

Abstract
The current “working model” for mammalian base excision repair involves two sub-pathways termed single-nucleotide base excision repair and long patch base excision repair that are distinguished by their repair patch sizes and the enzymes/co-factors involved. These base excision repair sub-pathways are designed to sequester the various DNA intermediates, passing them along from one step to the next without allowing these toxic molecules to trigger cell cycle arrest, necrotic cell death, or apoptosis. Although a variety of DNA-protein and protein-protein interactions are known for the base excision repair intermediates and enzymes/co-factors, the molecular mechanisms accounting for step-to-step coordination are not well understood. In this review, we explore the question of whether there is an actual step-to-step “hand-off” of the DNA intermediates during base excision repair in vitro. The results show that when base excision repair enzymes are pre-bound to the initial single-nucleotide base excision repair intermediate, the DNA is channeled from apurinic/apyrimidinic endonuclease 1 to DNA polymerase β and then to DNA ligase. In the long patch base excision repair sub-pathway, where the 5′-end of the incised strand is blocked, the intermediate after polymerase β gap filling is not channeled from polymerase β to the subsequent enzyme, flap endonuclease 1. Instead, flap endonuclease 1 must recognize and bind to the intermediate in competition with other molecules.
Keywords: DNA repair, DNA polymerase β, single-nucleotide base excision repair (SN BER), long patch base excision repair (LP BER), flap endonuclease 1 (FEN1), AP endonuclease 1 (APE1), uracil-DNA glycosylase (UDG), 5′-deoxyribose phosphate (5′-dRP)
Introduction and Background
Cellular genomic DNA suffers damage from a variety of physical and chemical agents, including ultraviolet light and ionization radiation, alkylating molecules and endogenous reactive oxygen species that accumulate in cells due to environmental stress and natural metabolic processes [1–3]. DNA damage, due to environmental factors and normal metabolic processes inside the cell, occurs at a rate of 1000 to 1000000 molecular lesions per cell per day [4–6]. While this constitutes only a small portion of the human genome's approximately 6 billion bases (3 billion base pairs), misrepaired or unrepaired lesions in critical genes (such as tumor suppression genes) can impede a cell's ability to carry out its function and appreciably increase the likelihood of tumor formation and other adverse conditions.
The single base lesion or strand break is the most common form of DNA damage occurring in the human genome. A DNA base can be lost through spontaneous hydrolysis or can be oxidized and/or alkylated during physiologic metabolism, and also can be modified by exogenous DNA damaging agents [2, 7, 8]. However, cells have evolved several specific pathways to remove such damage and maintain integrity of the genome. In mammalian cells, the primary means for correcting discrete small DNA base lesions is base excision repair (BER) [9– 12]. The current and widely accepted working model for mammalian BER is that the process involves two sub-pathways that are differentiated by repair patch size and enzymes involved [13–16]. These sub-pathways are termed “single-nucleotide BER” (SN BER) and “long-patch BER” (LP BER), as shown in Scheme 1. SN BER involves removal of a single damaged nucleotide that is replaced with an undamaged nucleotide through template-directed DNA synthesis to fill the single-nucleotide gap, whereas in LP BER, two or more nucleotides are replaced [13–18]. Both of the BER sub-pathways are processed sequentially, one step to the next, and are initiated either by enzymatic removal of a damaged base or by spontaneous chemical hydrolysis of the glycosidic bond connecting the damaged base to the sugar phosphate backbone [1, 2, 19, 20]. The resulting apurinic/apyrimidinic (AP) site is processed by AP endonuclease 1 (APE1) that incises the phosphodiester backbone 5′ to the abasic site, leaving a single-nucleotide gap with 3′-hydroxyl and 5′-deoxyribose phosphate at the gap margins [21, 22]. DNA polymerase β, a multifunctional enzyme consisting of the 8-kDa amino-terminal domain with deoxyribose phosphate (dRP) lyase activity [23, 24] and the 31-kDa carboxy-terminal domain with nucleotidyl transferase activity [25–27], then catalyzes template-guided gap filling and removal of the 5′-dRP group to generate the substrate for the final BER step, DNA ligation [17, 18, 23, 24, 28–31]. The ligation step is completed either by DNA ligase I or the XRCC1/DNA ligase III complex [32–34].
Scheme 1.
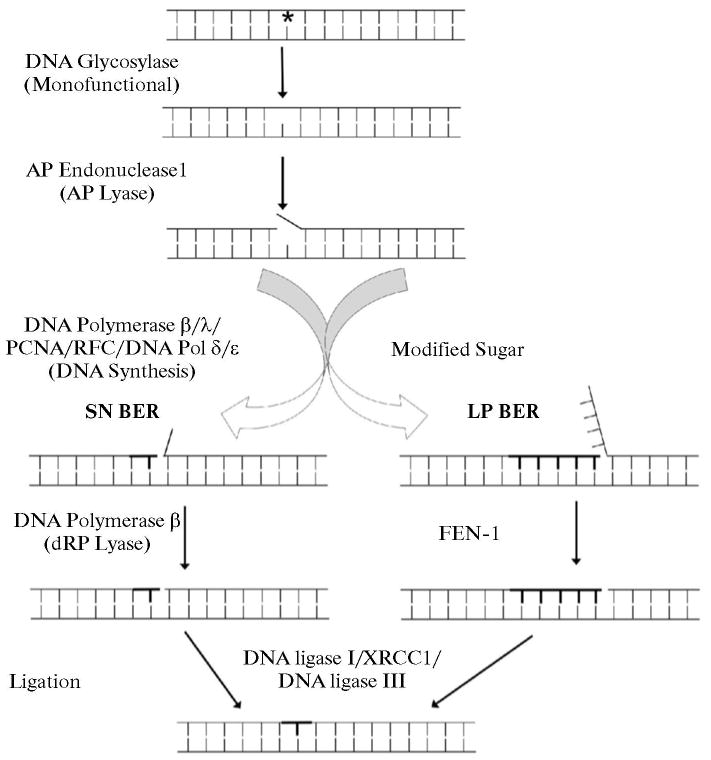
Working model illustrating the SN and LP BER sub-pathways for the repair of base damaged-DNA. First, a damaged-specific DNA glycosylase recognized and removed the damaged base from DNA. Then, the abasic DNA strand is incised by the endonucleolytic cleavage activity of APE1. After this step, the BER pathway can switch to either SN BER or LP BER, depending on the status (normal vs. oxidized/reduced) of the sugar group. Several DNA polymerases are involved in each sub-pathway. The 5′-dRP group in the SN BER sub-pathway is removed primarily by polymerase β, whereas the displaced DNA strand in the LP BER sub-pathway is cleaved by the flap endonuclease, FEN1. Finally, the nicked DNA is sealed by the DNA ligase I or by the XRCC1/DNA ligase III complex. Both sub-pathways operate in parallel, yet SN BER is considered to be 2- to 3-fold more active. Newly synthesized nucleotides are shown by bold lines.
The BER repair pathways and many of the BER enzymes are conserved in bacteria to humans, and these repair pathways have been reconstituted using purified natural and recombinant enzymes from various organisms [18, 28]. From biochemical and structural studies, a common theme emerged that the individual steps in BER are sequentially ordered. For example, it is only after damaged base removal that the AP site-containing DNA strand is recognized by APE1. DNA polymerase β then processes the gapped intermediate, generating the substrate for DNA ligase. Thus, to prevent exposure of the intermediates to harmful nuclease activities, recombination events or cell death signaling, it appears that the BER enzymes must coordinate with one another to efficiently receive the DNA substrates and pass the resulting DNA products along to the next enzyme in the sequence, just as a baton is passed from one individual to the next in a relay [35–37].
Biochemical and structural studies of BER enzymes suggested that BER intermediates could be passed sequentially from one enzyme to the next in a coordinated fashion [12, 37], in contrast to a model where all substrates and products are in equilibrium with free enzymes. To examine the step-to-step coordination in BER, we tested the hypothesis that DNA can be channeled from one step to the next during late steps in BER. Steps in a reconstituted SN BER system were examined including APE1 strand incision, DNA synthesis and dRP removal mediated by polymerase β, and ligation mediated either by DNA ligase I or XRCC1/DNA ligase III complex. This was accomplished by pre-loading the enzymes onto BER sub-strates and then conducting the repair incubation in the presence of a trap, such that any free enzyme or enzymes released from the DNA substrates during repair could no longer participate in the reactions. In the LP BER sub-pathway, we examined channeling of the DNA substrate from the DNA synthesis step to the FEN1 cleavage step. The results show that the BER intermediates of the SN BER sub-pathway could be channeled from APE1 to polymerase β and to DNA ligase. In contrast, in the LP BER sub-pathway, the DNA product after gap-filling by polymerase β was not channeled to FEN1 [38].
The design for these “single turnover” repair experiments is summarized in Scheme 2. Either an individual BER enzyme or a mixture of enzymes was first pre-incubated with the respective substrate DNA, and then the enzyme-substrate complex was mixed with a DNA trap plus an initiator for the reaction(s). After a brief incubation, products were recovered by gel electrophoresis and quantified. Scheme 2 illustrates the different types of incubations used: Type 1 involved individual BER enzymes and steps as shown; Types 2 and 3 involved mixtures of BER enzymes and more than one reaction product. In the pre-incubations, the substrate DNA concentrations were in the range of the dissociation constants for the respective enzymes (10 to 20 nM). The reaction products observed in the presence of the trap resulted from turnover of enzyme that was bound during the pre-incubation. The incubations were for 10 and 20 s, the shortest periods we could manage using manual methods, but much longer than the catalytic rate constants for each enzyme.
Scheme 2.

Illustration of different types of incubation protocols used in the single turnover experiments during SN BER. Purified human BER enzymes were pre-incubated with substrates either individually (Type 1) or as mixtures of enzymes (Types 2 and 3). The enzyme-substrate complexes formed during the pre-incubation were then mixed with a DNA trap plus initiator of the individual reaction. The trap was designed to remove any unbound free enzyme left in solution after the pre-incubation, along with any enzyme that might dissociate from the complex after initiation of the reaction. After addition of the trap and initiator, reaction mixtures were incubated for 10 and 20 s. In the Type 3 incubation, 32P-labeled dNTP was the labeled substrate, whereas in Types 1 and 2 incubations, 32P-labeled substrate DNA was used; the asterisks (*) indicate that the respective products were not measured. Figures illustrating the corresponding results are designated.
In LP BER, the DNA synthesis reaction may occur as a series of single-nucleotide gap filling steps in conjunction with FEN1 5′-tailoring (see Scheme 1) [39]. Accordingly, we also examined whether FEN1, can preload onto its substrate, conduct flap removal and then pass the product to the next step, gap-filling DNA synthesis [38].
APE1 Strand Incision Step
To examine the APE1-mediated incision step of the abasic site in BER, the reaction mixtures were assembled and incubated as shown in Scheme 2 and Fig. 1. The 32P-labeled DNA containing a lone uracil residue at position 15 was pretreated with uracil-DNA glycosylase (UDG) to generate an AP site-containing substrate. A 34-bp unlabeled DNA containing a synthetic AP site (tetrahydrofuran, THF) was used to trap any unbound APE1 in the reaction mixture. The reaction mixture was assembled on ice, and the reaction was initiated by transferring the reaction mixture to 30°C and adding a mixture of MgCl2 and ∼4000-fold excess DNA trap. In another set of control reaction mixtures, APE1 was pre-incubated with the trap first and the reaction was initiated as above. The results showed that APE1 pre-bound to the DNA substrate was able to incise the AP site (Fig. 1a); unbound APE1 in solution was quenched by the trap, as no increase in product formation was observed with a 20 s incubation (not shown). In contrast, when APE1 was first pre-incubated with the DNA trap and then the reaction was initiated by adding a mixture of DNA substrate and MgCl2, very little product was observed. This indicated that the trap was able to capture almost all of APE1 in the reaction mixture before it could bind and incise DNA substrate (Fig. 1b) [38].
Fig. 1.

Incision of the AP site-containing DNA by APE1 in the presence of a DNA trap. Schematic representations of 5′-end 32P-labeled AP site-containing DNA substrate (S) and DNA trap (T) are illustrated above the phosphorimage of the gels. The incision activity APE1 in the presence of a DNA trap was examined. The reaction mixture was assembled on ice, either with 30 nM APE1 and 10 nM 32P-labeled UDG-treated DNA (a), or with APE1 and DNA trap (b). Reactions were initiated by temperature jump and the addition of a mixture of the DNA trap and MgCl2 (a) or with the addition of 32P-labeled UDG-treated DNA, and MgCl2 (b), respectively. Samples were withdrawn after 10 s and analyzed. The positions of the 32P-labeled substrate and APE1-incised product are indicated.
Gap-Filling and dRP Lyase Steps
To examine these two polymerase β-mediated BER steps, reaction mixtures were assembled and incubated for 10 and 20 s. For DNA synthesis, a 34-mer nicked DNA was first prepared by annealing a 5′-end labeled 15-mer and 19-mer oligonucleotides to a complementary 34-mer DNA strand. This duplex DNA was pretreated with UDG, resulting in a single-nucleotide gapped DNA with 3′-OH and 5′-dRP groups at the gap margins, mimicking the APE1 incised BER intermediate. A 21-bp unlabeled gapped DNA was used to trap any unbound polymerase β in the reaction mixture.
To examine use of this BER intermediate by polymerase β in the DNA synthesis step, the reaction mixture was assembled on ice and the reaction was initiated by transferring the tubes to 30°C and adding a mixture of [α-32P]dCTP, MgCl2, and a ∼500-fold excess of trap. In another set of reaction mixtures, polymerase β was pre-incubated with the trap first and the reaction was initiated by adding a mixture of DNA substrate, dCTP and MgCl2. The reaction mixtures were incubated at 30°C, and samples were withdrawn for product analysis. The results showed that polymerase β pre-bound to the DNA substrate was able to incorporate one nucleotide (Fig. 2a, lane 2), i.e., incorporation of dCMP into DNA; unbound polymerase β in solution was trapped completely, as no further increase in dCMP incorporation was observed with a 20 s incubation (Fig. 2a, lane 3). In contrast, when polymerase β was first pre-incubated with the trap and then the reaction was initiated by adding a mixture of DNA substrate, dCTP and MgCl2, no incorporation of dCMP was observed. This indicated that the enzyme was productively bound during the pre-incubation and that the trap was able to capture all of the polymerase β in the reaction mixture (Fig. 2a, lanes 4 and 5).
Fig. 2.
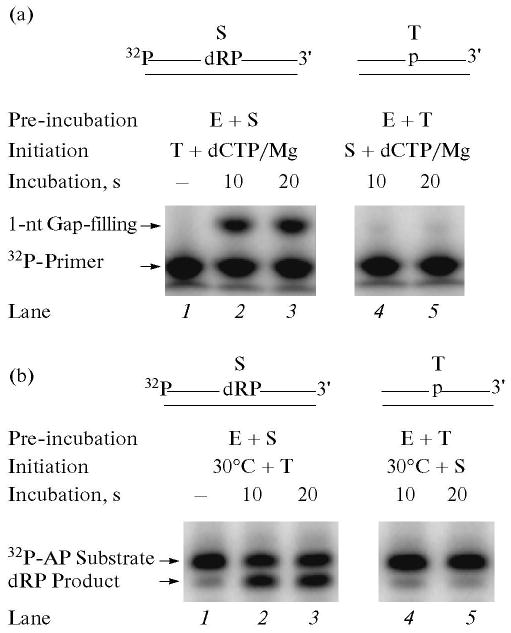
Gap-filling DNA synthesis and removal of the dRP group by polymerase β in the presence of a DNA trap. Schematic representations of 32P-labeled DNA substrate (S) and a DNA trap (T) are illustrated above the phosphorimage of the gels. E denotes polymerase β. (a) Gap-filling DNA synthesis by polymerase β in the presence of a DNA trap was examined. The reaction mixture was assembled on ice, either with 60 nM polymerase β and 20 nM 5′-end 32P-labeled UDG/APE1-treated DNA (lanes 1–3), or with polymerase β and DNA trap (lanes 4, 5). Reactions were initiated by temperature jump and the addition of a mixture of dCTP, DNA trap, and MgCl2 (lanes 1–3), or dCTP, 32P-labeled UDG/APE1-treated DNA, and MgCl2 (lanes 4, 5), respectively. Samples were withdrawn at 10 and 20 s and analyzed. The positions of the 32P-labeled primer and 1-nt gap-filling product are indicated. (b) For analyzing the dRP lyase activity of polymerase β in the presence of a DNA trap, the reaction mixture was assembled on ice, either with 60 nM polymerase β and 20 nM 3′-end 32P-labeled UDG/APE1-treated DNA (lanes 1–3), or polymerase β and the trap (lanes 4, 5). Reactions were initiated by temperature jump and addition of a DNA trap (lanes 1–3) or 32P-labeled substrate (lanes 4, 5), respectively. Samples were withdrawn at 10 and 20 s. The reaction products were stabilized by the addition of NaBH4, and the reaction products were analyzed. The positions of the 32P-labeled dRP substrate and the product are indicated.
Using the above protocol, we examined the 5′-dRP lyase step that also is catalyzed by polymerase β. In this case, the DNA substrate was 3′-end labeled. The removal of the dRP group from the substrate strand was monitored in a denaturing gel as a radiolabeled band migrating faster than the substrate. The reaction mixture was assembled on ice and the reaction was initiated by transferring the reaction mixture to 30°C and adding the trap. The results of this analysis showed that polymerase β pre-bound to the substrate excised the 5′-dRP group (Fig. 2b). The substrate was converted to product in the 10 s incubation, and there was no increase in product with the longer incubation (Fig. 2b, lanes 2 and 3). This indicated that the trap quenched free polymerase β in solution. To confirm the trapping of free enzyme, a reciprocal experiment was conducted where polymerase β was first pre-incubated with the trap, and the reaction was then initiated along with the addition of the radiolabeled substrate. No radiolabeled product above the background level was observed (Fig. 2b, lanes 4 and 5) [38].
Since polymerase β conducts both DNA synthesis and dRP lyase steps in the SN BER scheme, we next explored whether polymerase β bound to DNA could catalyze both steps before dissociating from the DNA. Since polymerase β is primarily a distributive enzyme and inserts one nucleotide at a time, our assumption was that if polymerase β incorporated dNMP first and then dissociated from the DNA, the trap in the reaction mixture would capture it. Hence, polymerase β would not be able to perform the next step in SN BER, i.e., the 5′-dRP-removal step. We tested the possibility of processive DNA synthesis and dRP lyase activities. A double-labeled 34-mer nicked DNA was prepared by annealing a 5′-end labeled 15-mer oligonucleotide and a 3′-end labeled 19-mer oligonucleotide to a complementary 34-mer DNA strand. The duplex DNA was pretreated with UDG, resulting in a single-nucleotide gapped DNA with 3′-OH and 5′-dRP groups at the gap margins and radiolabels on both ends.
The reaction mixture was assembled on ice as above, and the reactions were initiated by temperature jump along with addition of a mixture of dCTP, MgCl2 and a ∼500-fold excess of unlabeled trap DNA. The gap-filling and dRP lyase activities were measured in the same reaction mixture. Samples were withdrawn at 10 and 20 s, and the results shown in Fig. 3 revealed that polymerase β pre-bound to the DNA substrate incorporated one nucleotide and also excised the 5′-dRP group. In the control experiment, where polymerase β was pre-incubated first with the DNA trap, neither gap-filling nor 5′-dRP lyase activities were observed (Fig. 3b) [38].
Fig. 3.
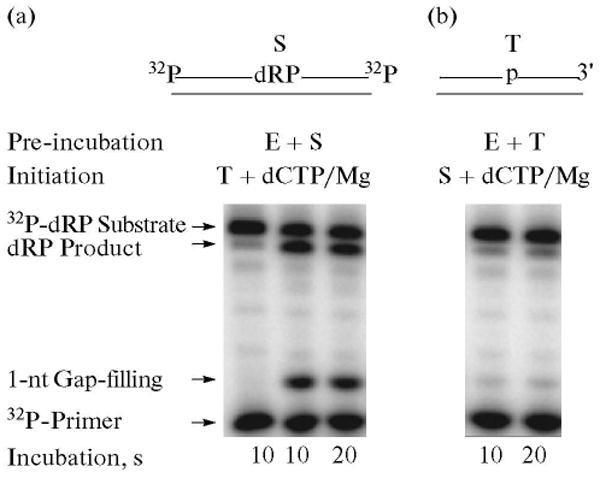
Gap-filling DNA synthesis and removal of the dRP group steps by polymerase β are concurrent. Schematic representations of a 32P-DNA substrate labeled at both ends (S) and the DNA trap (T) are illustrated above the phosphorimage of the gels. E denotes polymerase β. Double-labeled 34-bp DNA was prepared by annealing a 5′-end labeled 15-mer oligonucleotide and a 3′-end labeled 19-mer oligonucleotide to their complementary 34-mer DNA strand. The 19-mer oligonucleotide also contained a 5′-end phosphate and uracil. The duplex DNA was pretreated with UDG, resulting in a single-nucleotide gapped DNA with 3′-OH and 5′-dRP groups at the margins and radiolabels on both ends. Gap-filling DNA synthesis and dRP lyase reactions were performed by polymerase β in the presence of excess DNA trap. The repair reaction mixture was assembled on ice, either with polymerase β and 32P-labeled substrate (a), or polymerase βand DNA trap (b). Reactions were initiated by temperature jump and the addition of a mixture of dCTP, DNA trap, and MgCl2 (a), or dCTP, 32P-labeled substrate DNA, and MgCl2 (b), respectively. Samples were withdrawn at 10 and 20 s and analyzed. The positions of the 32P-Iabeled primer, 1-nt gap-filling DNA synthesis product, 32P-labeled dRP substrate and the dRP lyase product are indicated.
Kinetics of dRP Lyase Reaction
From the results in Fig. 3, the pre-bound polymerase β was able to perform both gap-filling synthesis and lyase without releasing the DNA substrate. However, from these experiments, it was not obvious which step occurred first. Previously, we had characterized the 5′-dRP lyase activity of polymerase β using steady-state kinetic methods [31], and it appeared that the DNA synthesis step was faster than the lyase step and presumably occurred first [30].
To further dissect kinetic features of the 5′-dRP lyase reaction, a quantitative pre-steady-state approach was undertaken. In the presence of high enzyme concentrations, time courses were biphasic (Fig. 4a). A rapid burst of product formation was followed by a linear slow phase. Extrapolation of the linear portion of the time course to the y-axis (t = 0) indicated that a burst of product formation occurred that was too fast to measure by manual sampling. The amplitude of this rapid unresolved phase was proportional to the concentration of enzyme in the reaction mixture (Fig. 4b). Normalizing the time courses for the differing enzyme concentrations indicates that the burst amplitude represented 70% of the added enzyme and that the linear phase corresponded to a turnover number of 0.12/min (Fig. 4c). Note that these time course experiments were performed at 15°C to limit the slope of the linear phase, thereby providing a better estimate of the extrapolated burst amplitude. The biphasic nature of the time course indicated that a step after chemistry (i.e., product release) limits the steady-state linear phase of the time course. The observation that 70% of the enzyme rapidly excised the dRP-group in the gap also indicates that only one molecule of polymerase β is bound/gap. If two or more molecules of polymerase β bound to a single-nucleotide gap, the burst amplitude would be ≤50%.
Fig. 4.
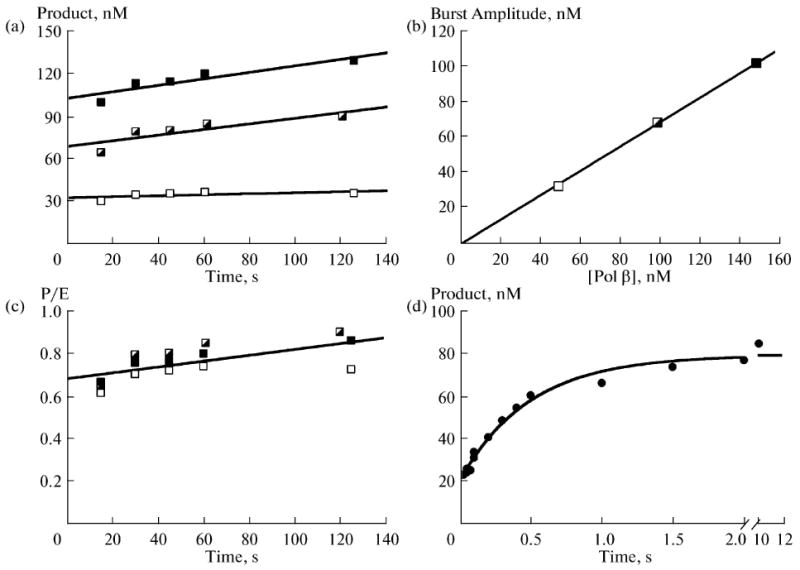
Transient-state kinetic analysis of polymerase β dRP lyase reaction. Time courses were determined. (a) Reactions were initiated by adding enzyme (50, open; 100, half-filled; or 150 nM, filled squares) to 500 nM DNA substrate, and product formation was determined at the indicated time points. (b) A secondary plot of the burst amplitudes (y-intercept) determined from an extrapolated linear fit for each enzyme concentration. The solid line is a linear fit of the data with a y-intercept of zero and slope of 0.69 nM product per 1 nM added enzyme. (c) A replot of the data in panel (a) normalized for enzyme concentration. Accordingly, the ordinate scale provides the number of enzyme turnovers and indicates that 0.7 nM product is formed during the burst, indicating that ∼70% of the added enzyme is productively bound. The turnover number for the linear phase is 0.12/min. (d) Single turnover analysis of the dRP lyase reaction. Polymerase β (1 μM) was rapidly mixed with 200 nM DNA substrate and time points collected. The time course exhibits two rapid phases: a fast phase that was too rapid to measure (amplitude ∼20 nM); and a slower exponential phase (kobs ∼ 120/min). Both of these phases were considerably more rapid that the linear phase determined in panel (c).
To measure the rate of the burst phase, single-turnover reactions were measured. In this situation, the enzyme concentration (500 nM after mixing) exceeds the substrate concentration (100 nM after mixing) and enzyme cycling (i.e., product release) does not confound the time course. The rapid rate of the burst required that time points be gathered with a rapid-mixing and quenching instrument [38]. The volatile nature of the NaBH4 quenching agent required that the reaction be stopped in the reaction collection tube rather than the quench syringe of the instrument. Accordingly, the earliest time point that could be collected safely and reliably was 15 ms. A typical time course is illustrated in Fig. 4d. Again, the time course was biphasic; a rapid phase (∼20 nM) was lost in the dead time of the instrument followed by a single-exponential time course of product formation (kobs = 120/min). When gap-filling DNA synthesis was measured under these identical conditions and with this same substrate, the rate of dNTP insertion was 7/min (not shown). Thus, the dRP lyase activity of polymerase β was at least 20-fold faster than the DNA synthesis activity. The biphasic nature of the time course (Fig. 4d) suggested that enzyme bound DNA substrate was partitioned between two forms: an active form that is poised for reaction (∼25%) and a population where the substrate is bound in an inappropriate conformation. Our previous crystallographic structures of polymerase β with substrate analogues indicated that the 5′-sugar-phosphate moiety binds in a non-productive mode that must undergo a conformational change before chemistry can occur (Figs. 5–7) [40, 41]. As shown in Fig. 6, the distance between the sugar C1′ and the Nε of K72, the residue found to be the sole Schiff base nucleophile in the dRP lyase reaction of polymerase β, is far too great for catalysis (10.1 Å) [40, 42]. Therefore, the non-catalytic structure indicates a requirement for the substrate rearrangement in order to attain a catalytically competent state shown in Fig. 7 [38, 40].
Fig. 5.
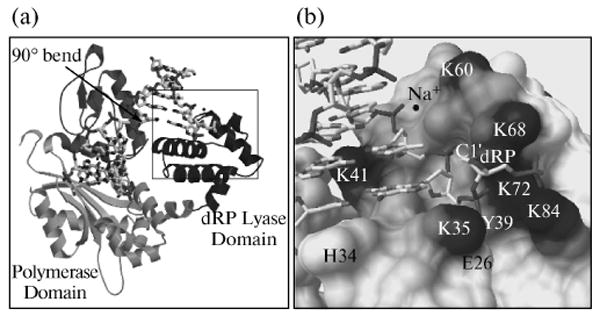
Global structure of polymerase β binary complex bound to DNA. (a) The conformation of the polymerase β binary complex bound to dRP-containing DNA (sugar ring in closed form) is shown. The amino-terminal 8-kDa lyase domain and the polymerase sub-domain are indicated. The structure is identical to that observed previously with gapped DNA [41], except that the downstream primer contains a 5′-THF phosphate (dRP, in boxed area). An arrow indicates the position of a 90° bend in the template strand. DNA is shown as a stick model. (b) A close-up view of the boxed area in (a).
Fig. 7.
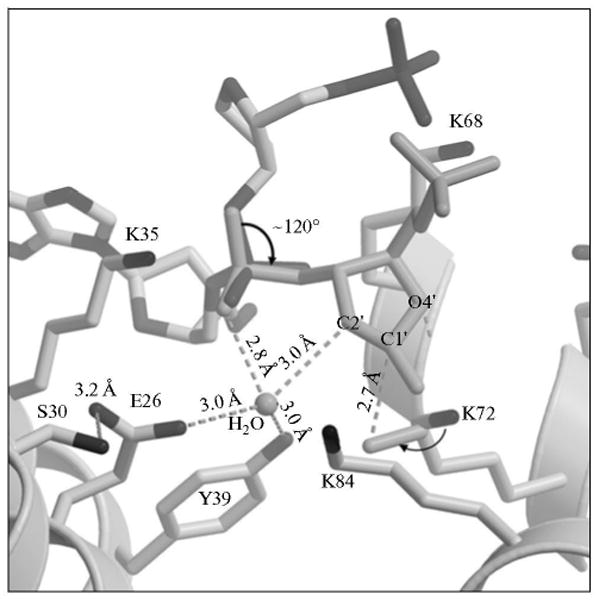
A model of a proposed conformational change in the dRP group. Key amino acid residues of the polymerase β lyase active site are indicated. Rotation of the sugar phosphate (thick arrow), along with additional small adjustments, adequately positions reactive groups necessary for catalysis. Dashed lines indicate distances between the catalytic residues and reactive groups. The polymerase β side chains and the sugar ring atoms are labeled. A water molecule in the active site is labeled. The attack of the K72 side chain nitrogen on the C1′ over 2.7 Å is illustrated.
Fig. 6.
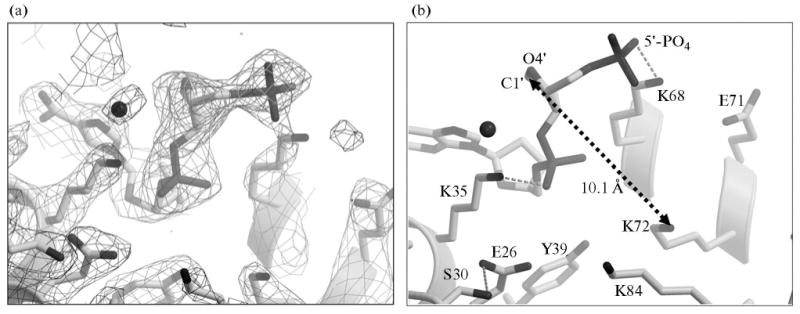
Electron density difference map. (a) 2Fobs–Fcalc electron density difference map contoured at 1s is shown in black for the polymerase β binary complex bound to DNA containing3dRP (sugar ring in closed form). Fobs – Fcalc electron density contoured at 3.3σ is shown for a simulated annealing omit map, with the dRP group omitted. (b) The key residues of the lyase domain that form a substrate-binding pocket are shown. Dashed arrow indicates the distance from dRP C1′ to K72 Nε. Other portions of the structure are excluded for clarity.
Coordination of APE1 Strand Incision and Gap-Filling Steps
To examine whether the APE1-incised BER intermediate is channeled to the next enzyme, polymerase β, we repeated assays for the incision and gap-filling synthesis steps with a 5′-end labeled AP site-containing DNA substrate. The incision activity of APE1 and the gap-filling activity of polymerase β were measured in the same reaction mixture using the Type 2 protocol in Scheme 2. The results shown in Fig. 8 revealed that APE1 pre-bound to the substrate was able to incise the DNA and then the product was channeled to polymerase β for gap-filling DNA synthesis (Fig. 8a). In a control experiment, where APE1 and polymerase β were pre-incubated first with the trap and then the reaction was initiated by adding a mixture of 32P-labeled DNA substrate, dCTP and MgCl2, gap-filling was not observed. These results showed that a portion of the BER intermediate after the APE1-incision step was channeled to polymerase β. This coordination between APE1 and polymerase β appears to be consistent with the complex formation and functional interaction between these enzymes and substrate DNA that was observed previously (Fig. 9) [43, 44].
Fig. 8.
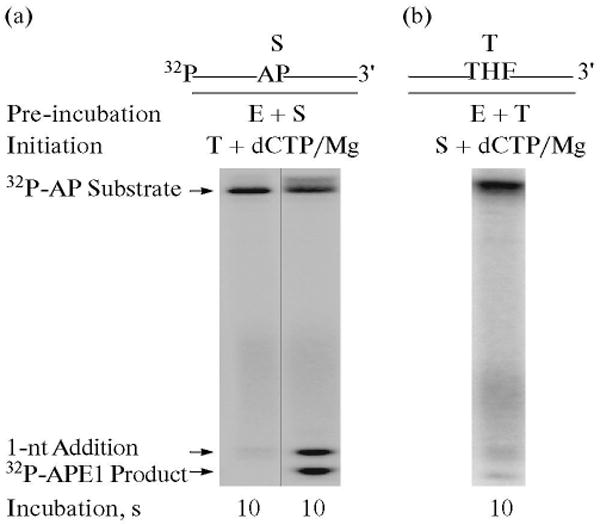
Combination of APE1-incision and gap-filling DNA synthesis steps. Schematic representations of 32P-labeled AP site-containing DNA substrate (S) and the DNA trap (T) are illustrated above the phosphorimage of the gels. The incision activity by APE1 and gap-filling DNA synthesis by polymerase β were examined in the presence of a DNA trap. The reaction mixture was assembled on ice, either with APE1, polymerase β and 32P-labeled substrate DNA (a), or with APE1, polymerase β and DNA trap (b). Reactions were initiated by temperature jump and addition of a mixture of the DNA trap and dCTP/MgCl2 (a), or 32P-labeled substrate DNA, and dCTP/MgCl2 (b), respectively. Samples were withdrawn at intervals and analyzed. The positions of the 32P-labeled substrate, APE1 incised product and 1-nt gap-filling product are indicated.
Fig. 9.
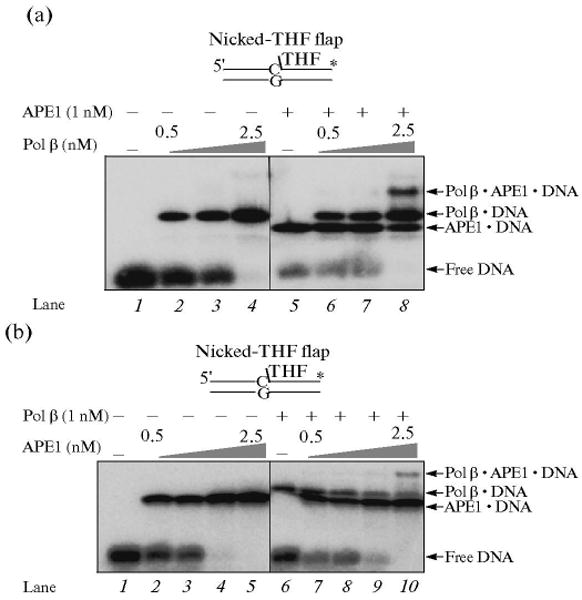
Formation of APE · polymerase β · DNA ternary complex. (a) Increasing concentrations of polymerase β were incubated with 5 nM nicked-THF flap substrate DNA in the absence (lanes 2–4) and presence of 1 nM APE1 (lanes 6–8). Lane 1 represents the binding mixture without enzyme. Lanes 2–4 correspond to mixtures containing increasing concentrations of polymerase β (0.5, 1, and 2.5 nM, respectively) and DNA. Lanes 6–8 indicate mixtures containing the same concentrations of polymerase β as lanes 2–4 along with 1 nM APE. Lane 5 indicates a binding mixture with APE and the DNA substrate. (b) Increasing concentrations of APE1 (0.5, 1, 2.5, and 5 nM, respectively) were incubated with 5 nm nicked-THF flap substrate DNA in the absence (lanes 2–5) and presence of 1 nM polymerase β (lanes 7–10). Lane 1 represents the reaction mixture without enzymes, whereas lane 6 corresponds to the mixture containing polymerase β and DNA substrate. The substrate DNA is schematically illustrated above the gel.
Ligation Step
To explore whether the nicked DNA after the gap-filling and dRP lyase steps in SN BER is channeled to DNA ligase, experiments were performed with a 5′-end labeled nicked DNA substrate. The strategy for study of the ligation step was according to the Type 1 protocol shown in Scheme 2. An unlabeled nicked DNA was used as a trap. The reaction mixture was assembled on ice with DNA ligase I and 32P-labeled nicked sub-strate, and then the reaction was initiated by adding a mixture of ATP, MgCl2, and ∼500-fold excess of unlabeled trap (nicked 30-mer DNA duplex). The reaction mixtures were transferred to 30°C for ligation to occur. After 10 and 20 s, samples were withdrawn and the reaction products analyzed. The results shown in Fig. 10 indicated that the substrate was converted into a fully ligated product. With the control experiment where DNA ligase I was first pre-incubated with the trap and the reaction then initiated, no ligated product was observed (Fig. 10, lanes 3 and 4). These results indicated that ligation occurred more rapidly than nicked DNA dissociation. Next, we asked whether other DNA ligases, such as DNA ligase III and T4 DNA ligase have a preference over DNA ligase I for use of the nicked substrate, as it had been reported that DNA ligase III is the preferred ligase in the SN BER path-way [45]. Using the same reaction conditions, we found that DNA ligase III and T4 ligase functioned with a similar outcome as that seen with DNA ligase I (Fig. 10, lanes 5, 6 and 9, 10, respectively) [38].
Fig. 10.
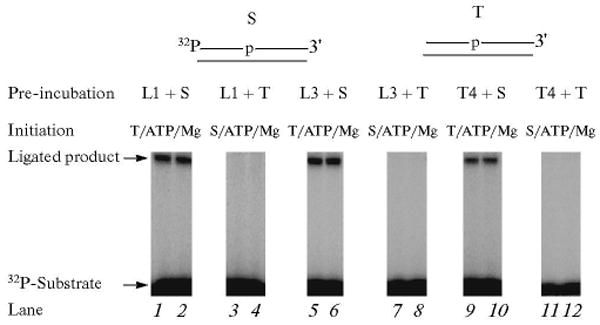
Analysis of the ligation step in the BER scheme using purified DNA ligases. Schematic representations of 32P-labeled nicked DNA substrate (S) and the DNA trap (T) are illustrated above the phosphorimage of the gels. The ligation reaction was performed with DNA ligase I (lanes 1–4), DNA ligase III (lanes 5–8) or T4 DNA ligase (lanes 9–12) in the presence of a DNA trap. The ligation reaction mixtures were assembled either with 32P-labeled DNA substrate and a DNA ligase or with DNA trap and a DNA ligase. Reactions were initiated by temperature jump and the addition of a mixture of ATP, MgCl2 and the DNA trap or ATP, MgCl2 and 32P-labeled DNA substrate, as indicated at the top of each lane. Samples were withdrawn at 10 and 20 s intervals and analyzed. The positions of the 32P-labeled primer and ligated product are indicated. L1, L3, and T4 denote DNA ligase I, DNA ligase III, and T4 DNA ligase, respectively.
Coordination of Gap-Filling, dRP Lyase and Ligation Steps
Thus far, we have demonstrated that the BER intermediates, after the base removal step, can be channeled from initial DNA binding to catalysis for the APE1, polymerase β 5′-dRP lyase and gap-filling DNA synthesis, when these steps were analyzed either individually or with APE1 and polymerase β together. To gain insight on whether the BER intermediates can be channeled throughout a complete BER reaction, we examined multiple steps in a single reaction mixture. The complete BER reaction was assembled as above, with UDG and APE1 pretreated DNA sub-strate, polymerase β and DNA ligase I (Scheme 2, Type 3). The reaction mixture was assembled on ice, and the reaction was initiated by transferring the tubes to 30°C and adding a mixture of [α-32P]dCTP, MgCl2, ATP, and ∼500-fold excess of the trap. The reaction mixtures were incubated at 30°C, and a sample was withdrawn after 10 s (Fig. 11). The results revealed that when the enzymes were pre-incubated with the sub-strate, a complete or ligated repaired product was observed (Fig. 11, lane 1). When the enzymes were first pre-incubated with the trap, the complete repair product was not observed, and similarly when ligase was not included, the gap-filling product was observed but not the complete product (lanes 2 and 3). This indicated that the initial BER intermediate was subjected to dRP lyase and gap-filling and then channeled to DNA ligase [38].
Fig. 11.
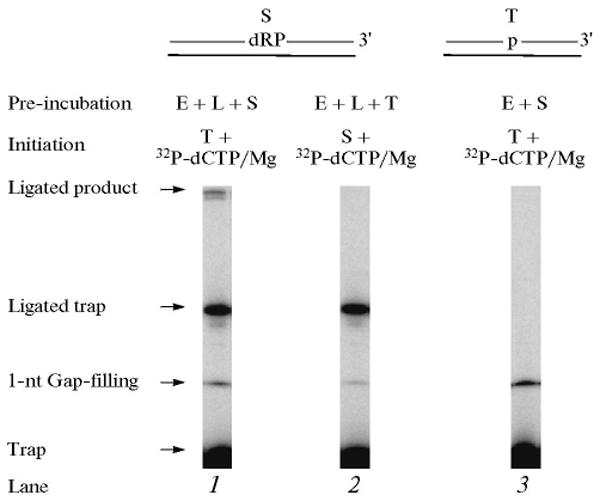
Combination of gap-filling, dRP lyase and ligation steps. Schematic representations of UDG/APE1-treated DNA substrate (S) and the DNA trap (T) are illustrated above the phosphorimage of the gels. A complete BER reaction mixture containing the pretreated DNA substrate or trap DNA, polymerase β, and DNA ligase I was assembled on ice. The reaction was initiated by temperature jump and the addition a mixture of [α-32P]dCTP, MgCl2, ATP, and DNA trap or [α-32P]dCTP, MgCl2, ATP, and DNA substrate as indicated at the top of each lane. Samples were withdrawn at 10 s and analyzed. The positions of the 1-nt gap-filling product, ligated complete BER product, free trap and the ligated trap are indicated. E and L denote polymerase β and DNA ligase I, respectively.
“Hit and Run” Mechanism in the LP-BER Sub-Pathway
Earlier, a working model for LP-BER was proposed where polymerase β conducts strand displacement DNA synthesis (two or more nucleotides) leaving a long DNA flap; FEN1 then removes the DNA flap, and a DNA ligase seals the resulting nick. However, Liu et al. [39] recently suggested an alternate “Hit and Run” mechanism for LP BER (Scheme 3). In this model, polymerase β fills the initial one-nucleotide gap in LP BER, leaving a lesion-containing flap. FEN1 then removes the flap plus one nucleotide in the damaged strand to create a 1-nt gap, and finally, the gap is filled and sealed by polymerase β and DNA ligase, respectively. Furthermore, polymerase β and FEN1 can cooperate with each other in their 1-nt gap-filling and gap forming activities, respectively, to form a longer repair patch [39]. Alternatively, if DNA ligase dominates, the LP BER gap size will be limited (Scheme 3) by the ligation activity. Here, we explored the possibility in LP BER that the DNA product, after gap-filling by polymerase β, was passed along to FEN1 for flap removal.
Scheme 3.
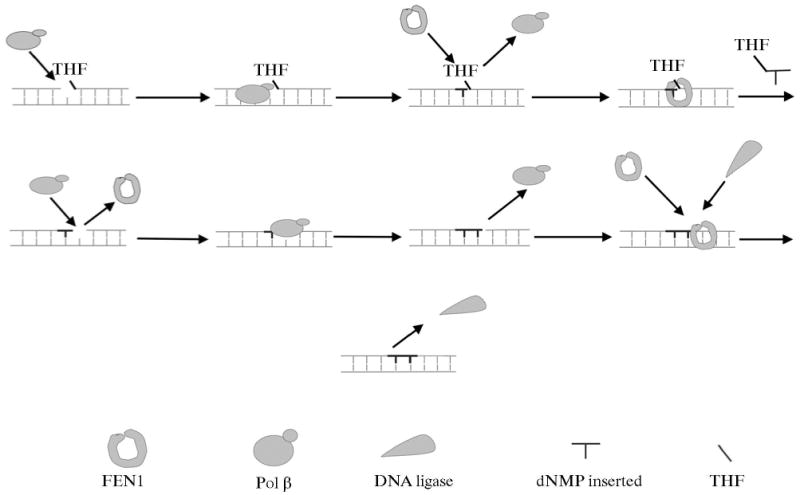
The Hit and Run mechanism of polymerase β/FEN1-mediated long LP BER. AP endonuclease 1 makes an endonucleolytic cleavage at the 5′-side of an abasic site resulting in a 5′-dRP in a one-nucleotide-gapped DNA. If the dRP moiety is reduced or oxidized, polymerase β employs its polymerase activity to fill the gap, creating a nicked-reduced/oxidized sugar flap (bold lines), because the polymerase β dRP lyase cannot remove the modified sugar residue. Polymerase β rapidly dissociates from the nicked-THF flap product permitting FEN1 access, which cleaves the THF flap, resulting in a 1-nt gap. Polymerase β then fills the gap (new nucleotide shown in bold lines), producing a nick. Subsequent FEN1 removal of one nucleotide from the 5′-end of the nick leads to another 1-nt gap for polymerase β to fill. Alternating FEN1 cleavage and polymerase β DNA synthesis shown as enzyme binding, catalysis, and dissociation (Hit and Run), leads to removal of the modified dRP moiety plus nucleotides (bold lines), and results in replacement of 2 or more nucleotides (long patch) until DNA ligase seals the nick and terminates LP-BER. FEN1 and DNA ligation competition is expected to control the length of the LP BER patch.
The repair reaction mixture was assembled as above with the APE1 pretreated THF-containing DNA substrate, polymerase β and FEN1. The reaction was initiated by adding [α-32P]dCTP, dATP, dGTP, TTP, MgCl2, and ∼500-fold excess of unlabeled DNA trap. In another set of reaction mixtures, polymerase β and FEN1 were mixed with the trap first, and then the reaction was initiated by adding the DNA substrate, [α-32P]dCTP, dATP, dGTP, TTP, and MgCl2. After 10 s, samples were withdrawn for product analysis. The assumption tested was that polymerase β will incorporate one nucleotide, and the product will be passed to FEN1 for flap cleavage. FEN1 cleavage will create a one-nucleotide gap for polymerase β to insert the second nucleotide in LP BER. Interestingly, the results of this analysis showed that polymerase β pre-bound to the DNA substrate incorporated only one nucleotide, whether or not FEN1 was in the pre-incubation (Fig. 12). We explained these results as follows: once polymerase β filled the one-nucleotide gap, it dissociated from the DNA and was captured by the trap. It was possible that the nicked-flap DNA product thus formed was passed to FEN1 for cleavage of the THF-flap structure and creating the one-nucleotide gapped DNA for polymerase β to insert the second nucleotide in the LP BER gap. Since polymerase β was trapped prior to formation of the second nucleotide gap, we did not expect to observe a second nucleotide insertion into the DNA (Fig. 12). To our surprise, however, in an experiment where a 3′-end labeled THF-containing strand was used, we failed to observe cleavage of the THF-flap (data not shown). These results indicated that the product after gap-filling DNA synthesis was not passed to FEN1. Another explanation could be that the trap captured FEN1 even before its DNA substrate was formed. In other words, the enzymes were not pre-assembled on the BER substrate prior to the polymerase β gap-filling step. To further investigate this point, experiments were repeated in the presence of DNA ligase. The results of this analysis revealed gap-filling incorporation of only one nucleotide without formation of ligated product (Fig. 13). Thus, after gap-filling by polymerase β, the BER intermediate still contained the THF flap and was not a substrate for DNA ligase. These results confirmed that the THF-flap product formed after gap-filling DNA synthesis by polymerase β was not channeled to FEN1. Instead, FEN1 action on this intermediate involves binding by free FEN1 in solution to the product of the polymerase β gap-filling reaction. This obviously cannot occur in the presence of a trap [38].
Fig. 12.
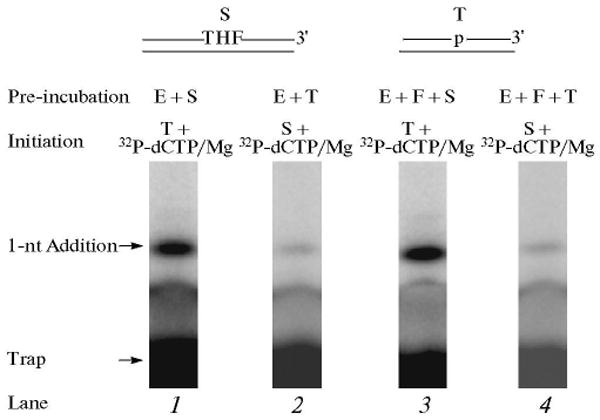
DNA synthesis step in LP BER. Schematic representations of APE1-treated THF-DNA substrate (S), a LP BER intermediate, and the DNA trap (T) are illustrated above the phosphorimage of the gels. E and F denote polymerase β and FEN1, respectively. The DNA synthesis reaction was performed by polymerase β alone or polymerase β and FEN1 in the presence of excess DNA trap. The repair reaction mixture was assembled on ice, either with polymerase β alone (lane 1) and substrate DNA or with polymerase β and FEN1 (lane 3) and substrate DNA, respectively. The reactions were initiated by temperature jump and the addition of a mixture of [α-32P]dCTP, dATP, dGTP, TTP, DNA trap, and MgCl2 (lanes 1 and 3). In another set of reaction mixtures, polymerase β or polymerase β and FEN1 were mixed with the DNA trap first, and the reactions were initiated by temperature jump and adding a mixture of [α-32P]dCTP, dATP, dGTP, TTP, substrate DNA, and MgCl2 (lanes 2 and 4), respectively. Reaction mixtures were incubated for 10 s and analyzed. The positions of the 1-nt addition product and free-labeled trap are indicated.
Fig. 13.
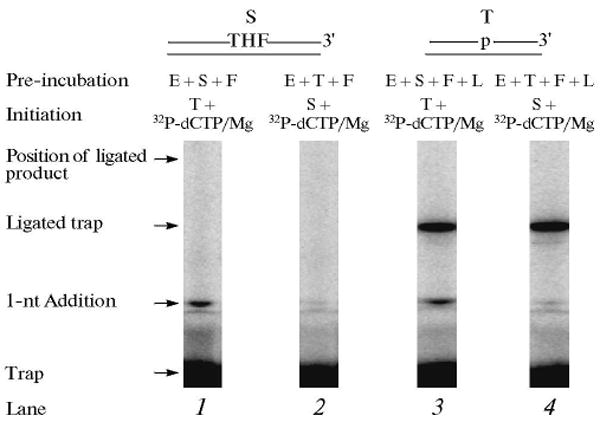
Gap-filling, FEN1 cleavage and ligation steps in LP BER. Schematic representations of APE1-treated THF-DNA substrate (S), LP BER intermediate, and the DNA trap (T) are illustrated above the phosphorimage of the gels. E, F, and L denote polymerase β, FEN1, and DNA ligase I, respectively. The repair reaction mixture was assembled on ice, either with polymerase β, FEN1 substrate DNA (lane 1) or with polymerase β, FEN1, DNA ligase I, and substrate DNA (lane 3). The reactions were initiated by the addition of a mixture of [α-32P]dCTP, dATP, dGTP, TTP, ATP, DNA trap, and MgCl2 (lanes 1 and 3). In another set of reaction mixtures, polymerase β and FEN1 or polymerase β, FEN1 and DNA ligase I were pre-incubated with the DNA trap first, and the reactions were initiated by adding a mixture of [α-32P]dCTP, dATP, dGTP, TTP, ATP, substrate DNA, and MgCl2 (lanes 2 and 4). Reaction mixtures were incubated for 10 s and analyzed. The positions of 1-nt gap-filling product, ligated BER product, free-labeled trap, and the ligated labeled trap are indicated.
Concluding Remarks
Based on structural studies of APE1-DNA cocrystals by Tainer and his associates [35, 36], it had been suggested that enzymatic steps in BER may involve recognition of a product–enzyme complex by the next enzyme in the pathway, rather than binding to an intermediate that is free in solution. Wilson and Kunkel [37] referred to these coordinated events of processing DNA repair intermediates as “passing the baton.” For the first time, we tested the hypothesis that the mammalian BER intermediates can be channeled from one step to the next.
Purified human BER enzymes APE1, polymerase β and DNA ligase I when pre-bound to their respective DNA substrates, were able to conduct their respective enzymatic reactions in the presence of a DNA trap. A new finding here was that mixtures of these enzymes could conduct a sequence of BER reactions in the presence of a DNA trap when the enzymes were pre-bound to the initial BER intermediate. This indicated that substrates and products of the BER steps could be handed off from one enzyme to the next during the SN BER process, reducing the possibility that a sequestered intermediate could trigger the DNA damage surveillance system. The possibility that a physical assembly of BER enzymes is recruited to the site of a BER lesion is consistent with the rapid recruitment of the enzymes studied here to sites of DNA damage in living cells [46–48]. On the other hand, recruitment in cells does not always correlate with hand-off for purified enzymes, since FEN1 and polymerase β did not exhibit hand off in the in vitro studies reported here, but these two enzymes are recruited to sites of LP BER intermediates in living cells [47]. Perhaps accessory proteins might influence the behavior of the purified enzymes, but this possibility has not yet been studied.
Another new finding in the present work was the very rapid enzymatic processing of the 5′-dRP lyase step of SN BER. In addition, the order of the gap-filling and lyase steps was found to be different from our previous understanding [30, 31]. The 5′-dRP lyase rate constant was much faster (at least 20-fold) than the DNA polymerase gap-filling rate constant. As found earlier for the gap-filling reaction by polymerase β, the product release step for the 5′-dRP lyase reaction is rate-limiting under steady-state reaction conditions [38].
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences (Research Projects Z01-ES050158 and Z01-ES050159).
Footnotes
The article is published in the original.
References
- 1.Lindahl T. DNA repair enzymes. Annu Rev Bio-chem. 1982;51:61–87. doi: 10.1146/annurev.bi.51.070182.000425. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 3.Loeb LA, Preston BD. Mutagenesis by apurinic/apyrimidinic sites. Annu Rev Genet. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 4.Drinkwater NR, Miller EC, Miller JA. Estimation of apurinic/apyrimidinic sites and phosphotriesters in deoxyribonucleic acid treated with electrophilic carcinogens and mutagens. Biochemistry. 1980;19:5087–5092. doi: 10.1021/bi00563a023. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura J, Swenberg JA. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 1999;59:2522–2526. [PubMed] [Google Scholar]
- 6.Roberts KP, Sobrino JA, Payton J, Mason LB, Turesky RJ. Determination of apurinic/apyrimidinic lesions in DNA with high-performance liquid chromatography and tandem mass spectrometry. Chem Res Toxicol. 2006;19:300–309. doi: 10.1021/tx0502589. [DOI] [PubMed] [Google Scholar]
- 7.Horton JK, Prasad R, Hou E, Wilson SH. Protection against methylation-induced cytotoxicity by DNA polymerase beta-dependent long patch base excision repair. J Biol Chem. 2000;275:2211–2218. doi: 10.1074/jbc.275.3.2211. [DOI] [PubMed] [Google Scholar]
- 8.Ward JE. DNA repair in higher eucaryotes. In: Nivkoloff JA, Hoekstra MF, editors. DNA Damage and Repair. Totowa, NJ: Humana Press; 1998. pp. 65–84. [Google Scholar]
- 9.Kubota Y, Nash RA, Klungland A, Schar P, Barnes DE, Lindahl T. Reconstitution of DNA base excision-repair with purified human proteins: Interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 10.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 11.Wilson DM, 3rd, Thompson LH. Life without DNA repair. Proc Natl Acad Sci U S A. 1997;94:12754–12757. doi: 10.1073/pnas.94.24.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson SH. Mammalian base excision repair and DNA polymerase beta. Mutat Res. 1998;407:203–215. doi: 10.1016/s0921-8777(98)00002-0. [DOI] [PubMed] [Google Scholar]
- 13.Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J Biol Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- 14.Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: Reconstitution with purified proteins and requirement for DNase IV (FEN1) EMBO J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fortini P, Pascucci B, Parlanti E, Sobol RW, Wilson SH, Dogliotti E. Different DNA polymerases are involved in the short-and long-patch base excision repair in mammalian cells. Biochemistry. 1998;37:3575–3580. doi: 10.1021/bi972999h. [DOI] [PubMed] [Google Scholar]
- 16.Biade S, Sobol RW, Wilson SH, Matsumoto Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J Biol Chem. 1998;273:898–902. doi: 10.1074/jbc.273.2.898. [DOI] [PubMed] [Google Scholar]
- 17.Singhal RK, Prasad R, Wilson SH. DNA polymerase beta conducts the gap-filling step in uracil-initiated base excision repair in a bovine testis nuclear extract. J Biol Chem. 1995;270:949–957. doi: 10.1074/jbc.270.2.949. [DOI] [PubMed] [Google Scholar]
- 18.Dianov G, Price A, Lindahl T. Generation of single-nucleotide repair patches following excision of uracil residues from DNA. Mol Cell Biol. 1992;12:1605–1612. doi: 10.1128/mcb.12.4.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mosbaugh DW, Bennett SE. Uracil-excision DNA repair. Prog Nucl Acids Res Mol Biol. 1994;48:315–370. doi: 10.1016/s0079-6603(08)60859-4. [DOI] [PubMed] [Google Scholar]
- 20.Slupphaug G, Eftedal I, Kavli B, Bharati S, Helle NM, Haug T, Levine DW, Krokan HE. Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry. 1995;34:128–138. doi: 10.1021/bi00001a016. [DOI] [PubMed] [Google Scholar]
- 21.Doetsch PW, Helland DE, Haseltine WA. Mechanism of action of a mammalian DNA repair endonuclease. Biochemistry. 1986;25:2212–2220. doi: 10.1021/bi00356a054. [DOI] [PubMed] [Google Scholar]
- 22.Doetsch PW, Cunningham RP. The enzymology of apurinic/apyrimidinic endonucleases. Mutat Res. 1990;236:173–201. doi: 10.1016/0921-8777(90)90004-o. [DOI] [PubMed] [Google Scholar]
- 23.Matsumoto Y, Kim K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science. 1995;269:699–702. doi: 10.1126/science.7624801. [DOI] [PubMed] [Google Scholar]
- 24.Piersen CE, Prasad R, Wilson SH, Lloyd RS. Evidence for an imino intermediate in the DNA polymerase beta deoxyribose phosphate excision reaction. J Biol Chem. 1996;271:17811–17815. doi: 10.1074/jbc.271.30.17811. [DOI] [PubMed] [Google Scholar]
- 25.Casas-Finet JR, Kumar A, Morris G, Wilson SH, Karpel RL. Spectroscopic studies of the structural domains of mammalian DNA beta-polymerase. J Biol Chem. 1991;266:19618–19625. [PubMed] [Google Scholar]
- 26.Kumar A, Abbotts J, Karawya EM, Wilson SH. Identification and properties of the catalytic domain of mammalian DNA polymerase beta. Biochemistry. 1990;29:7156–7159. doi: 10.1021/bi00483a002. [DOI] [PubMed] [Google Scholar]
- 27.Kumar A, Widen SG, Williams KR, Kedar P, Karpel RL, Wilson SH. Studies of the domain structure of mammalian DNA polymerase beta: Identification of a discrete template binding domain. J Biol Chem. 1990;265:2124–2131. [PubMed] [Google Scholar]
- 28.Dianov G, Lindahl T. Reconstitution of the DNA base excision-repair pathway. Curr Biol. 1994;4:1069–1076. doi: 10.1016/s0960-9822(00)00245-1. [DOI] [PubMed] [Google Scholar]
- 29.Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 30.Srivastava DK, Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH. Mammalian abasic site base excision repair: Identification of the reaction sequence and rate-determining steps. J Biol Chem. 1998;273:21203–21209. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- 31.Prasad R, Beard WA, Strauss PR, Wilson SH. Human DNA polymerase beta deoxyribose phosphate lyase: Substrate specificity and catalytic mechanism. J Biol Chem. 1998;273:15263–15270. doi: 10.1074/jbc.273.24.15263. [DOI] [PubMed] [Google Scholar]
- 32.Prasad R, Singhal RK, Srivastava DK, Molina JT, Tomkinson AE, Wilson SH. Specific interaction of DNA polymerase beta and DNA ligase I in a multiprotein base excision repair complex from bovine testis. J Biol Chem. 1996;271:16000–16007. doi: 10.1074/jbc.271.27.16000. [DOI] [PubMed] [Google Scholar]
- 33.Prigent C, Satoh MS, Daly G, Barnes DE, Lindahl T. Aberrant DNA repair and DNA replication due to an inherited enzymatic defect in human DNA ligase I. Mol Cell Biol. 1994;14:310–317. doi: 10.1128/mcb.14.1.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caldecott KW, McKeown CK, Tucker JD, Ljungquist S, Thompson LH. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol Cell Biol. 1994;14:68–76. doi: 10.1128/mcb.14.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected] Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 36.Parikh SS, Mol CD, Hosfield DJ, Tainer JA. Envisioning the molecular choreography of DNA base excision repair. Curr Opin Struct Biol. 1999;9:37–47. doi: 10.1016/s0959-440x(99)80006-2. [DOI] [PubMed] [Google Scholar]
- 37.Wilson SH, Kunkel TA. Passing the baton in base excision repair. Nature Struct Biol. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- 38.Prasad R, Shock DD, Beard WA, Wilson SH. Sub-strate channeling in mammalian base excision repair pathways: Passing the baton. J Biol Chem. 285:40479–40488. doi: 10.1074/jbc.M110.155267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Beard WA, Shock DD, Prasad R, Hou EW, Wilson SH. DNA polymerase beta and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J Biol Chem. 2005;280:3665–3674. doi: 10.1074/jbc.M412922200. [DOI] [PubMed] [Google Scholar]
- 40.Prasad R, Batra VK, Yang XP, Krahn JM, Pedersen LC, Beard WA, Wilson SH. Structural insight into the DNA polymerase beta deoxyribose phosphate lyase mechanism. DNA Repair (Amst) 2005;4:1347–1357. doi: 10.1016/j.dnarep.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 41.Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: Evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 42.Deterding LJ, Prasad R, Mullen GP, Wilson SH, Tomer KB. Mapping of the 5′-2-deoxyribose-5-phosphate lyase active site in DNA polymerase beta by mass spectrometry. J Biol Chem. 2000;275:10463–10471. doi: 10.1074/jbc.275.14.10463. [DOI] [PubMed] [Google Scholar]
- 43.Liu Y, Prasad R, Beard WA, Kedar PS, Hou EW, Shock DD, Wilson SH. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J Biol Chem. 2007;282:13532–13541. doi: 10.1074/jbc.M611295200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong D, Demple B. Modulation of the 5′-deoxyribose-5-phosphate lyase and DNA synthesis activities of mammalian DNA polymerase beta by apurinic/apyrimidinic endonuclease 1. J Biol Chem. 2004;279:25268–25275. doi: 10.1074/jbc.M400804200. [DOI] [PubMed] [Google Scholar]
- 45.Tomkinson AE, Chen L, Dong Z, Leppard JB, Levin DS, Mackey ZB, Motycka TA. Completion of base excision repair by mammalian DNA ligases. Prog Nucl Acids Res Mol Biol. 2001;68:151–164. doi: 10.1016/s0079-6603(01)68097-8. [DOI] [PubMed] [Google Scholar]
- 46.Lavrik OI, Prasad R, Sobol RW, Horton JK, Ackerman EJ, Wilson SH. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate: Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J Biol Chem. 2001;276:25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- 47.Zolghadr K, Mortusewicz O, Rothbauer U, Kleinhans R, Goehler H, Wanker EE, Cardoso MC, Leonhardt H. A fluorescent two-hybrid assay for direct visualization of protein interactions in living cells. Mol Cell Proteomics. 2008;7:2279–2287. doi: 10.1074/mcp.M700548-MCP200. [DOI] [PubMed] [Google Scholar]
- 48.Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:13738–13743. doi: 10.1073/pnas.0406048101. [DOI] [PMC free article] [PubMed] [Google Scholar]