

Abstract
The central nervous system of Drosophila melanogaster contains an α-bungarotoxin-binding protein with the properties expected of a nicotinic acetylcholine receptor. This protein was purified 5800-fold from membranes prepared from Drosophila heads. The protein was solubilized with 1% Triton X-100 and 0.5 m sodium chloride and then purified using an α-cobratoxin column followed by a lentil lectin affinity column. The purified protein had a specific activity of 3.9 μmol of 125I-α-bungarotoxin binding sites/g of protein. The subunit composition of the purified receptor was determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis. This subunit profile was identical with that revealed by in situ labeling of the membrane-bound protein using the photolyzable methyl-4-azidobenzoimidate derivative of 125I-α-bungarotoxin. The purified receptor reveals two different protein bands with molecular masses of 42 and 57 kDa. From sedimentation analysis of the purified protein complex in H2O and D2O and gel filtration, a mass of 270 kDa was calculated. The receptor has a s20,w of 9.4 and a Stoke's radius of 7.4 nm. The frictional coefficient was calculated to be 1.7 indicating a highly asymmetric protein complex compatible with a transmembrane protein forming an ion channel. The sequence of a peptide obtained after tryptic digestion of the 42-kDa protein allowed the specific identification of the Drosophila Dα5 subunit by sequence comparison. A peptide-specific antibody raised against the Dα5 subunit provides further evidence that this subunit is a component of an α-bungarotoxin binding nicotinic acetylcholine receptor from the central nervous system of Drosophila.
Nicotinic acetylcholine receptors (nAChRs)1 are key elements of fast cholinergic synaptic transmission (1, 2). They are part of a large superfamily of ligand-gated ion channels comprising 5HT3, GABAA, and GABAC, glycine, and invertebrate anionic glutamate receptors. nAChRs are widely distributed in both vertebrates and invertebrates. In addition to their occurrence in the highly specialized tissue of the electrical organ of certain species of fish (Torpedo californica, Electrophorus electricus), nAChRs are mostly found at the vertebrate neuromuscular junction and the nervous system of both vertebrates and invertebrates (3, 4).
Structurally, nAChRs are formed by assembly of their subunits into a pentameric membrane protein complex. Subunits carrying two conserved adjacent cysteines in the amino-terminal domain in the vicinity of the acetylcholine binding site are classified as α-subunits, while the remainder of the subunit types are designated non-α or -β. The individual subunits have a large amino-terminal domain, which includes the binding site for the ligand, followed by four membrane spanning domains M1–M4. The M2 segments of the five subunits form the lining of the ion channel, which is transiently permeable to cations (Na+, K+, Ca2+) upon binding of the ligand acetylcholine (ACh). Between M3 and M4 a large intracellular loop exists, which contains several phosphorylation sites required for receptor stabilization (for reviews, see Refs. 3, 5, and 6).
Biochemical analysis of receptors subunits composition in a variety of species as well the information obtained from the sequencing of the genome of humans, mice, the nematode Caenorhabditis elegans and the fruit fly Drosophila melanogaster indicates a large number of genes encoding nicotinic acetylcholine receptor subunits. Nicotinic acetylcholine receptors from muscle have an invariant subunit composition ((α1)2(β1)δγ (fetal) or ∊). Neuronal nicotinic acetylcholine receptors are composed of α and non-α-subunits with a stoichoimetry of (α)2(non-α)3. The large number of genes encoding nAChR subunits in the nervous system can give rise to an array of functionally distinct nAChRs. Since the vertebrate nervous system also contains homomeric nAChRs (e.g. (α7)5) the entire range of subunit combinations in the formation of physiological diverse nAChRs is currently unknown. The physiological significance of this subunit diversity, as well as the mechanism governing expression and receptor assembly, are important issues that need to be addressed.
The model organism D. melanogaster offers many of the molecular and genetic tools to study these questions in an in vivo system. In Drosophila, as in insects in general, expression of nAChRs is restricted to the nervous system (7). Unlike in the mammalian central nervous system, nAChRs are part of a major neurotransmitter system in insects with 900–1000 fmol of 125I-α-bungarotoxin binding sites/mg of protein. The sequence of the Drosophila genome predicts seven α- and three non-α-subunits. The characterization of four of the α-subunits (ALS, SAD, Dα3, and Dα4) (8–10) as well as two of the β-subunits (ARD and SBD) (11, 12) was previously carried out through screening with homologous probes of vertebrate nAChR cDNAs. The existence of the remaining three α-subunits Dα5 (13, 14) and Dα6 and Dα7) (14) as well as Dβ3 (15) was recently confirmed by reverse transcription-PCR based on the Drosophila genome information (16).
Little information is currently available about the nature of the receptor subtypes assembled from the various subunit genes in Drosophila. Immunoprecipitation experiments indicate that the ALS and SAD subunits might be part of a receptor complex while Dα3 and ARD might be part of a different receptor subtype (17). It should be noted, however, that it has not been possible to reconstitute functional nAChRs from Drosophila in a heterologous system with any of these subunit combinations tested so far (18, 19). Purification and/or affinity labeling with several ligands in situ allowed the identification of several proteins but no correlation was established between the known Drosophila nAChR subunits and the proteins detected (20–22). In this paper we show that after a 5800-fold purification of nAchRs from Drosophila heads two major bands with masses of 42 and 57 kDa can be identified. The sequence of a peptide obtained after tryptic digestion and microbore HPLC of the 42-kDa protein allowed the specific identification of the Drosophila Dα5 subunit as part of this receptor.
Materials and Methods
Chemicals
Protease inhibitors pepstatin A, leupeptin, and antipain were obtained from Bachem (King of Prussia, PA); benzamidine, bacitracin, iodoacetamide, phenylmethylsulfonyl fluoride, aprotinin, soybean trypsin inhibitor, and ovomucoid trypsin inhibitor were purchased from Sigma as well as carbamylcholine, nicotine-tartrate, d-tubocurarine, hexamethonium chloride, atropine sulfate, physostigmine, acetylcholine bromide, and deoxycholate. Methyl-α-d-mannopyranoside was obtained from Pfanstiehl Laboratories (Waukegan, IL). α-Bungaro-toxin, κ-toxin, and Naja naja siamensis crude venom were purchased from Miami Serpentarium Laboratory (Punta Gordia, FL). Na125I was received from PerkinElmer Life Sciences. Trypsin was obtained from Roche Applied Science.
Drosophila Cultures
The wild-type Oregon R strain of D. melanogaster was used in all experiments. Flies were raised on a standard cornmeal agar medium (23) in polypropylen boxes at 23 ± 2 °C and 65% relative humidity. Flies were harvested in a cold room (2 °C) and stored frozen at −75 °C.
Preparation of 125I-α-Bungarotoxin
α-Bungarotoxin was iodinated as described (33). The initial specific activity was in the range of 40–80 Ci/mmol. For competitive binding assays monoiodinated α-bungaro-toxin was prepared by chromatography on CM-Sephadex essentially as described previously (24). The specific activity was adjusted by addition of unlabeled α-bungarotoxin to a range of 60–120 Ci/mmol.
Buffers and Solutions
The following buffers and solutions were used: assay buffer, 50 mm NaCl, 3 mm NaN3, 1% Triton X-100, 0.01% cytochrome c, 10 mm NaPi, pH 7.3; washing buffer, 3 mm NaN3, 0.1% Triton X-100, 10 mm NaPi, pH 7.3; homogenization buffer, 1% (w/v) sucrose, 5 mm EDTA, 5 mm EGTA, 3 mm NaN3, 5 mm NaPi, pH 7.3; solubilization buffer, 2% Triton X-100, 1 m NaCl, 10 mm EDTA, 10 mm EGTA, 3 mm NaN3, 5 mm NaPi, pH 7.3; dialysis buffer 1, 1 mm EDTA, 3 mm NaN3,l mm phenylmethylsulfonyl fluoride (PMSF), 1 mm benzamidine, 1 mm NaPi, pH 7.3; dialysis buffer 2,100 mm NaCl, 1 mm EDTA, 3 mm NaN3 10 mm NaPi, pH 7.3; column buffer 1,1m NaCl, 1% Triton X-100, 3 mm NaN3, 1 mm EDTA, 1 mm EGTA, 10 mm NaPi, pH 7.3; column buffer 2, 100 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 3 mm NaN3,10 mm NaPi, pH 7.3; column buffer 3, 50 mm Tris-Cl, pH 8.5, 0.1% deoxycholate, 100 mm NaPi, 3 mm NaN3, 1 mm EDTA; buffer 4, 50 mm Tris-Cl, pH 8.5, 0.1% deoxycholate, 1 m NaCl, 100 mm NaPi, 3 mm NaN3, 1 mm EDTA; Lentil lectin column storage buffer, 50 mm Tris-Cl, pH 7.3, 1 mm MnCl2, 1 mm CaCl2, 1% Triton X-100, 2% mannopyranoside, 3 mm NaN3; protease inhibitor, 10 mm iodoacetamide; protease inhibitor mixture 1, 20 KIU/ml aprotinin, 1 μg/ml antipain, 5 mm benzamidine, 100 μg/ml bacitracin, 2 μg/ml leupeptin, 1 mm Na2S2O5, 2 μg/ml pepstatin A, 5 mg/ml soybean trypsin inhibitor, 1 μg/ml trypsin inhibitor, 1 mm PMSF; protease inhibitor mixture 2, same as protease inhibitor mixture 1, except aprotinin is omitted; protease inhibitor mixture 3, 5 mm benzamidine, 1 mm Na2S2O5, 1 mm PMSF; blotting buffer, 80% 25 mm Tris-Cl, pH 8.3, 192 mm glycine, 20% methanol (v/v).
Determination of 125I-α-Bungarotoxin Binding Activity
Samples of membrane-bound and solubilized binding protein were incubated, if not stated otherwise, with 5 nm 125I-α-bungarotoxin in a final volume of 0.3 ml of assay buffer for 60 min at 23 °C. At the end of the incubation period the assay mixture was collected on a double thickness of DEAE-filter disk, which was prewetted with washing buffer, using a filtration manifold apparatus (Earle Sandbek, Airville, PA). The filter disks were washed twice with 4 ml each of washing buffer, dried, and counted in a LKB γ-counter. Nonspecific binding was determined by preincubating samples in the presence of d-tubocurarine at a final concentration of 10−4 m for 20 min. All binding data represent the mean of triplicate determinations.
The IC50 for several cholinergic ligands was determined by preincubating the respective receptor fraction with the various ligand at 10 different concentrations for each ligand for 20 min followed by incubation with 5 nm 125I-α-bungarotoxin for 1 h.
Protein Determination
A modification of the Lowry protein assay procedure, which allows determination of micrograms of proteins in the presence of detergents, was used (25).
Protein Precipitation
Protein precipitation of membrane proteins was carried out as described (26).
SDS-Polyacrylamide Gel Electrophoresis
Electrophoresis was carried out in 10% polyacrylamide (acrylamide:bisacrylamide 37.5:1) according to the procedure of Laemmli (27). The following prestained molecular weight markers were used: 2-macroglobulin (Mr = 180,000), β-galactosidase (Mr = 116,000), fructose-6-phosphate kinase (Mr = 84,000), pyruvate kinase (Mr = 58,000), fumarase (Mr = 48,500), lactic dehydrogenase (Mr = 36,500), and triosephosphate isomerase (Mr = 26,600)
Preparation of the Methyl-4-azidobenzoimidate Derivative of 125I-α-Bungarotoxin
The derivatized α-bungarotoxin was synthesized essentially as described by (28) with the following modifications. A solution containing 200–400 nm monoiodo-125I-α-bungarotoxin, 60 mm NaCl, and 3.3 mm NaPi, pH 7.3, was adjusted to pH 9.5 by the addition of 20 mg Na2CO3 and 25 mg NaHCO3. All subsequent steps were conducted in the dark using a Wratten no. 2 safe light. At 0, 2, 4, and 7 h, 1 mg of methyl-4-azidobenzoimidate was added to the toxin, and the mixture was stirred continuously at 4 °C for a total of 22 h. Cytochrome c (1 mg) was then added as a carrier and the mixture was purified on a Sephadex G-25 column (0.7 cm × 30 cm), which was equilibrated and eluted with 3.3 mm NaPi buffer, pH 7.3. 0.5-ml fractions were collected, and the first major peak fractions were pooled and stored in the dark at 4 °C in the presence of 0.02% NaN3. Methyl-4-azidobenzoimidate was synthesized according to published procedure (29).
Autoradiography
Gels were soaked for 2 h in a solution containing 0.1% (v/v) glycerol, 10% (v/v) acetic acid, dried on filter paper in a gel dryer, and exposed for autoradiography using Kodak XAR film and Cronex intensifying screens (DuPont Co.).
Sucrose Density Gradient Centrifugation
Linear 5–20% sucrose gradients were prepared in either H2O or D2O containing 500 mm NaCl, 1% Triton X-100, 1 mm EDTA, 3 mm NaN3, 10 mm NaPi, pH 7.3. Centrifugation was carried out essentially as described by Martin and Ames (49), using a Beckman SW 50.1 rotor. The standards used were β-galactosidase (s20,w = 15.9), catalase (s20,w = 11.3), and alcohol dehydrogenase (s20,w = 7.6).
Gel Filtration
Gel filtration was carried out using a siliconized glass column (50 × 1 cm) with Sepharose 4B in the same buffer used for gradient centrifugation. The Stoke's radii of the protein markers used were thyroglobulin (8.5 nm), β-galactosidase (6.9 nm), ferritin (6.1 nm), catalase (5.2 nm), and alcohol dehydrogenase (4.5 nm).
Preparation of Affinity Resins
α-Cobratoxin was purified from the crude venom of N. naja siamensis as described (30). Purity was assessed by SDS-polyacrylamide gel electrophoresis in 15% polyacrylamide gels and by sequencing using 15 degradation cycles on an Applied Biosystems 477A protein sequencer. α-Cobratoxin was coupled to Separose 4B (2 ml of gel/mg of toxin) as described by March et al. (31) without the 2 m urea wash step. The obtained affinity gel had a capacity of about 35 pmol of 125I-α-bungarotoxin binding sites/ml of gel.
Lentil lectin was purified essentially as described (32) and its purity analyzed by SDS-polyacrylamide gel electrophoresis on 10% polyacrylamide. Coupling was carried out at a ratio of 1:1.8 (ml of gel/mg of lectin) as described for α-cobratoxin. When used as the second affinity column this gel had a capacity of 35 pmol of 125I-α-bungarotoxin binding sites per ml of gel.
Photoaffinity Labeling
Photoaffinity labeling of the membrane-bound and solubilized receptor was carried out as described (28, 29) in the presence of protease inhibitor mixture 1. The labeling reaction was equilibrated in the dark for 40 min on ice before radiation with a small UV lamp (330 nm) for 40 min.
Purification of the Acetylcholine Receptor
Unless otherwise noted all steps were carried out at 0–4 °C. Heads from Drosophila were essentially prepared as described (33) with the modification that an equal amount of powdered dry ice was added during both the disjoining and sieving step to assure that the tissue was kept well frozen. The isolated heads were stored overnight in a deep freezer at −75 °C to allow the dry ice to sublimate.
Membranes for receptor purification were prepared by suspending heads at a concentration of 100 mg of heads/ml in homogenization buffer containing 10 mm iodoacetamide in addition to protease inhibitor mixture 1. The heads were homogenized using a VirTis-23 homogenizer. The homogenate was centrifuged for 10 min at 2000 × g in a GSA rotor (Sorvall), and the supernatant was filtered through a double layer of cheesecloth. The pellet was resuspended to 100 mg of heads/ml of homogenization buffer plus protease inhibitor mixture 1 and homogenized and centrifuged as described above. The two supernatants were combined and diluted by one volume of the homogenization buffer and then centrifuged for 40 min at 17,000 × g (GSA rotor). The upper pink part of the pellet was resuspended to 160 mg of heads/ml of homogenization buffer plus inhibitors and homogenized in a Teflon pestle-glass homogenizer using 30 strokes at 1700 rpm at 0 °C. This membrane fraction was used for subsequent solubilization and purification. If not used immediately it was stored in 50-ml plastic tubes in liquid nitrogen. Receptors were solubilized by diluting the membrane fraction with an equal volume of solubilization buffer containing 10 mm iodoacetamide and protease inhibitor mixture 1. The suspension was homogenized with 30 strokes at 1700 rpm in a Teflon pestle-glass homogenizer and centrifuged for 30 min at 235,000 × g (Type Ti45 rotor, Beckman). The supernatant was dialyzed in Spectrapor 2 tubing for 1–2 h against two changes (5 liters of each) of dialysis buffer until the conductivity was equivalent a 100 mm NaCl solution.
The dialyzed preparation was subjected to affinity chromatography on an α-cobratoxin column. Two columns (2.5 × 6-cm gel bed in a 60-ml plastic syringe) were washed with 60 ml of column buffer 1 and protease inhibitor mixture 2. The dialysate was then applied to the column. The column was washed with 120 ml of column buffer 2. The receptor was eluted from the column with 120 ml of column buffer 2 containing 200 mm carbamylcholine, and the entire 120 ml of eluate was applied immediately to the lentil lectin affinity column as described below. The cobratoxin column was regenerated by washing with 60 ml of column buffer 1 containing 1 m carbamylcholine followed by 120 ml of column buffer 2 without the protease inhibitors and stored at 4 °C for reuse.
The lentil lectin column (2.5 × 3-cm gel bed) was washed with 100 ml of column buffer 2, and the eluate from the α-cobratoxin column was applied. The column was washed with 30 ml of column buffer 1 and then with 30 ml of column buffer 2, both containing protease inhibitor mixture 2 and than 15 ml of column buffer 3 containing protease inhibitor mixture 3. The receptor was eluted by incubating the column for 30 min with 15 ml of column buffer 3 containing 15% methyl-α-D-mannopyranoside and protease inhibitor mixture 3 and then eluting with the same buffer until a total of 30 ml had been collected. The lentil lectin column was washed with 60 ml of column buffer 1 containing 20% methyl-α-D-mannopyranoside followed by 50 ml of lentil lectin column storage buffer and stored at 4 °C for later use.
Final purification was achieved by concentrating the eluate from the lentil lectin column in Amicon 8050 and Amicon 8010 concentrators using a Diaflo Ultrafiltraton membrane YM100 (Amicon Division, W. R. Grace & Co.) under pressure of 20 p.s.i. argon to a volume of 0.5–1 ml on ice.
Sequence Analysis
Proteins separated by SDS-gel electrophoresis were blotted on nitrocellulose membrane at 20 mA for 16 h. Transfer was verified by briefly staining the membrane with 0.1% Ponceau S in 1% acetic acid. Tryptic digestion of the membrane was carried out according to Aebersold et al. (34) in 100 mm Tris-Cl, pH 8.2/acetonitrile 95:5 (v/v) containing 0.2% Ca2+ overnight. Tryptic digest were extracted from the membrane by acidification with 10% trifluoroacetic acid. Extracts were separated by microbore HPLC on a Brownlee Aquapore RP-300 column (2.1 × 100 mm) in the following buffer system: Buffer A, 0.1% trifluoroacetic acid in water, Buffer B: 0.09 trifluoroacetic acid in acetonitrile/H2O, 70:30 (v/v) using a gradient of 0–100% Buffer B at 25 °C at a flow rate of 100 μl/min. Eluting peptides were monitored at 220 nm using a Waters 490 detector and collected manually. Fractions were either lyophylized or loaded directly on a glass fiber filter for sequencing using an Applied Biosystems 477A automated sequencer coupled to an on-line 120A HPLC system.
Generation of Dα5 Antibodies
A peptide-specific antibody was generated in rabbits against the peptide NH2-CSDTSSERKHQILSD-VELK-COOH corresponding to amino acids 650–664 of the Dα5 sequence. The production and affinity purification of the antibody was carried out by BioSynthesis, Inc. (Lewisville, TX).
Western Blot Analysis
Aliquots of protein fractions to be analyzed were separated by SDS-gel electrophoresis on 8% acrylamide gels, followed by blotting to a polyvinylidene difluoride membrane. The membrane was incubated overnight in blocking solution, incubated with Dα5 antibody (1:500 dilution), washed, and incubated with horseradish peroxidase-conjugated anti-rabbit antibody (1:8000) (Bio-Rad). For antibody visualization the Supersignal West Pico chemiluminescent substrate kit (Pierce) was used.
Results
Photoaffinity Labeling of the Acetylcholine Receptor Protein
Derivatization of 125I-α-bungarotoxin with methyl-4-azidobenzo-imidate had no noticeable effect on the ability of the toxin to bind to the membrane bound α-bungarotoxin-binding protein. The dissociation kinetics of the derivatized toxin by unlabeled α-bungarotoxin prior to photoactivation was also unaffected displacing more than 80% of the derivatized toxin in 2 h. After photoactivation more than 80% of the photolyzable toxin derivative bound irreversible to the membrane and could not be displaced by a 1000-fold excess of unlabeled α-bungarotoxin (data not shown). As seen in Fig. 1 labeling of the membrane fraction from Drosophila heads allows detection of two major bands with masses of 42 and 57 kDa (after correction for the mass of the toxin). Preincubation of the membranes with the cholinergic ligands α-bungarotoxin (α-Bgtx), α-cobratoxin (α-Cbtx), κ-toxin, Ach, or nicotine (Nic) prevented labeling of the two major bands of 42 and 57 kDa as well as three very weak bands of 78 and 26 kDa.
Fig. 1. Affinity labeling of membrane bound receptor with the 4-azidobenzoimidate derivative of 125I-α-bungarotoxin.
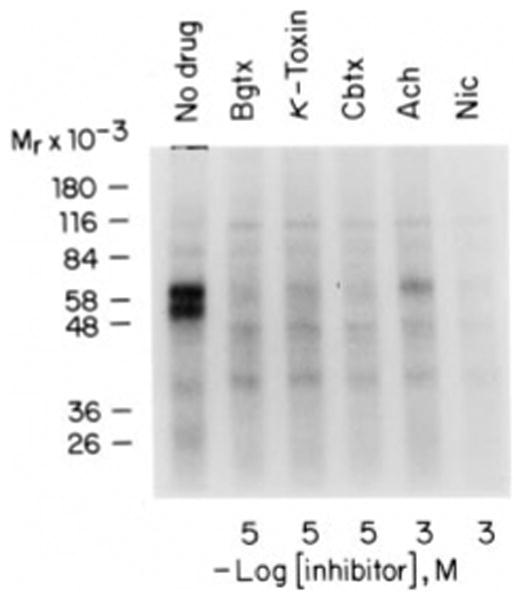
Membranes prepared from Drosophila heads were preincubated for 20 min with the ligands α-bungarotoxin (Bgtx), κ-toxin, α-cobratoxin (Cbtx), Ach, and nicotine (Nic) at the concentrations indicated. Affinity labeling was carried out with the 4-azidobenzoimidate derivative of 125I-α-bungarotoxin. After affinity labeling membranes were collected by centrifugation and subjected to SDS-polyacrylamide gel electrophoresis using a 10% polyacrylamide gel. An autoradiogram of the dried gel is shown. Cross-linking with α-bungarotoxin increases the molecular mass of each band by about 8 kDa.
Fig. 2 shows the results if the affinity labeling is carried out after preincubation of the membrane fraction with either increasing concentrations of Ach (Fig. 2A) or increasing concentrations of α-bungarotoxin (Fig. 2B). The increase in the concentration of unlabeled α-bungarotoxin resulted in a uniform decrease of labeling of both major bands. However, increasing the concentration of acetylcholine abolished labeling first of the 42-kDa band at 10−4 m, while a concentration of 10−3 m was required to prevent labeling of the 57-kDa band. A similar result was obtained with nicotine at 10−6 and 10−5 m, respectively (data not shown). These results clearly indicated the presence of at least two independent binding sites for cholinergic ligands on two different subunits.
Fig. 2. Affinity labeling of membrane bound binding site with the 4-azidobenzoimidate derivative of 125I-α-bungarotoxin in the presence of increasing amounts of acetylcholine (A) and α-bungarotoxin (B).
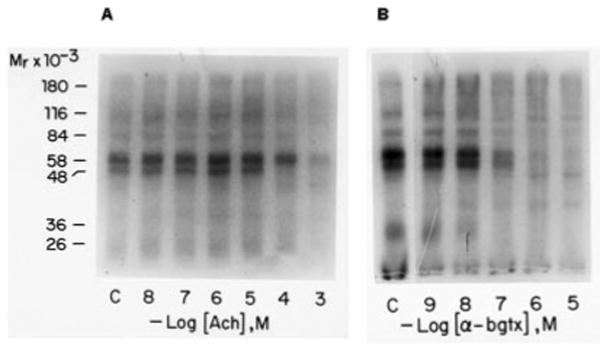
A, membranes prepared from Drosophila heads were preincubated with increasing amounts of acetylcholine for 20 min in the presence of 1 μm of the cholinesterase inhibitor physostigmine. The presence of the inhibitor had no effect on the binding of 125I-α-bungarotoxin or its photoactivatable derivative (data not shown). B, membranes prepared from Drosophila heads were preincubated with increasing amounts of α-bungarotoxin (α-bgtx). Photoaffinity labeling was carried out in both experiments as described under experimental procedures. Following labeling membranes were collected by centrifugation and analyzed by SDS-polyacrylamide gel electrophoresis. An autoradiogram of the dried gel is shown. Cross-linking with α-bungarotoxin increases the molecular mass of each band by about 8 kDa.
Purification of the Acetylcholine Receptor Protein
We developed a purification procedure for the acetylcholine receptor protein from the central nervous system of D. melanogaster (see Table I). Heads from Drosophila were prepared as the first step of the procedure because more than 50% of the α-bungarotoxin-binding protein is localized in this tissue. Pulverized dry ice was added during the head isolation to keep the tissue well frozen and to minimize proteolytic degradation. Homogenization and all subsequent steps were carried out at temperatures between 0–4 °C, and an assortment of protease inhibitors were added to prevent, or at least minimize, proteolytic degradation. Analysis of the purified receptor fraction isolated using only PMSF, EDTA, and EGTA as protease inhibitor resulted in only a smear or no polypeptide at all (data not shown).
Table I. Purification of the acetylcholine receptor protein.
This semi-preparative purification is based on a starting amount of 28g of heads (300 g of flies).
| Fraction | 125I-α-Bungarotoxin binding | Recovery | Protein | Specific activity | Purification fold |
|---|---|---|---|---|---|
| pmol | % | mg | μmol/g protein | ||
| Homogenate | 2281 | 100 | 3326 | 0.00067 | |
| Triton X-100 extract | 453 | 19 | 273 | 0.00166 | 2.4 |
| α-Cobratoxin column eluate | 210 | 9.2 | 1.3 | 0.156 | 232 |
| Lectin column eluate | 106 | 4.6 | 0.056 | 1.9 | 2830 |
| YM100 concentrate | 45 | 2.0 | 0.0113 | 3.9 | 5820 |
Enrichment of the α-bungarotoxin-binding protein was achieved by preparing a membrane fraction by differential centrifugation. This step allowed removal of chitin and large debris. The membrane fraction obtained contains 60–70% of the starting activity of the α-bungarotoxin-binding protein.
We and others (35–37) have encountered difficulties in solubilizing the α-bungarotoxin-binding protein from Drosophila. Solubilization was most effectively accomplished using 1% Triton X-100 in the presence of 500 mm NaCl but in agreement with the previous investigators only about 20% of the starting binding activity remains in the supernatant of the high speed spin (235,000 × g for 45 min).
The ionic strength of the supernatant from the high speed spin had to be reduced to about 100 mm NaCl equivalent because otherwise the α-bungarotoxin-binding protein would not bind to the α-cobratoxin affinity column. Dialysis of the supernatant in Spectraphore 2 against 15 volumes of dialysis buffer for 1 h was sufficient to lower the NaCl concentration.
After passage of the solubilized extract over the α-cobratoxin column about 50% of the α-bungarotoxin-binding protein bound to the column. Repeated application did not improve this result. Analysis of the crude solubilized fraction by gel filtration on Sepharose 4B revealed that about 50% of α-bungarotoxin-binding protein activity eluted in the void volumes and did not subsequently bind to the α-cobratoxin colum (data not shown). Sedimentation analysis of the solubilized receptor in a 5–20% sucrose gradient revealed the presence of two peaks of 125I-α-bungarotoxin binding activity with sedimentation coefficients of ∼9 and 13 S (data not shown). The 13 S component was not sensitive to sulfhydryl reducing agents and represented the material that did not bind to the affinity column (data not shown).
At this stage of the purification it was not possible to wash the affinity column in higher than 100 mm NaCl with out eluting large amounts of α-bungarotoxin-binding protein. About 60–80% of the fraction absorbed could be eluted with 200 mm carbamylcholine. The yield was not improved by using higher, up to 1 m, concentrations of carbamylcholine.
The fraction eluted from the α-cobratoxin column bound quantitatively to the lectin affinity column in the presence of carbamylcholine indicating that the receptor is a glycoprotein and the binding sites for agonist and lectin on the receptor are different. Lectins from ordinary lentils (lens culinaris) were chosen as affinity ligand because the α-bungarotoxin-binding protein could not be eluted from a concanavalin A affinity column, while the recovery from the lentil lectin affinity column was about 50%. Washing the column with buffer containing 1 m NaCl was possible without loss in binding activity. The lectin affinity column step resulted in an additional 10-fold purification.
Final purification was accomplished using an Amicon concentrator with a YM100 membrane resulting in a further 2-fold purification.
Hydrodynamic Properties of the Acetylcholine Receptor Protein
The purified α-bungarotoxin-binding protein was subjected to gel filtration on a Sepharose 4B column in the presence of suitable marker proteins. The receptor protein eluted as a sharp symmetrical peak (Fig. 3A). The Stoke's radius was calculated to be 7.4 nm.
Fig. 3.
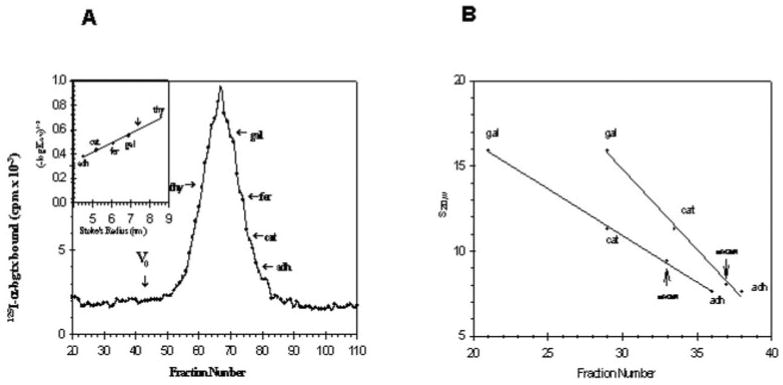
A, determination of Stoke's radius of purified receptor. Gel filtration of the purified receptor is shown. Purified receptor and marker proteins were applied to a column (1 × 50 cm) of Separose 4B as described under “Materials and Methods.” The flow rate was 7.5 ml/h, and 400-μl fractions were collected. The void volume V0 of the column was determined using blue dextran. Specific binding of 125I-α-bungarotoxin to aliquots to each fraction was determined with the DEAE filter assay. thy, thyroglobulin; gal, β-galactosidase; fer, ferritin; cat, catalase, adh, alcohol dehydrogenase). The inset shows the data plotted according to Siegel and Monty (40). The vertical arrow marks the position of the peak of the purified binding protein. B, determination of the apparent s20,w values for the receptor protein in H2O and D2O. Linear sucrose gradients (5–20%) were prepared in H2O and D2O containing 10 mm NaPi, pH 7.3, 500 mm NaCl, 1% Triton X-100, 1 mm EDTA, 3 mm NaN3. 200 μl of sample containing protease inhibitors was loaded onto 4.8 ml of gradient. Centrifugation was carried out in a Beckman SW 50.1 rotor at 50,000 rpm for 5 h at 4 °C. After centrifugation 100-μl fractions were collected. Aliquots of each fractions were used to determine the peaks of α-bungarotoxin binding activity and internal standards. The vertical arrows indicate the peak of α-bungarotoxin binding activity. The internal standards used were β-galactosidase (s20,w = 15.9), bovine liver catalase (s20,w = 11.3), and yeast alcohol dehydrogenase (s20,w = 7.6).
Since the receptor exhibited properties of an integral membrane protein, it is most likely that large amounts of the detergent are bound to it during solubilization, as has been reported for other receptors (38–40). The contribution of detergent to the mass of the receptor protein can be determined by sedimentation analysis in H2O and D2O in the presence of suitable marker proteins, which do not bind detergents (41). The purified α-bungarotoxin-binding protein exhibited an apparent sedimentation coefficient at 9.4 in H2O but only 8.2 in D2O. This indicates the presence of tightly bound detergent. The data from this analysis allowed us to calculate some of the physical properties of the receptor, which are summarized in Table II. The partial specific volume v was calculated to be 0.768 ml/g, which is considerably higher than that of an average soluble protein (= 0.73 ml/g). Using the Stoke's radius, s20,w, and the partial specific volume, the molecular weight corrected for the contribution of detergent was calculated to be 270,000. The frictional ratio was determined to be 1.72 indicating a highly asymmetric protein complex, compatible with the notion that it is a transmembrane protein.
Table II. Physical properties of the acetylcholine receptor.
| Partial specific volume υ | 0.768 ml/g |
| s20,w | 9.2 S |
| Stoke's radius | 7.4 nm |
| Mr of receptor-Triton X-100 complexa | 330,000 |
| Mr of receptorb | 270,000 |
| f/f0 | 1.72 |
The molecular weight and frictional ratio were calculated according to Siegel and Monty (40) using the following formula,
where a = Stokes radius, N = Avogadro's number, η = viscosity of water at 20 °C, ρ = density of water at 20 °C, and υ = partial specific volume.
These calculations are based on the assumption that the υ observed represents an average of the contributions made by protein (0.735 ml/g) and tightly bound Triton X-100 (0.94 ml/g).
The frictional ratio was calculated from Siegel and Monty (40).
Subunit Composition
Analysis of about 1.5 μg of purified receptor protein by SDS-polyacrylamide gel electrophoresis followed by either Coomassie Blue staining (Fig. 4, lane 2E2) or silver staining (Fig. 4, lane 2E1) revealed two major bands with masses of 42 and 57 kDa. These two bands were observed consistently in five independent purifications. In addition three bands larger than 57 kDa are barely visible. Occurrence of these bands varied in between different purifications.
Fig. 4. SDS-polyacrylamide gel electrophoresis of the main fractions obtained during the receptor protein purification.
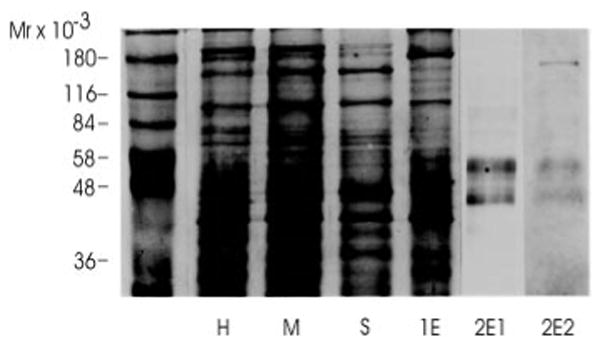
Aliquots of the main fractions obtained during the purification were analyzed by SDS-polyacrylamide gel electrophorsis on a 10% polyacrylamide gel. The equivalent of 10 μg of protein was loaded in lanes H, M, S, and 1E. 1.5 μg of protein was loaded in lanes 2E1 and 2E2. Following electrophoresis proteins were visualized by silver staining with the exception of lane 2E2, which was stained with Coomassie Blue. H, head homogenate; M, membrane fraction; S, solubilized binding protein fraction; 1E, eluate from the cobratoxin affinity column; 2E1 and 2E2, eluates of the lentil lectin affinity column after concentration.
To establish that the polypeptides observed after SDS-polyacrylamide electrophoresis were part of the receptor, we carried out sedimentation analysis of the purified protein using a 5–20% sucrose gradient. Fractions collected were analyzed for 125I-α-bungarotoxin binding activity. The binding protein sedimented as a single symmetrical peak (Fig. 5). Fractions containing the leading edge (fraction 1), the peak fraction (fraction 2), and the trailing edge (fraction 3) were pooled as indicated in Fig. 5. The protein was precipitated and analyzed by SDS-polyacrylamide electrophoresis. Polypeptides of 42 and 57 kDa correlated in intensity with the peak fraction (fraction 2).
Fig. 5. Analysis of the purified binding protein fraction.
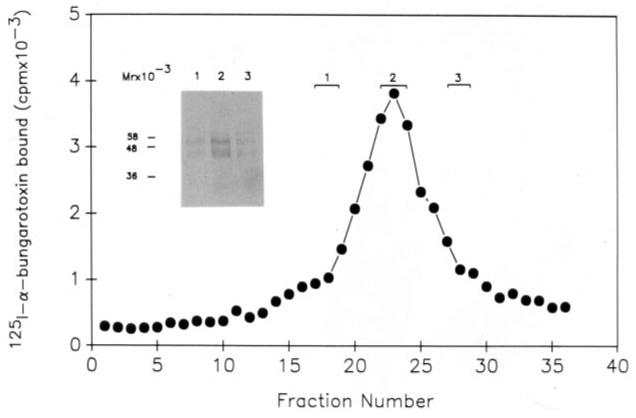
26 μg of purified binding protein fraction was layered on a sterile 5–20% sucrose gradient as described under “Materials and Methods.” Centrifugation was carried out in a Beckman SW 50.1 rotor at 45,000 rpm for 12 h at 4 °C. Fractions of 110 μl were collected from the gradient and analyzed for α-bungarotoxin binding activity. Fractions obtained from the gradient were pooled as indicated in the figure and the proteins in the three pools precipitated quantitatively as described under “Materials and Methods.” The precipitated proteins from the three pools were analyzed by SDS-polyacrylamide gel electrophoresis corresponding to lanes 1, 2, and 3 on the inserted panel and visualized by silver staining.
Pharmacological Properties
Competitive binding studies with various cholinergic ligands using both the membrane bound and the purified receptor protein were carried out, and the results are shown in Table III. Nicotine and d-tubocurarine showed the greatest potency in displacing 125I-α-bungarotoxin followed by acetylcholine, while atropine and hexamethonium had considerable less affinity to the receptor protein. Comparison of the IC50 of these ligands for membrane-bound and purified receptor protein indicated a slight decrease of the IC50 for d-tubocurarine after purification, while the IC50 for atropine has increased. In general, however, the pharmacological specificity of the receptor protein appeared to be preserved during the purification.
Table III. IC50 of various cholinergic ligands for binding to the receptor.
Binding of 125I-α-bungarotoxin was determined as described under “Materials and Methods” except that the samples were preincubated with competitors for 20 min.
| Ligand | Membrane-bound | Purified receptor |
|---|---|---|
| Nicotine | 4.5 × 10−7 m | 5.0 × 10−7 m |
| d-Tubocurarine | 2.6 × 10−6 m | 7.4 × 10−7 m |
| Acetylcholine | 2.8 × 10−6 m | 3.6 × 10−6 m |
| Atropine | 5.7 × 10−5 m | 1.6 × 10−3 m |
| Hexamethonium | 1.3 × 10−3 m | |
| Decamethonium | 2.0 × 10−4 m |
Sequence Analysis
An attempt to sequence the 44- and 57-kDa protein bands after SDS-gel electrophoresis followed by electroblotting onto Immobilon gave no sequencing signal indicating that the amino termini of the proteins in the two bands are blocked. Consequently, we digested each band with trypsin after blotting onto nitrocellulose membrane followed by separation and collection by microbore HPLC. The sequence of two major peptides designated M55 (WKPDVLMY) and M27 (AD-EGFDGTYQTN) derived from the 44-kDa protein band was determined. A sequence comparison of these two sequences with the sequence of the known acetylcholine receptor genes from the Drosophila data base were carried out as shown in Fig. 6. The sequence of the M55 only matched the acetylcholine receptor subunits Dα5, Dα6, and Dα7, while the sequence of M27 peptide matched uniquely only the Dα5 subunit, strongly supporting that this subunit is part of a neuronal nicotinic acetylcholine receptor from Drosophila. The sequence relationship of the M55 and M27 peptide to the Drosophila Dα5 and the remaining Drosophila nAChR subunits is shown in Fig. 7 in a dendrogram.
Fig. 6. Sequence alignment of the predicted proteins of the acetylcholine receptor genes from Drosophila.

The protein sequences were obtained from the Swiss Protein Data Base and aligned. The Dα5-derived peptide designated M55 matches amino acids 394–402, while M27 matches amino acids 404–415, of the predicted Dα5 sequence. The sequence alignment was obtained using the AlignX program of the Vector NTI 8 program package (Invitrogen) using the Drosophila nAChR protein sequences from the Swiss Protein Data Base.
Fig. 7. Dendrogram of the Drosophila AChR subunit proteins.
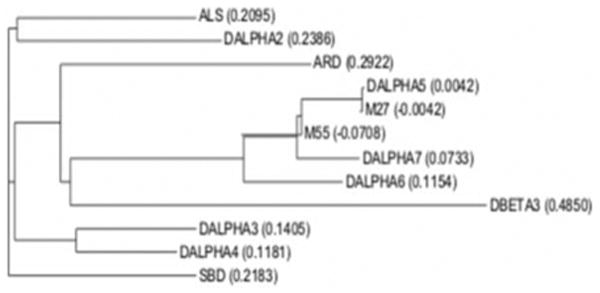
A nearest neighbor dendrogram of the Drosophila AChR subunit proteins including peptides M55 and M27 obtained by sequencing peptides derived from the 42-kDa protein is shown. This dendrogram clearly demonstrates that peptide M27 is closely related to the Dα5, Dα6, and Dα7 subunits, while peptide M27 is only related to Dα5. The dendrogram was obtained using the AlignX program of the Vector NTI 8 program package (Invitrogen) using the Drosophila nAChR protein sequences from the Swiss Protein Data Base.
Western Blot Analysis
We raised a peptide-specific polyclonal antibody against the Dα5 subunit and analyzed the membrane fraction from Drosophila heads, the fraction eluted from the α-cobratoxin column, and the purified receptor fraction by Western blot analysis. As can be seen in Fig. 8 in all three fractions a band of 42 kDa can be detected. This band is co-migrating with the 42-kDa band detectable after silver staining the purified receptor fraction, further supporting that the Dα5 subunit is expressed in the central nervous system of Drosophila and is part of a nicotinic acetylcholine receptor.
Fig. 8. Western blot analysis.
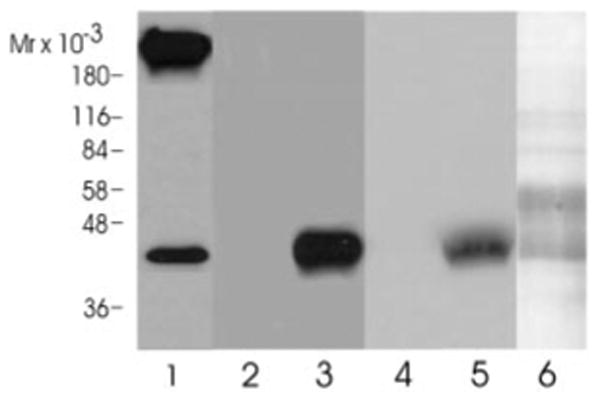
Aliquots of the crude membrane fraction (lane 1), the α-cobratoxin eluate (lanes 2 and 3), and the lentil lectin column eluate (lanes 4 and 5) were analyzed by SDS-gel electrophoresis and immunoblotted onto a polyvinylidene difluoride membrane. Blots were probed either with the preimmunserum (lanes 2 and 4) or the affinity purified Dα5 antibody (1:500 dilution). Bound antibodies were visualized with a horseradish peroxidase-conjugated anti-rabbit antibody (Bio-Rad) using an enhanced chemiluminescence Western blot kit (Pierce). The antibody detected a protein of 42 kDa in lanes 1, 3, and 5. Lane 6 shows a silver stain of the purified receptor fraction that was run on the gel in parallel to the samples in lanes 4 and 5.
Discussion
Purification of membrane proteins is subject to potential artifacts due to proteolysis or co-purification of functional unrelated proteins. For this reason, prior to purification, we wanted to investigate the subunit structure of the receptor in its membrane environment by crosslinking 125I-α-bungarotoxin to the receptor. We chose to derivatize mono-125I-α-bungaro-toxin with the heterobifunctional cross-linking agent 4-methyl-azidobenzoimidate. The derivatized toxin still maintains a positive charge on the lysine-ε-amino groups essential for toxin binding. A similar cross-linking agent (ethyl-N-5-azido-2-nitro-benzylaminoacetamide) had been used previously with success for labeling correctly the subunits of the nicotinic acetylcholine receptor from T. californica and rat diaphragm (42). The results of our photoaffinity labeling experiments reveal two major bands of 42 and 57 kDa with additional weakly labeled bands of 102, 68, and 26 kDa. Using chemical cross-linking of 125I-α-bungarotoxin with a carbodiimide Schloss et al. (20) detected a polypeptide of 42 kDa. Tomizawa et al. (22) reported the specific labeling of a 66-kDa polypeptide with [3H]azido-neonicotinoid. Using a neonicotinoid affinity colum, Tomizawa et al. (21) reported the purification of a nAChR from Drosophila with a subunit composition of 61, 66, and 69 kDa. Using a similar neonicotinoid ligand these authors eluted from an α-bungarotoxin affinity column a nAChR with identical subunit composition. However, when elution of this affinity column was carried out with either carbachol or d-tubocurarine, no receptor subunits could be identified. The experiment described by these authors is difficult to interpret because they solubilized the receptor with Triton X-100 in the presence of 100 mm NaCl and in the absence of any protease inhibitors other then PMSF. At this NaCl concentration we and others (37) found it difficult to solubilize any substantial amount of 125I-α-bungarotoxin binding activity. Since these authors did not characterize the purified receptor any further, additional experiments are required to establish the identity of the proteins observed in their purified receptor fraction.
Our experiments demonstrate that after 5800-fold purification the receptor revealed by SDS-polyacrylamide gel electrophoresis had the same subunit profile as demonstrated by photoaffinity labeling. In addition, photoaffinity labeling of the α-cobratoxin column and lentil lectin column fractions demonstrated that the two bands originally labeled in the membrane in situ purified without changes in size through the purification procedure.
Gel filtration and sedimentation analysis demonstrated that AChR from Drosophila was similar to other nAChRs. The observed sedimentation coefficient of 9.2 S is close to the sedimentation coefficient for the receptor from T. californica (9 S). The calculated mass of 270 kDa is also similar to other nAChRs (for reviews, see Refs. 5, 6, 43, and 44).
The pharmacological profile of the receptor resembles the pharmacology of AchR of the cockroach (Planetaria americana) (45). Most notably, κ-toxin binds to the Drosophila receptor and competes for the binding of α-bungarotoxin. A comparison of the IC50 for membrane-bound and purified receptor indicates that almost no changes in the pharmacological properties occurred during purification with the exception of a 4-fold decrease in the IC50 for d-tubocurarine.
Our sequence analysis has so far focused only on the 42-kDa protein band, demonstrating that it contains the Dα5 protein. This subunit appears to be a major protein in this band although we cannot eliminate the possibility that other subunits co-migrate. Grauso et al. (14) using rapid amplification of cDNA ends-PCR determined the 5′- and 3′-end of the Dα5 mRNA. Based on this information they postulated a putative protein of 807 amino acids in length and a mass of about 91 kDa. The protein has a long (270 amino acids), and for nAChRs, unusual, leader sequence. Since the receptor is a glycoprotein we would expect the 42-kDa band to migrate abnormally, not reflecting its correct molecular mass, as has been seen for the receptor subunits from T. californica. In addition, rapid amplification of cDNA ends-PCR experiments reported by us (13) are in disagreement with the findings of Grauso et al. (14) regarding the length of the Dα5 mRNA. In our experiment we have found an identical 3′-end of the Dα5 mRNA, but the 5′-end, to be 600 nucleotides shorter then reported by Grauso et al. (14). This would shorten the putative open reading frame by almost 200 amino acids, resulting in a protein with an approximate mass of 69 kDa.
The recent demonstration of the occurrence of the three subunits Dα5 (13, 14) and Dα6 and Dα7 (14, 46) adds complexity to the possible subunit combinations expressed in nAChRs in Drosophila, in particular because the homologues of these subunits are known to form homomeric receptors in mammalian systems (e.g. see Ref. 47). Chamaon et al. (17, 48) probed the structure of the Drosophila nicotinic acetylcholine receptor with antibodies raised against the ALS, Dα2, Dα3, ARD, and SBD subunits. Based on their analysis, they propose the occurrence of three distinct receptor subtypes composed of either ALS, SBD, Dα2 or Dα3, ARD, or ARD, SBD with the remaining subunits for each pentameric complex to be determined. Based on the data presented in this paper it is clear that the Dα5 subunit is a major subunit of the nicotinic acetylcholine receptor in Drosophila and should be considered in all models of the receptor structure.
Acknowledgments
We thank Dr. Shirley Raps, Dr. Maria Figueiredo-Pereira, Dr. Peter Lipke, and Judy Zhu for critical reviews of the manuscript.
Footnotes
This work was supported by NIGMS/National Institutes of Health (NIH) Grant 1SO6 GM 60654, Professional Staff Congress-City University of New York Grants 41373-23 03 and 41373-23 04, a Hunter College Presidential Award (to T. S. G.), and “Research Centers in Minority Institutions” Award RR-03037 from the National Center for Research Resources, NIH. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: nAChR, nicotinic acetylcholine receptor; ACh, acetylcholine; HPLC, high performance liquid chromatography; PMSF, phenylmethylsulfonyl fluoride.
References
- 1.Changeux JP, Edelstein SJ. Neuron. 1998;21:959–980. doi: 10.1016/s0896-6273(00)80616-9. [DOI] [PubMed] [Google Scholar]
- 2.Changeux J, Edelstein SJ. Curr Opin Neurobiol. 2001;11:369–377. doi: 10.1016/s0959-4388(00)00221-x. [DOI] [PubMed] [Google Scholar]
- 3.Karlin A, Akabas MH. Neuron. 1995;15:1231–1244. doi: 10.1016/0896-6273(95)90004-7. [DOI] [PubMed] [Google Scholar]
- 4.Lindstrom J. Mol Neurobiol. 1997;15:193–222. doi: 10.1007/BF02740634. [DOI] [PubMed] [Google Scholar]
- 5.Sargent PB. Annu Rev Neurosci. 1993;16:403–443. doi: 10.1146/annurev.ne.16.030193.002155. [DOI] [PubMed] [Google Scholar]
- 6.Stroud RM, McCarthy MP, Shuster M. Biochemistry. 1990;29:11009–11023. doi: 10.1021/bi00502a001. [DOI] [PubMed] [Google Scholar]
- 7.Leech CA, Sattelle DB. EXS. 1993;63:81–97. doi: 10.1007/978-3-0348-7265-2_5. [DOI] [PubMed] [Google Scholar]
- 8.Bossy B, Ballivet M, Spierer P. EMBO J. 1988;7:611–618. doi: 10.1002/j.1460-2075.1988.tb02854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawruk E, Schloss P, Betz H, Schmitt B. EMBO J. 1990;9:2671–2677. doi: 10.1002/j.1460-2075.1990.tb07452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schulz R, Sawruk E, Mulhardt C, Bertrand S, Baumann A, Phannavong B, Betz H, Bertrand D, Gundelfinger ED, Schmitt B. J Neurochem. 1998;71:853–862. doi: 10.1046/j.1471-4159.1998.71020853.x. [DOI] [PubMed] [Google Scholar]
- 11.Hermans-Borgmeyer I, Zopf D, Ryseck R, Hovemann B, Betz H, Gundelfinger E. EMBO J. 1986;5:1503–1508. doi: 10.1002/j.1460-2075.1986.tb04389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawruk E, Udri C, Betz H, Schmitt B. FEBS Lett. 1990;273:177–181. doi: 10.1016/0014-5793(90)81078-3. [DOI] [PubMed] [Google Scholar]
- 13.Ma D, Ma JG, Pierzchala M, Schmidt-Glenewinkel T. 31st Annual Meeting of the Society of Neuroscience, San Diego, CA; November 10–15; Washington, D.C.. 2001. Abstract 809.9. [Google Scholar]
- 14.Grauso M, Reenan RA, Culetto E, Sattelle DB. Genetics. 2002;160:1519–1533. doi: 10.1093/genetics/160.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lansdell SJ, Millar NS. J Neurochem. 2002;80:1009–1018. doi: 10.1046/j.0022-3042.2002.00789.x. [DOI] [PubMed] [Google Scholar]
- 16.Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE, Richards S, Ashburner M, Henderson SN, Sutton GG, Wortman JR, Yandell MD, Zhang Q, Chen LX, Brandon RC, Rogers YH, Blazej RG, Champe M, Pfeiffer BD, Wan KH, Doyle C, Baxter EG, Helt G, Nelson CR, Gabor GL, Abril JF, Agbayani A, An HJ, Andrews-Pfannkoch C, Baldwin D, Ballew RM, Basu A, Baxendale J, Bayraktaroglu L, Beasley EM, Beeson KY, Benos PV, Berman BP, Bhandari D, Bolshakov S, Borkova D, Botchan MR, Bouck J, Brokstein P, Brottier P, Burtis KC, Busam DA, Butler H, Cadieu E, Center A, Chandra I, Cherry JM, Cawley S, Dahlke C, Davenport LB, Davies P, de Pablos B, Delcher A, Deng Z, Mays AD, Dew I, Dietz SM, Dodson K, Doup LE, Downes M, Dugan-Rocha S, Dunkov BC, Dunn P, Durbin KJ, Evangelista CC, Ferraz C, Ferriera S, Fleischmann W, Fosler C, Gabrielian AE, Garg NS, Gelbart WM, Glasser K, Glodek A, Gong F, Gorrell JH, Gu Z, Guan P, Harris M, Harris NL, Harvey D, Heiman TJ, Hernandez JR, Houck J, Hostin D, Houston KA, Howland TJ, Wei MH, Ibegwam C, Jalali M, Kalush F, Karpen GH, Ke Z, Kennison JA, Ketchum KA, Kimmel BE, Kodira CD, Kraft C, Kravitz S, Kulp D, Lai Z, Lasko P, Lei Y, Levitsky AA, Li J, Li Z, Liang Y, Lin X, Liu X, Mattei B, McIntosh TC, McLeod MP, McPherson D, Merkulov G, Milshina NV, Mobarry C, Morris J, Moshrefi A, Mount SM, Moy M, Murphy B, Murphy L, Muzny DM, Nelson DL, Nelson DR, Nelson KA, Nixon K, Nusskern DR, Pacleb JM, Palazzolo M, Pittman GS, Pan S, Pollard J, Puri V, Reese MG, Reinert K, Remington K, Saunders RD, Scheeler F, Shen H, Shue BC, Siden-Kiamos I, Simpson M, Skupski MP, Smith T, Spier E, Spradling AC, Stapleton M, Strong R, Sun E, Svirskas R, Tector C, Turner R, Venter E, Wang AH, Wang X, Wang ZY, Wassarman DA, Weinstock GM, Weissenbach J, Williams SM, Woodage T, Worley KC, Wu D, Yang S, Yao QA, Ye J, Yeh RF, Zaveri JS, Zhan M, Zhang G, Zhao Q, Zheng L, Zheng XH, Zhong FN, Zhong W, Zhou X, Zhu S, Zhu X, Smith HO, Gibbs RA, Myers EW, Rubin GM, Venter JC. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 17.Chamaon K, Smalla KH, Thomas U, Gundelfinger ED. J Neurochem. 2002;80:149–157. doi: 10.1046/j.0022-3042.2001.00685.x. [DOI] [PubMed] [Google Scholar]
- 18.Lansdell SJ, Schmitt B, Betz H, Sattelle DB, Millar NS. J Neurochem. 1997;68:1812–1819. doi: 10.1046/j.1471-4159.1997.68051812.x. [DOI] [PubMed] [Google Scholar]
- 19.Schulz R, Bertrand S, Chamaon K, Smalla KH, Gundelfinger ED, Bertrand D. J Neurochem. 2000;74:2537–2546. doi: 10.1046/j.1471-4159.2000.0742537.x. [DOI] [PubMed] [Google Scholar]
- 20.Schloss P, Mayser W, Gundelfinger ED, Betz H. Neurosci Lett. 1992;145:63–66. doi: 10.1016/0304-3940(92)90204-k. [DOI] [PubMed] [Google Scholar]
- 21.Tomizawa M, Latli B, Casida JE. J Neurochem. 1996;67:1669–1676. doi: 10.1046/j.1471-4159.1996.67041669.x. [DOI] [PubMed] [Google Scholar]
- 22.Tomizawa M, Casida JE. Neurosci Lett. 1997;237:61–64. doi: 10.1016/s0304-3940(97)00811-2. [DOI] [PubMed] [Google Scholar]
- 23.Lewis E. Drosophila Inform Serv. 1960;34:117–118. [Google Scholar]
- 24.Blanchard SG, Quast U, Reed K, Lee T, Schimerlik MI, Vandlen R, Claudio T, Strader CD, Moore HP, Raftery MA. Biochemistry. 1979;18:1875–1883. doi: 10.1021/bi00577a005. [DOI] [PubMed] [Google Scholar]
- 25.Peterson GL. Methods Enzymol. 1983;91:95–119. doi: 10.1016/s0076-6879(83)91014-5. [DOI] [PubMed] [Google Scholar]
- 26.Wessel D, Flugge UI. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 27.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 28.Ji I, Shin J, Ji TH. Anal Biochem. 1985;151:348–349. doi: 10.1016/0003-2697(85)90186-1. [DOI] [PubMed] [Google Scholar]
- 29.Ji TH. J Biol Chem. 1977;252:1566–1570. [PubMed] [Google Scholar]
- 30.Cooper D, Reich E. J Biol Chem. 1972;247:3008–3013. [PubMed] [Google Scholar]
- 31.March SC, Parikh I, Cuatrecasas P. Anal Biochem. 1974;60:149–152. doi: 10.1016/0003-2697(74)90139-0. [DOI] [PubMed] [Google Scholar]
- 32.Hayman MJ, Crumpton MJ. Biochem Biophys Res Commun. 1972;47:923–930. doi: 10.1016/0006-291x(72)90581-5. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt-Nielsen BK, Gepner JI, Teng NN, Hall LM. J Neurochem. 1977;29:1013–1029. doi: 10.1111/j.1471-4159.1977.tb06505.x. [DOI] [PubMed] [Google Scholar]
- 34.Aebersold RH, Leavitt J, Saavedra RA, Hood LE, Kent SB. Proc Natl Acad Sci U S A. 1987;84:6970–6974. doi: 10.1073/pnas.84.20.6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dudai Y. Biochim Biophys Acta. 1978;539:505–517. doi: 10.1016/0304-4165(78)90084-3. [DOI] [PubMed] [Google Scholar]
- 36.Rudloff E. Exp Cell Res. 1978;111:185–190. doi: 10.1016/0014-4827(78)90248-3. [DOI] [PubMed] [Google Scholar]
- 37.Schloss P, Hermans-Borgmeyer I, Betz H, Gundelfinger ED. EMBO J. 1988;7:2889–2894. doi: 10.1002/j.1460-2075.1988.tb03146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meunier JC, Olsen RW, Changeux JP. FEBS Lett. 1972;24:63–68. doi: 10.1016/0014-5793(72)80827-5. [DOI] [PubMed] [Google Scholar]
- 39.Reynolds JA, Tanford C. Proc Natl Acad Sci U S A. 1976;73:4467–4470. doi: 10.1073/pnas.73.12.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siegel LM, Monty KJ. Biochem Biophys Res Commun. 1965;19:494–499. doi: 10.1016/0006-291x(65)90152-x. [DOI] [PubMed] [Google Scholar]
- 41.Clarke S. J Biol Chem. 1975;250:5459–5469. [PubMed] [Google Scholar]
- 42.Nathanson NM, Hall ZW. J Biol Chem. 1980;255:1698–1703. [PubMed] [Google Scholar]
- 43.Conti-Tronconi BM, Raftery MA. Annu Rev Biochem. 1982;51:491–530. doi: 10.1146/annurev.bi.51.070182.002423. [DOI] [PubMed] [Google Scholar]
- 44.Role LW. Curr Opin Neurobiol. 1992;2:254–262. doi: 10.1016/0959-4388(92)90112-x. [DOI] [PubMed] [Google Scholar]
- 45.Pinnock RD, Lummis SC, Chiappinelli VA, Sattelle DB. Brain Res. 1988;458:45–52. doi: 10.1016/0006-8993(88)90494-5. [DOI] [PubMed] [Google Scholar]
- 46.Lansdell SJ, Millar NS. J Neurochem. 2004;90:479–489. doi: 10.1111/j.1471-4159.2004.02499.x. [DOI] [PubMed] [Google Scholar]
- 47.McGehee DS, Role LW. Annu Rev Physiol. 1995;57:521–546. doi: 10.1146/annurev.ph.57.030195.002513. [DOI] [PubMed] [Google Scholar]
- 48.Chamaon K, Schulz R, Smalla KH, Seidel B, Gundelfinger ED. FEBS Lett. 2000;482:189–192. doi: 10.1016/s0014-5793(00)02057-3. [DOI] [PubMed] [Google Scholar]
- 49.Martin RG, Ames BN. J Biol Chem. 1961;236:1372–1379. [PubMed] [Google Scholar]