to develop ‘epigenetic imaging’ probes that are expected to allow the comprehensive assessment of the pharmacokinetics of epigenetic probes, the expression levels of particular regulators, receptor occupancy, and potentially even enzymatic activity in vivo in live animals, including within the CNS (Reid et al. 2009; Hooker et al. 2010). For example, Hooker et al. (2010) characterized the pharmacokinetics and distribution of the benzamide-based HDAC inhibitor MS-275 in the brain using positron emission tomography (PET) through carbon-11 labeling. These studies demonstrated that [(11)C]MS-275 had poor brain penetration when administered intravenously to non-human primates, providing important information to guide further studies using this probe in the CNS and a path forward for using PET to assess brain penetration of novel HDAC inhibitors that can be labeled using carbon-11.
1.4.3 Pharmacodynamic Assessment of Epigenetic Probes
Besides ADME/PK issues, a second major issues associated with the use of small-molecule probes that to date, in the context of targeting epigenetic mechanisms involved in memory processes and other behaviors, has also received little attention is the issue of the relevant pharmacodynamic markers for correlating target engagement in vivo with measurable PK properties and functional readouts. In the case of HDACs, while a number of studies have published that either intra-brain or systemic administration of HDAC inhibitors increases histone acetylation, there is little consensus on the doses used, routes of administration (intra-brain or systemically) or vehicles. Additionally, the methods used for analyzing the acetylation states of histones have varied as have the particular histone modification sites that have been measured. In light of the studies by Choudhary et al. (2009) described above, which have revealed more than 3,600 specific lysine acetylation sites on 1,750 proteins in human cell lines, the choice of a relevant pharmacodynamic marker for small-molecule probes studies and conclusions drawn from it with respect to mechanism need to be carefully considered particularly with long-term chronic administration of drug or genetic manipulations spanning days to months.
1.5 Genetic Studies of HDAC Isoforms Involved in Memory Formation
To complement the use of small-molecule probes described above, as summarized in Fig. 2, recent studies using gain-of-function and loss-of-function mouse models revealed that neuron-specific (from the Mapt gene locus) overexpression of HDAC2, but not that of HDAC1, decreased dendritic spine density, synapse number, synaptic plasticity and memory formation (Guan et al. 2009). Here the coordinated morphological, biochemical, and physiological changes indicate that HDAC2 plays a key role in regulating gene expression programs involved in the establishment of the neurocircuitry involved in memory formation. In support of this notion, conversely, Hdac2 deficiency resulted in increased synapse number and memory facilitation. These findings are similar to what has been observed with chronic treatment (e.g. 7–21 days) with HDAC inhibitors in mice. Notably, the reduced synapse number and learning impairment of HDAC2-overexpressing mice could be ameliorated by chronic treatment with the HDAC inhibitor SAHA. Correspondingly, treatment with SAHA failed to further facilitate memory formation in Hdac2-deficient mice. Collectively, these data suggest that HDAC2 functions as a memory suppressor and that targeting HDAC2-mediated neuroplasticity selectively may provide a means to treat cognitive disorders (Guan et al. 2009).
Figure 2. Evidence for HDAC2 as a memory suppressor.
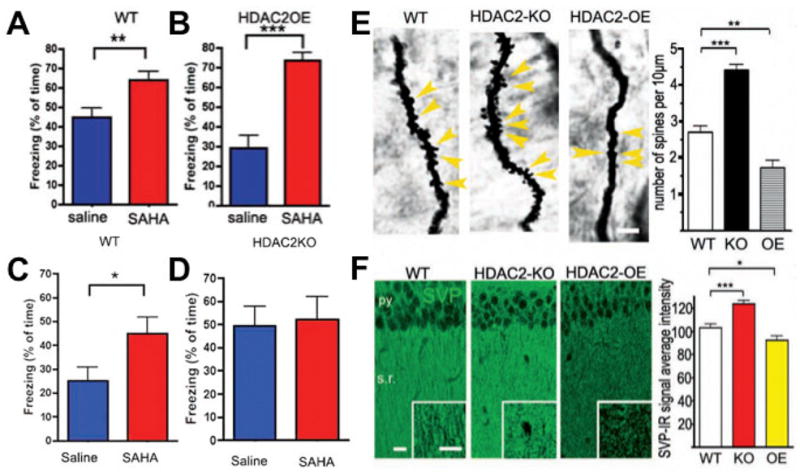
[adapted from Guan et al. 2009]. (A) SAHA treatment enhances contextual fear conditioning in wildtype (WT) mice when administered systemically for 10 days (25 mg/kg) and restores fear conditioning to WT levels in the HDAC2-overexpression (OE) mice. (C) Enhancement of fear conditioning in WT littermates but (D) no further enhancement of fear conditioning in HDAC2-knockout (KO) mice. (E) Golgi staining and quantification of enhanced synaptic spine density in HDAC2-KO mice and reduced spine density in HDAC2-OE mice in the CA1 region of hippocampus. (F) Synaptophysin staining and quantification of enhanced synaptic density in HDAC2-KO mice and reduced density in HDAC2-OE in the CA1 region of hippocampus.
Recent studies, described in the accompanying review from Wood et al. have also revealed a role for the class I HDAC3 in long-term memory for object location as well as long-term memory for a familiar object (McQuown et al. 2011). While the role of HDAC3 in other types of memory and models of neurodegeneration remain important open questions, this exciting finding provides evidence for multiple possible roles of HDACs in neuroplasticity. Of note, Nr4a2, which as discussed above, was shown to be required for the memory enhancing effects of selective loss of HDAC3 in the hippocampus (McQuown et al. 2011), has been shown to interact with the CoREST corepressor complex (Saijo et al. 2009). Since CoREST is a major component of HDAC2 complexes in neurons (Ballas et al. 2001; Lakowski et al. 2006), these findings suggesting a possible point of cross-talk between HDAC3- and HDAC2-mediated regulation of memory formation.
It remains important to keep in mind a number of considerations when comparing genetic models to the effects of administering small-molecule probes. First, even with conditional deletion of genes using Cre/Lox-mediated approaches with crossing to cell-type specific Cre drivers or viral delivery of Cre recombinase that the kinetics of the manipulation are often of a much more prolonged nature (months to days) compared to a pharmacological approach with small molecules. Second, with a mutation in a gene involved in epigenetic regulation, resulting in most case in the deletion of that gene, the entire protein is removed. In contrast, a small-molecule probe will only interact directly with a limited number of amino acids in a target potentially causing a selective loss of a subset of many possible functions. As discussed above, given that many epigenetic regulators, particularly HDACs are part of large macromolecular protein complexes with multiple domains and protein-protein interactions (Bantscheff et al. 2011), these differences between a complete loss-of-function(s) and small-molecule perturbations may result in very different phenotypic effects. Understanding these differences will also be informed by gaining insight into the pharmacokinetics and other properties of the small-molecule probes as described below in more detail.
1.6 Cross-talk Between Epigenetic Mechanisms in Neurons
Besides potential for cross-talk between different HDAC isoform-containing co-repressor complexes, while histone acetlyation has emerged as a critical regulator of neuronal gene expression involved in phenotypic and behavioral plasticity, there is growing evidence from a variety of model systems for functional cross-talk between different types of histone post-translational modifications. In addition, many chromatin remodeling complex components have been shown to contain one or more chromatin-binding modules, such as the acetyl-lysine-binding bromodomain and methyl-lysine-binding chromodomain, that in a combinatorial fashion can provide an important mechanism for epigenetic control of gene regulation through controlling protein localization (Ruthenberg et al. 2007; Oliver et al. 2010). In light of these findings, Borrelli et al. (2008) have proposed a step-wide process for consolidating epigenetic information in neurons. According to this model, successive, and interconnected, histone post-translational modifications lead to a transition from an “unlocked” chromatin state characterized by dynamic and charged post-translational modifications, such as phosphorylation and acetylation, to a “locked” fully committed state that can lead to either gene expression or silencing, which is characterized by non-charged, stable modifications of both histone and DNA by methylation. The precise mechanisms that determine the transition between chromatin states and how these mechanisms ultimately integrate information coming from experience-dependent neural activity will require continued study and integration of research findings at multiple levels.
1.7 Unanswered Questions and Future Directions
The role established for HDACs in long-term memory formation, and other types of long lasting changes in behavioral plasticity, exemplifies how epigenetic mechanisms have the dual capacity to elicit both plasticity and stability in the nervous system. Like any nascent field, there remains many unanswered questions and critical needs in order to better understand role of epigenetic mechanisms in the brain a number of which we summarize here.
1.7.1 Understanding the Substrates of Chromatin-Modifying Enzymes
Despite their original namesake, which was based upon the first known substrate, HDACs are more accurately considered as general lysine deacetylases since their substrate specificity almost definitely extends beyond histones. While this is known to be the case for HDAC6, which functions as a tubulin deacetylase (Hubbert et al. 2002; Haggarty et al. 2003), the extent of this remains largely unknown for each family member. In support of this notion, high-resolution mass spectrometry studies in non-neuronal tissues have identified 3,600 lysine acetylation sites on a total of 1,750 proteins (Choudhary et al 2009). Analysis of these acetylation targets has suggested a key role for acetylation in the regulation of large macromolecular complexes involved in diverse cellular processes, such as chromatin remodeling, cell cycle, splicing, nuclear transport, and actin nucleation. Since the levels of many of these non-histone acetylation sites changed in response to the deacetylase inhibitors SAHA and MS-275, this suggests the need for caution when interpreting effects of HDAC inhibitors on neuroplasticity and behavior as being caused by alteration of histone acetylation and epigenetic regulation. In support of this notion, a role for the acetylation of the transcription factor NF-kappaB in memory formation has been reported (Yeh et al. 2004). By extenstion, the substrates of other histone-modifying enzymes, such as the methyltransferases, also need to be understood in greater detail.
1.7.2 Understanding Relevant Target Genes
The target genes of HDACs involved in mediating normal learning and memory and memory formation enhancement by HDAC inhibitor treatment need to be understood in greater detail in order to discern those changes in gene expression that are necessary and sufficient to affect memory processes. As part of these efforts, it will be important to understand whether the effect of acute HDAC inhibitor treatment that converts short-term memories into long-term and persistent memories involves the same molecular and cellular mechanisms as those operating with chronic HDAC inhibitor treatment over multiple days. Such studies would provide insight into the specific genomic loci and epigenetic mechanisms that are operating and thereby potentially provide additional targets for therapeutic intervention. In particular, the determination of whether the modulation of a single HDAC target gene is sufficient to alter the effects of HDAC inhibition, as has been recently shown to be the case for novel object memory in HDAC3-floxed mice by siRNA-mediated silencing of Nr4a2 (McQuown et al. 2011), needs to be explored for different types of memory processes, different types of genetic perturbations, and by different pharmacological probes of HDACs and other chromatin-modifying enzymes.
1.7.3 Modulation of HDACs in a Cell-Type and Circuit-Specific Manner
In the case of the role of HDAC2 as a memory suppressor, since the genetic loss-of-function of HDAC2 was not brain-specific, the use of floxed alleles crossed with Cre recombinase-drivers expressed in different brain regions and cell types should further clarify the contribution of HDAC2 and other HDAC family members to memory-related neuroplasticity (Montgomery et al. 2009). However, such studies will still not prove that it is the deacetylase activity per se of HDAC2 that mediates these effects because of the known role of HDACs to recruit other co-repressor complex members. To do so will require knock-in mutations of key catalytic site residues or viral-mediated approaches ideally in combination with the use of selective inhibitors of the deacetylase activity.
1.7.4 Understanding the Macromolecular Machinery
At the most basic level, we still have a far from complete understanding of chromatin-modifying and chromatin-remodeling complexes within the brain and the extent to which these macromolecular complexes may vary or be dynamically regulated over developmental time or within different cell types and brain regions in response to experience-dependent neuronal activity (Bantscheff et al. 2011). Knowledge in this area would provide greater insight into the molecular mechanisms involved in chromatin-mediated neuroplasticity. Given the existence of a variety of other post-translational modifications of chromatin (Borrelli et al. 2008; Ruthenburg et al. 2007; Gupta et al., 2010), there are bound to be additional targets that can be modulated to have beneficial effects on neuroplasticity in the brain. Related to this, our understating higher-order chromatin structures in post-mitotic neurons and the extent, if any, of change over developmental time and in response to experience remains very limited. One intriguing example of these types of higher-order changes in chromatin structure is made evident by the use of the fluorescent DNA binding dye, TO-PRO-3, which was shown to be reveal unique arrangements of chromatin in the nuclei of medium spiny neurons and interneurons that allowed their identification within the striatum (Matamales et al. 2009). These findings suggest that a key aspect of cell identity and different classes of neurons in the adult nervous system may involve dramatically different arrangement of chromatin within nuclei.
1.7.5 Developing Novel Small-Molecule Probes
The dynamic and combinatorial nature of epigenetic mechanisms means that the ability to manipulate the underlying molecular mechanisms in the nervous system provides a particular challenge. To address this challenge, developing a new generation of small-molecule probes with well-characterized in vitro selectivity, which are blood-brain barrier penetrant, and have suitable pharmacological properties is critically needed to move the field forward and to help further evaluate the potential for enhancing memory and other process of chromatin-mediated neuroplasticity processes pharmacologically.
Although the clinical use of phenylbutyrate, valproate, and SAHA demonstrates that non-selective HDAC inhibitors are, in general, well-tolerated in animals and humans, it is likely the case that compounds with selectivity for different family members will present less untoward side effects in patients and perhaps have better efficacy for use as potential treatment of neurodegenerative and neuropsychiatric disorders. In the case of HDACs, those inhibitors that do exist are thought all to act as substrate competitive inhibitors. Identifying new structural classes and potentially allosteric inhibitors may provide improved selectivity and further insight into different modes of regulation. Toward this end, Marcaurelle et al. (2010) has recently described a new class of macrocyclic HDAC inhibitors that show mixed inhibition kinetics and the ability to alter histone H4K12 acetylation levels in mouse neurons. Beyond the deacetylase activity of HDAC complexes, it may be possible to selectively target specific protein-protein interactions between co-repressor complex members. For example, in the case of HDAC3 its deacetylase activity is critically dependent upon the presence of the deacetylase activating domain of NCOR, suggesting that selectively disrupting this interaction may inhibit HDAC3 deacetylase activity. In the case of HDAC2, there are also key interactions with proteins, such as members of the mSIN3 family, which could potentially be targeted with small molecules.
1.8 Conclusion: Cajal’s Second Legacy & Human Chromatinopathies
While it is well recognized that Ramón y Cajal’s pioneering investigation on the structure of neurons and their connectivity provided a foundation for modern neuroscience, it is not well known that at the beginning of the 20th Century Cajal then turned his attention toward providing the first detailed monograph “El núcleo de las células piramidales del cerebro humano y de algunos mamíferos” on the structure and function of the neuronal nucleus at the level of light microcopy (Cajal, 1910). As reviewed in Lafarga et al. (2009), using adaptations of Golgi’s reduced silver nitrate preparation, Cajal made detailed drawings of what we now recognize as the nucleolus, nuclear matrix, nuclear speckles of splicing factors, Cajal (accessory) body, and the double nuclear membrane of pyramidal neurons from the human cerebral cortex. Remarkably, Cajal’s drawings of pyramidal neuron nuclei depicted “neutrophil granules” or nuclear foci that bear a striking resemblance to what are now known as sites of transcriptionally active chromatin containing acetylated histone proteins. Now, over a century latter with technology and resolution that even Cajal could not have imagined, the field of neuroepigenetics is poised to understand neuronal nuclear structure and function, including the exact nature of the genomic loci corresponding to these “neurotrophil granules” and how these structures change in response to experience-dependent input into the nervous system.
Lastly, beyond in vitro models and animal models, clues to the overall importance of epigenetic mechanisms in neuroplasticity and cognition are provided by human genetics. Here, there is an emerging recognition of the existence a variety disorders that may be termed ‘chromatinopathies’ due to the presence of altered epigenetic state either as a cause or consequence of disease pathophysiology (Gräff and Mansuy 2009; van Bokhoven and Kramer 2010). A causal role for epigenetic dysregulation in disorders with cognitive phenotypes is best demonstrated by a number of monogenic, Mendelian disorders in which mutations have been identified in chromatin regulators (Table 3). These include histone acetlyation, as in the case of Rubinstein-Taybi syndrome and mutations in the gene encoding the Creb Binding Protein (CBP), histone methylation, as in the case of Kleefstra subtelomeric deletion syndrome that causes loss of expression of the gene encoding Euchromatic Histone Methyltransferase 1 (EHMT1), and DNA methylation, as in the case of MeCP2 mutations in Rett syndrome. With advances in the ability to perform whole exome and genome sequencing that are facilitating the mapping and identification of human disease causing genes, it can be anticipated that additional mutations in genes involved in epigenetic mechanisms will be identified. In light of these developments, and what might called Cajal’s second legacy, the burgeoning field of neuroepigenetics promises to provide a long-lasting impact on our understanding of memory and other forms of neuroplasticity mediated through chromatin.
Table 3.
List of human cognitive disorders with epigenetic dysregulation due to mutations or alterations in chromatin-modifying, binding, or regulatory processes.
| OMIM | Disorder | Type | Cause |
|---|---|---|---|
| 180849 | RUBINSTEIN-TAYBI SYNDROME 1 | HAT: Histone Acetylation | Mutations in the gene encoding CREB-binding protein (CREBBP) |
| 613684 | RUBINSTEIN-TAYBI SYNDROME 2 | HAT: Histone Acetylation | Mutations in the gene encoding EP300 |
| 300624 | FRAGILE X MENTAL RETARDATION SYNDROME | DNA Methylation | Mutations in the gene encoding FMR1 (CGG repeat expansion) |
| 303600 | COFFIN-LOWRY SYNDROME | Histone Phosphorylation | Mutations in the gene encoding RSK2, ribosomal protein S6 kinase, 90kDa polypeptide 3 (RPS6KA3) |
| 610253 | KLEEFSTRA SYNDROME, 9q34 .3 SUBTELOMERIC DELETION SYNDROME | HMT: Histone methylation | Mutations in EHMT1 histone methyltransferase |
| 117550 | SOTOS SYNDROME | HMT: Histone Methylation | Mutations in the gene encoding NSD1 (nuclear receptor binding SET domain protein 1) histone methyltransferase, |
| 300263 | SIDERIUS X-LINKED MENTAL RETARDATION SYNDROME, PHF8 | HDM: Histone Demethylation | Mutations in the gene encoding PHF8 (PHD finger protein 8), a JmjC domain containing histone demethylase |
| 300534 | MENTAL RETARDATION, X-LINKED, SYNDROMIC, KDM5C | HDM: Histone Demethylation | Mutations in the gene encoding KDM5C (JARID1C), a JmjC domain containing histone demethylase |
| 301040 | ALPHA-THALASSEMIA/MENTAL RETARDATION SYNDROME, NONDELETION TYPE, X-LINKED, ATRX | ATP-dependent Chromatin Remodeling | Mutations in ATRX component of SWI/SNF-like remodeling complex |
| 300166 | MICROPHTHALMIA, SYNDROMIC 2, BCOR | PcG group HMT and Jmjc Domain Containing HDM Chromatin Remodeling Complexes | Mutation sin the BCL6 corepressor gene (BCOR) |
| 312750 | RETT SYNDROME | MDB: DNA Methylation | Mutations in the gene encoding methyl-CpG-binding protein-2 (MECP2) |
| 242860 | IMMUNODEFICIENCY-CENTROMERIC INSTABILITY-FACIAL ANOMALIES SYNDROME | DMT: DNA Methylation | Mutations in the gene encoding DNA methyltransferase-3B (DNMT3B) |
| 105830 | ANGELMAN SYNDROME (15q11-q13) | DNA Methylation (imprinting) | Loss of maternal contribution of 15q11-q13 genes |
| 17627 | PRADER-WILLI SYNDROME (15q11-q13) | DNA Methylation (imprinting) | Loss of paternal contribution of 15q11-q13 genes |
Abbreviations: DNMT, DNA methyltransferase; HAT, histone acetyltransferases; HMT, histone methyltransferase; HDM, histone demethylases; Jmjc, Jumonji C; MBD, methylyated DNA binding proteins; PcG, Polycomb group; PHD, plant homeodomain; PHF, PHD finger protein
Neuroepigenetic research aims to understand the role of chromatin in brain plasticity.
Histone deacetylases (HDAC) are critical regulators of neuroplasticity.
Epigenetic mechanisms may provide new targets for therapeutic development.
Certain human diseases can be classified as æchromatinopathiesÆ.
Acknowledgments
We would like to thank members of the Haggarty and Tsai laboratories, as well as the Stanley Center for Psychiatric Research for their contributions to the critical questions and work ongoing. S.J.H is supported through funding from the NIH (R01DA028301, R01DA030321, RC1AG035711) and the Stanley Medical Research Institute. L.H.T. is supported through funding from the NIH (R01DA028301, RC1AG035711, RO1NS051874, PO1AG027916), the Stanley Medical Research Institute, and the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Anamika K, Krebs AR, Thompson J, Poch O, Devys D, Tora L. Lessons from genome-wide studies: an integrated definition of the coactivator function of histone acetyl transferases. Epigenetics Chromatin. 2010;3:18. doi: 10.1186/1756-8935-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME, Burger C, Moniwa M, Davie JR, Bowers WJ, Federoff HJ, Rose DW, Rosenfeld MG, Brehm P, Mandel G. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–365. doi: 10.1016/s0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dümpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol. 2011;29:255–265. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- Barrett RM, Wood MA. Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn Mem. 2008;15:460–467. doi: 10.1101/lm.917508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci. 2006;7:137–151. doi: 10.1038/nrn1846. [DOI] [PubMed] [Google Scholar]
- Berndsen CE, Denu JM. Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr Opin Struct Biol. 2008;18:682–689. doi: 10.1016/j.sbi.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieliauskas AV, Pflum MK. Isoform-selective histone deacetylase inhibitors. Chem Soc Rev. 2008;37:1402–1413. doi: 10.1039/b703830p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozon B, Davis S, Laroche S. Regulated transcription of the immediate-early gene Zif268: mechanisms and gene dosage-dependent function in synaptic plasticity and memory formation. Hippocampus. 2002;12:570–577. doi: 10.1002/hipo.10100. [DOI] [PubMed] [Google Scholar]
- Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6:238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ. Distribution of histone deacetylases 1–11 in the rat brain. J Mol Neurosci. 2007;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- Bredy TW, Barad M. The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn Mem. 2008;15:39–45. doi: 10.1101/lm.801108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. 2007;14:268–276. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajal SR. El núcleo de las células piramidales del cerebro humano y de algunos mamíferos. Trab Lab Invest Biol. 1910;8:27–62. [Google Scholar]
- Chawla S, Vanhoutte P, Arnold FJ, Huang CL, Bading H. Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J Neurochem. 2003;85:151–159. doi: 10.1046/j.1471-4159.2003.01648.x. [DOI] [PubMed] [Google Scholar]
- Chou CJ, Herman D, Gottesfeld JM. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J Biol Chem. 2008;283:35402–35409. doi: 10.1074/jbc.M807045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Owens GC, Makarenkova H, Edelman DB. HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS One. 2010;5:e10848. doi: 10.1371/journal.pone.0010848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Colón-Cesario WI, Martínez-Montemayor MM, Morales S, Félix J, Cruz J, Adorno M, Pereira L, Colón N, Maldonado-Vlaar CS, Peña de Ortiz S. Knockdown of Nurr1 in the rat hippocampus: implications to spatial discrimination learning and memory. Learn Mem. 2006;13:734–744. doi: 10.1101/lm.407706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Orsi SA, Moore AN. Histone deactylase inhibition combined with behavioral therapy enhances learning and memory following traumatic brain injury. Neuroscience. 2009;163:1–8. doi: 10.1016/j.neuroscience.2009.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dompierre JP, Godin JD, Charrin BC, Cordelières FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fass DM, Butler JE, Goodman RH. Deacetylase activity is required for cAMP activation of a subset of CREB target genes. J Biol Chem. 2003;278:43014–43019. doi: 10.1074/jbc.M305905200. [DOI] [PubMed] [Google Scholar]
- Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- Fiorentini A, Rosi MC, Grossi C, Luccarini I, Casamenti F. Lithium improves hippocampal neurogenesis, neuropathology and cognitive functions in APP mutant mice. PLoS One. 2010;5:e14382. doi: 10.1371/journal.pone.0014382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Foley AG, Gallagher HC, Murphy KJ, Regan CM. Pentyl-4-yn-valproic acid reverses age-associated memory impairment in the Wistar rat. Neurobiol Aging. 2004;25:539–546. doi: 10.1016/j.neurobiolaging.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Fontán-Lozano A, Romero-Granados R, Troncoso J, Múnera A, Delgado-García JM, Carrión AM. Histone deacetylase inhibitors improve learning consolidation in young and in KA-induced-neurodegeneration and SAMP-8-mutant mice. Mol Cell Neurosci. 2008;39:193–201. doi: 10.1016/j.mcn.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Francis YI, Fà M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, Arancio O. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer’s disease. J Alzheimers Dis. 2009;18:131–139. doi: 10.3233/JAD-2009-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräff J, Mansuy IM. Epigenetic dysregulation in cognitive disorders. Eur J Neurosci. 2009;30:1–8. doi: 10.1111/j.1460-9568.2009.06787.x. [DOI] [PubMed] [Google Scholar]
- Grozinger CM, Schreiber SL. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem Biol. 2002;9:3–16. doi: 10.1016/s1074-5521(02)00092-3. [DOI] [PubMed] [Google Scholar]
- Guan Z, Giustetto M, Lomvardas S, Kim JH, Miniaci MC, Schwartz JH, Thanos D, Kandel EE. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, Paylor RE, Lubin FD. Histone methylation regulates memory formation. J Neurosci. 2010;30:3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain selective small molecule inhibitor of HDAC6-mediated tubulin deacetylation. Proc Natl Acad Sci USA. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty SJ, Wong JC, Koeller KM, Butcher RA, Schreiber SL. Multidimensional chemical genetic analysis of diversity-oriented synthesis-derived deacetylase inhibitors using cell-based assays. Chem Biol. 2003;10:383–396. doi: 10.1016/s1074-5521(03)00095-4. [DOI] [PubMed] [Google Scholar]
- Hammarlund-Udenaes M, Friden M, Syvanen S, Gupta A. On the rate and extent of drug delivery to the brain. Pharm Res. 2008;25:1737–50. doi: 10.1007/s11095-007-9502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooker JM, Kim SW, Alexoff D, Xu Y, Shea C, Reid A, Volkow N, Fowler JS. Histone deacetylase inhibitor, MS-275, exhibits poor brain penetration: PK studies of [C]MS-275 using Positron Emission Tomography. ACS Chem Neurosci. 2010;1:65–73. doi: 10.1021/cn9000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Ito H, Yoshimura N, Kurosawa M, Ishii S, Nukina N, Okazawa H. Knock-down of PQBP1 impairs anxiety-related cognition in mouse. Hum Mol Genet. 2009;18:4239–4254. doi: 10.1093/hmg/ddp378. [DOI] [PubMed] [Google Scholar]
- Jones MW, Errington ML, French PJ, Fine A, Bliss TV, Garel S, Charnay P, Bozon B, Laroche S, Davis S. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat Neurosci. 2001;4:289–296. doi: 10.1038/85138. [DOI] [PubMed] [Google Scholar]
- Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2010;35:870–880. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapska E, Kaczmarek L. A gene for neuronal plasticity in the mammalian brain: Zif268/Egr-1/NGFI-A/Krox-24/TIS8/ZENK? Prog Neurobiol. 2004;74:183–211. doi: 10.1016/j.pneurobio.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacombe O, Videau O, Chevillon D, Guyot AC, Contreras C, Blondel S, Nicolas L, Ghettas A, Benech H, Thevenot E, Pruvost A, Bolze S, Kraczkowski L, Prévost C, Mabondzo A. In-Vitro Primary Human and Animal Cell-Based Blood-Brain Barrier Models as a Screening Tool in Drug Discovery. Mol Pharm. 2011 doi: 10.1021/mp1004614. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Lafarga M, Casafont I, Bengoechea R, Tapia O, Berciano MT. Cajal’s contribution to the knowledge of the neuronal cell nucleus. Chromosoma. 2009;118:437–443. doi: 10.1007/s00412-009-0212-x. [DOI] [PubMed] [Google Scholar]
- Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, Sambucini S, Bottomley MJ, Lo Surdo P, Carfí A, Koch U, De Francesco R, Steinkühler C, Gallinari P. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A. 2007;104:17335–17340. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowski B, Roelens I, Jacob S. CoREST-like complexes regulate chromatin modification and neuronal gene expression. J Mol Neurosci. 2006;29:227–239. doi: 10.1385/JMN:29:3:227. [DOI] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Lattal KM, Barrett RM, Wood MA. Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav Neurosci. 2007;121:1125–131. doi: 10.1037/0735-7044.121.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, O’Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Li S, Murakami Y, Wang M, Maeda K, Matsumoto K. The effects of chronic valproate and diazepam in a mouse model of posttraumatic stress disorder. Pharmacol Biochem Behav. 2006;85:324–331. doi: 10.1016/j.pbb.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Liu X, Chen C, Smith BJ. Progress in brain penetration evaluation in drug discovery and development. J Pharmacol Exp Ther. 2008;325:349–356. doi: 10.1124/jpet.107.130294. [DOI] [PubMed] [Google Scholar]
- MacDonald JL, Roskams AJ. Histone deacetylases 1 and 2 are expressed at distinct stages of neuro-glial development. Dev Dyn. 2008;237:2256–2267. doi: 10.1002/dvdy.21626. [DOI] [PubMed] [Google Scholar]
- Maguschak KA, Ressler KJ. Beta-catenin is required for memory consolidation. Nat Neurosci. 2008;11:1319–1326. doi: 10.1038/nn.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcaurelle LA, Comer E, Dandapani S, Duvall JR, Gerard B, Kesavan S, Lee MD, 4th, Liu H, Lowe JT, Marie JC, Mulrooney CA, Pandya BA, Rowley A, Ryba TD, Suh BC, Wei J, Young DW, Akella LB, Ross NT, Zhang YL, Fass DM, Reis SA, Zhao WN, Haggarty SJ, Palmer M, Foley MA. An aldol-based build/couple/pair strategy for the synthesis of medium- and large-sized rings: discovery of macrocyclic histone deacetylase inhibitors. J Am Chem Soc. 2010;132:16962–16976. doi: 10.1021/ja105119r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Matamales M, Bertran-Gonzalez J, Salomon L, Degos B, Deniau JM, Valjent E, Hervé D, Girault JA. Striatal medium-sized spiny neurons: identification by nuclear staining and study of neuronal subpopulations in BAC transgenic mice. PLoS One. 2009;4:e4770. doi: 10.1371/journal.pone.0004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuown SC, Barrett RM, Matheos DP, Post RJ, Rogge GA, Alenghat T, Mullican SE, Jones S, Rusche JR, Lazar MA, Wood MA. HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci. 2011;31:764–774. doi: 10.1523/JNEUROSCI.5052-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RL, Hsieh J, Barbosa AC, Richardson JA, Olson EN. Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc Natl Acad Sci U S A. 2009;106:7876–7881. doi: 10.1073/pnas.0902750106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. S-nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature. 2008;455:411–415. doi: 10.1038/nature07238. [DOI] [PubMed] [Google Scholar]
- Oliver SS, Denu JM. Dynamic Interplay between Histone H3 Modifications and Protein Interpreters: Emerging Evidence for a “Histone Language”. Chembiochem. 2010 Oct 29; doi: 10.1002/cbic.201000474. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Loinsigh ED, Gherardini LM, Gallagher HC, Foley AG, Murphy KJ, Regan CM. Differential enantioselective effects of pentyl-4-yn-valproate on spatial learning in the rat, and neurite outgrowth and cyclin D3 expression in vitro. J Neurochem. 2004;88:370–379. doi: 10.1111/j.1471-4159.2004.02158.x. [DOI] [PubMed] [Google Scholar]
- Peña de Ortiz S, Maldonado-Vlaar CS, Carrasquillo Y. Hippocampal expression of the orphan nuclear receptor gene hzf-3/nurr1 during spatial discrimination learning. Neurobiol Learn Mem. 2000;74:161–178. doi: 10.1006/nlme.1999.3952. [DOI] [PubMed] [Google Scholar]
- Racki LR, Narlikar GJ. ATP-dependent chromatin remodeling enzymes: two heads are not better, just different. Curr Opin Genet Dev. 2008;18:137–144. doi: 10.1016/j.gde.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read K, Braggio S. Assessing brain free fraction in early drug discovery. Expert Opinion. Review. 2010;6:1–8. doi: 10.1517/17425250903559873. [DOI] [PubMed] [Google Scholar]
- Reid AE, Hooker J, Shumay E, Logan J, Shea C, Kim SW, Collins S, Xu Y, Volkow N, Fowler JS. Evaluation of 6-([(18)F]fluoroacetamido)-1-hexanoicanilide for PET imaging of histone deacetylase in the baboon brain. Nucl Med Biol. 2009;36:247–258. doi: 10.1016/j.nucmedbio.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Nestler EJ. Epigenetic mechanisms in drug addiction. Trends Mol Med. 2008;14:341–350. doi: 10.1016/j.molmed.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Ricobaraza A, Cuadrado-Tejedor M, Pérez-Mediavilla A, Frechilla D, Del Río J, García-Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology. 2009;34:1721–1732. doi: 10.1038/npp.2008.229. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, Glass CK. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvi BR, Cassel JC, Kundu TK, Boutillier AL. Tuning acetylation levels with HAT activators: therapeutic strategy in neurodegenerative diseases. Biochim Biophys Acta. 2010;1799:840–853. doi: 10.1016/j.bbagrm.2010.08.012. [DOI] [PubMed] [Google Scholar]
- Smith BC, Hallows WC, Denu JM. Mechanisms and molecular probes of sirtuins. Chem Biol. 2008;15:1002–1013. doi: 10.1016/j.chembiol.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanko DP, Barrett RM, Ly AR, Reolon GK, Wood MA. Modulation of long-term memory for object recognition via HDAC inhibition. Proc Natl Acad Sci U S A. 2009;106:9447–9452. doi: 10.1073/pnas.0903964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerfield SG, Read K, Begley DJ, Obradovic T, Hidalgo IJ, Coggon S, Lewis AV, Porter RA, Jeffrey P. Central nervous system drug disposition: the relationship between in situ brain permeability and brain free fraction. J Pharmacol Exp Ther. 2007;322:205–213. doi: 10.1124/jpet.107.121525. [DOI] [PubMed] [Google Scholar]
- Sugo N, Oshiro H, Takemura M, Kobayashi T, Kohno Y, Uesaka N, Song WJ, Yamamoto N. Nucleocytoplasmic translocation of HDAC9 regulates gene expression and dendritic growth in developing cortical neurons. Eur J Neurosci. 2010;31:1521–1532. doi: 10.1111/j.1460-9568.2010.07218.x. [DOI] [PubMed] [Google Scholar]
- Toledo EM, Inestrosa NC. Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer’s disease. Mol Psychiatry. 2010;15:272–285. doi: 10.1038/mp.2009.72. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Neve RL, Nestler EJ. Sustained chromatin regulation in hippocampus is a mouse model of depression and antidepressant action: a role for HDAC5. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Tummino PJ, Copeland RA. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry. 2008;47:5481–5492. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- van Bokhoven H, Kramer JM. Disruption of the epigenetic code: an emerging mechanism in mental retardation. Neurobiol Dis. 2010;39:3–12. doi: 10.1016/j.nbd.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AM, Lombardi TL, Abel T. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem. 2005;12:111–119. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Attner MA, Oliveira AM, Brindle PK, Abel T. A transcription factor-binding domain of the coactivator CBP is essential for long-term memory and the expression of specific target genes. Learn Mem. 2006;13:609–617. doi: 10.1101/lm.213906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009;14:51–59. doi: 10.1038/sj.mp.4002099. [DOI] [PubMed] [Google Scholar]
- Yeh SH, Lin CH, Gean PW. Acetylation of nuclear factor-kappaB in rat amygdala improves long-term but not short-term retention of fear memory. Mol Pharmacol. 2004;65:1286–1292. doi: 10.1124/mol.65.5.1286. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Matsuyama A, Komatsu Y, Nishino N. From discovery to the coming generation of histone deacetylase inhibitors. Curr Med Chem. 2003;10:2351–2358. doi: 10.2174/0929867033456602. [DOI] [PubMed] [Google Scholar]