

Abstract
Darunavir and tipranavir are two inhibitors that are active against multi-drug resistant (MDR) HIV-1 protease variants. In this study, the in vitro inhibitory efficacy was tested against a MDR HIV-1 protease variant, MDR 769 82T, containing the drug resistance mutations of 46L/54V/82T/84V/90M. Crystallographic and enzymatic studies were performed to examine the mechanism of resistance and the relative maintenance of potency. The key findings are as follows: (i) The MDR protease exhibits decreased susceptibility to all nine HIV-1 protease inhibitors approved by the U.S. Food and Drug Administration (FDA), among which darunavir and tipranavir are the most potent; (ii) the threonine 82 mutation on the protease greatly enhances drug resistance by altering the hydrophobicity of the binding pocket; (iii) darunavir or tipranavir binding facilitates closure of the wide-open flaps of the MDR protease; and (iv) the remaining potency of tipranavir may be preserved by stabilizing the flaps in the inhibitor-protease complex while darunavir maintains its potency by preserving protein main chain hydrogen bonds with the flexible P2 group. These results could provide new insights into drug design strategies to overcome multi-drug resistance of HIV-1 protease variants.
Keywords: darunavir, tipranavir, multi-drug resistant HIV-1 protease, x-ray crystallography
1. Introduction
The emergence of drug resistance during antiretroviral therapy is a key concern for HIV patients. In patients undergoing combination antiretroviral therapy, protease inhibitors are important contributors to the decrease in the number of circulating virus particles. In total, nine HIV-1 protease inhibitors have been approved by the U.S. Food and Drug Administration (FDA). In the 99 amino acid residue HIV-1 protease, 36 mutation loci have been identified which lead to various levels of resistance to protease inhibitor treatment [1,2]. The current HIV-1 protease inhibitors are designed with a hydroxyl group to mimic the transition state of the substrate's scissile peptide bond. Due to the structural similarity of inhibitors, the mutations in HIV-1 protease are commonly associated with cross-resistance to the other inhibitors [3].
The clinical multi-drug resistant (MDR) HIV-1 strain 769 was isolated by Palmer et al. from patients failing protease inhibitor-containing antiretroviral regimens, and the protease of strain 769, MDR 769, is resistant to all protease inhibitors tested [4]. As observed in the crystal structure previously solved by our group, the flaps of MDR 769 are further apart compared to the distance of wild-type (WT) HIV-1 protease flaps [5]. The threonine mutation at residue 82 of the MDR HIV-1 protease (MDR 769 82T) alters the hydrophobicity of the P1 and P1' binding pockets and could further enhance cross-drug resistance. The uncomplexed MDR 769 82T crystal structure adopts the wide-open flap conformation as reported earlier in MDR 769 [5] [6].
According to virologic response studies, darunavir and tipranavir show a higher genetic barrier to resistance [7]. Both inhibitors have been used to treat patients infected with protease inhibitor-resistant viral strains and have effectively inhibited a range of MDR protease isolates [8,9,10]. Based on the scoring function of the HIV Drug Resistance Database (http://hivdb.stanford.edu), MDR 769 82T has low resistance to darunavir and high resistance to tipranavir as well as the other seven protease inhibitors [1,2].
The structural study of the inhibitor-bound MDR HIV-1 protease facilitates the understanding of drug resistance mechanisms. The aim of this study is to test the in vitro inhibitory potency of darunavir and tipranavir against MDR 769 82T and to determine the mechanism of overcoming resistance by examining the binding conformation, key contacts, and the stability of inhibitor-protease complexes. The protease inhibition assays demonstrate the decreased susceptibility of MDR 769 82T to all the tested inhibitors and confirm that 82T severely enhances drug resistance. Compared to other protease inhibitors, the higher resistance barrier of darunavir is due to maintaining main chain hydrogen bonds by inhibitor flexibility while the higher resistance barrier of tipranavir is due to tight flap binding.
2. Materials and methods
2.1 Protein expression and purification
Table 1 lists the protein sequences of MDR 769, MDR 769 82T, and WT (NL4-3) HIV-1 protease. Active MDR 769 and MDR 769 82T, and inactive MDR769 82T genes were codon optimized for E. coli expression with the software DNA 2.0 [11], synthesized by GENEART, Inc. (Regensburg, Germany), and inserted into the pET21b plasmid. The inactive MDR 769 82T protease had an active site mutation, D25N, to eliminate catalytic activity. To prevent auto-proteolyses, the Q7K mutation was introduced into the active MDR genes. The protein expression, purification, and refolding procedures were described earlier [12]. The proteases prepared for crystallization were concentrated to 1.5 mg/ml using Amicon concentrators with 5 kDa molecular mass cut-off (Millipore Corporation, Billerica, MA).
Table 1.
Sequences of the HIV-1 proteases.
| HIV-1 protease | Sequencesa |
|---|---|
| NL4-3 | PQITLWKRPL VTIKIGGQLK EALLDTGADD TVLEEMNLPG RWKPKMIGGI GGFI PVNIIGRNLL TQIGCTLNF |
| MDR 769 | PQITLWKRPI VTIKIGGQLK EALLDTGADD TVLEEVNLPG RWKPKLIGGI GGFV PANVIGRNLM TQIGCTLNF |
| MDR 769 82T | PQITLWKRPI VTIKIGGQLK EALLDTGADD TVLEEVNLPG RWKPKLIGGI GGFV PTNVIGRNLM TQIGCTLNF |
The drug resistant mutations are in bold and polymorphic changes are underlined.
2.2 Protease inhibition assays
The HIV-1 protease inhibitors, requested from the NIH AIDS Research and Reference Reagent Program, and HIV-1 protease Forster Resonance Energy Transfer (FRET) substrate I, purchased from AnaSpec, Inc. (Fremont, CA), were used in the half-maximal inhibitory concentration (IC50) determination experiments. The fluorescence emitted by substrate cleavages was monitored with a microplate reader (SpectraMax M5, Molecular Devices, Sunnyvale, CA) at a 340 nm excitation wavelength with an emission wavelength of 490 nm. The HIV-1 protease reaction buffer was adjusted to pH 4.7 [0.1 M sodium acetate, 1.0 M sodium chloride, 1.0 mM ethylenediaminetetraacetic acid (EDTA), 1.0 mM DTT, 10% dimethylsulfoxide (DMSO), and 1 mg/ml bovine serum albumin (BSA)]. In the reaction buffer containing 5 μM FRET substrate, the concentration of all proteases used in enzyme assays was adjusted to a substrate cleavage velocity of 5 Relative Fluorescence Units (RFU)/min. The final HIV-1 protease concentration was approximately 7 nM. The protease inhibitor was serially diluted in DMSO from 10 μM to 0.013 nM. The active proteases and inhibitors were pre-incubated at 37°C for 20 min prior to fluorescence signal monitoring. An inhibitor-free control was tested as the background fluorescence signal. The progress of the reaction was monitored over 20 min sampling at 1 min intervals. The FRET data were plotted with the software SoftMax Pro V5.2 (Molecular Devices, Sunnyvale, CA) to determine the IC50 values. The difference in Gibbs free energy (δδG) was calculated with the formula δδG = −RTln[IC50(MDR)/IC50(WT)]. The resistance score for each inhibitor was calculated based on information provided by the Stanford HIV Drug Resistance Database [2].
2.3 Differential scanning fluorimetry
Differential Scanning Fluorimetry (DSF) was used to detect the change in protein melting temperature change after ligand binding [13]. A modified DSF procedure is described as follows. The HIV-1 protease was diluted in 100 mM sodium acetate and 150 mM sodium chloride at pH 5.0 to reach a final concentration of 0.4 mg/ml. The protease solution was pre-incubated with 500 μM protease inhibitors. 5×SYPRO orange dye (Sigma-Aldrich, Inc., St. Louis, MO) was added to the protease-inhibitor mixture. 100 μl of the stained solution was heated from 25°C to 82°C. During the heating process, the fluorescence signal was read using a microplate reader (SpectraMax M5, Molecular Devices, Sunnyvale, CA) with the excitation and emission wavelength at 492 nM and 610 nM, respectively.
2.4 Crystallization and diffraction data collection
Darunavir or tipranavir was co-crystallized with the MDR769 82T inactive protease by the hanging drop vapor diffusion method. The expression level of active MDR protease was generally lower than the expression level of the inactive enzyme. The 82T mutation enhances MDR 769 expression and facilitates crystallization [14], [6]. The protease and the inhibitor were pre-mixed at a molar ratio of 1:20 before crystallization set-up. The protease-inhibitor mixture was then mixed at 2:1 v/v ratio with the precipitant (0.1 M MES and 2.4 M ammonium sulfate at pH 6.0). Co-crystals grew within one week to a suitable size for x-ray diffraction. Diffraction data were collected at the Life Sciences Collaborative Access Team (LS-CAT) Sector 21, at the Advanced Photon Source (APS), Argonne National Laboratory (Argonne, IL), and processed with the program suite HKL-2000 [15].
2.5 Structure refinement and analysis
The darunavir-MDR 769 82T and tipranavir-MDR 769 82T co-crystal structures were determined at a resolution of 2.87 Å and 1.24 Å, respectively. Molecular replacement was performed with the CCP4 program Molrep-autoMR [16]. A previously solved HIV-1 protease structure was used as a search model for molecular replacement. The models of darunavir and tipranavir were built in COOT [17] using a ligand finding algorithm where the molecular formulas of the inhibitors were provided in SMILES representation. The protease-ligand model was further refined using Refmac5. The tipranavir molecules were refined in two orientations with half-occupancy while the darunavir molecule was refined in one orientation with full-occupancy. Restrained anisotropic refinement was used for the tipranavir-MDR 769 82T co-crystal structure. The structure was validated using Procheck V3.4.4 [18]. The model of tipranavir or darunavir complexed with MDR 769 82T was analyzed for molecular interaction using LIGPLOT V4.5.3 [19]. The structures were deposited in the Protein Data Bank. The PDB IDs for darunavir-MDR 769 82T and tipranavir-MDR 769 82T structures are 3SO9 and 3SPK, respectively.
3. Results
3.1 Darunavir and tipranavir presented high barrier of resistance compared to the other protease inhibitors
The nine FDA-approved HIV-1 protease inhibitors were effective against the WT HIV-1 protease, NL4-3, at low nanomolar or sub-nanomolar concentration while the MDR 769 82T was resistant to all nine inhibitors (Table 2). The resistance ranged from 11-fold to 2,600-fold as compared with the inhibition level against NL4-3. Although 11–13 fold resistance was observed, darunavir and tipranavir were the two most potent inhibitors against MDR 769 82T. Based on the IC50 values, the difference in Gibbs free energy (ΔΔG) values calculated with the IC50 of MDR 769 82T and WT HIV-1 protease were −1.48 kcal/mol and −1.58 kcal/mol for darunavir and tipranavir, respectively.
Table 2.
IC50 measurements and fold resistance.
| Inhibitor | IC50 (nM) | Fold resistance | Predicted resistance scorea | ||||
|---|---|---|---|---|---|---|---|
| WT (NL4-3) | MDR 769 | MDR 769 82T | MDR 769 | MDR 769 82T | MDR 769 | MDR 769 82T | |
| darunavir | 0.26 ±0.024 | 0.74 ±0.20 | 2.76 ±0.16 | 2.8 | 11 | 10 | 10 |
| tipranavir | 0.24 ±0.18 | 0.63 ±0.17 | 3.02 ±0.10 | 2.6 | 13 | 24 | 64 |
| lopinavir | 0.28 ±0.27 | 0.50 ±0.060 | 3.18 ±0.23 | 1.8 | 11 | 76 | 76 |
| amprenavir | 0.43 ±0.044 | 4.82 ±0.90 | 7.87 ±0.16 | 11 | 18 | 86 | 86 |
| atazanavir | 0.19 ±0.071 | 2.90 ± 0.16 | 10.8 ±0.40 | 15 | 57 | 98 | 103 |
| nelfinavir | 1.58 ±0.85 | 109 ±5.0 | 448 ±65 | 69 | 2.8×102 | 184 | 184 |
| ritonavir | 0.34 ±0.15 | 60.7 ±3.5 | 237 ±5.6 | 180 | 7.0×102 | - | - |
| saquinavir | 0.50 ±0.017 | 294 ±9.4 | 1.30×103 ±87 | 590 | 2.6×103 | 114 | 114 |
| indinavir | 0.47 ±0.19 | 119 ± 13 | 307 ±29 | 250 | 6.5×102 | 128 | 128 |
The resistance scores for darunavir, tipranavir, lopinavir, amprenavir, atazanavir, saquinavir, and indinavir were predicted based on the protease inhibitor plus ritonavir co-administration. The larger scores indicate higher predicted resistance.
The 82T mutation of the MDR protease greatly enhanced drug resistance. The fold resistance of the MDR 769 (1.8–590 fold) was approximately 2.6–6.1 times lower compared to that of MDR 769 82T. The 82T mutation caused relatively larger differences in the fold resistance of darunavir, tipranavir, and lopinavir, and the fold resistance was elevated 3.9, 5.0, and 6.1 times, respectively. The IC50 values are not fully correlated with predicted values based on the scoring of inhibitor resistance mutations from the HIV Drug Resistance Database since the 82T mutation has not been reported previously as darunavir resistance mutation [20].
3.2 Darunavir or tipranavir induced flap closure of the multi-drug resistant HIV-1 protease
The binding of tipranavir or darunavir effectively closed the wide-open flap of the MDR protease and exhibited a conserved binding mode in the MDR protease relative to that in the wild-type protease. The tipranavir-MDR 769 82T complex crystallized in the hexagonal space group P61, and the crystal structure was determined at 1.24 Å resolution. The darunavir-MDR 769 82T complex crystallized in the orthorhombic space group P212121, and the crystal structure was determined at 2.87 Å resolution. The diffraction and refinement statistics are shown in Table 3. Compared with the binding of darunavir or tipranavir to the WT HIV-1 protease (PDB ID: 3BVB and 2O4P), the butyl group of tipranavir and the P1', P2, and P2' groups of darunavir adopted different conformations relative to their binding to WT HIV-1 protease (Fig. 1A, 1B).
Table 3.
Crystallographic statistics.
| Dataset | Tipranavir-MDR 769 82T co-crystal | Darunavir-MDR 769 82T co-crystal |
|---|---|---|
| Data collection | ||
| Space group | P61 | P212121 |
| Wavelength (Å) | 0.979 | 0.979 |
| Cell constants (Å) | a=63.12 b=63.12 c=83.54 | a=28.90 b=66.38 c=90.84 |
| Resolution range (Å) | 29.52–1.24 | 30.00–2.87 |
| Number of unique reflections | 51613 | 10515 |
| Completeness (%) | 96.7 | 98.2 |
| Redundancy | 9.1 | 4.4 |
| Mean I/σ (I) | 13.4 | 7.1 |
| Rmerge a | 0.084 | 0.142 |
| Refinement | ||
| Rwork (%)b | 17.40 | 21.65 |
| Rfree (%)b | 22.70 | 25.62 |
| Number of atoms | ||
| Ligand | 42 | 38 |
| Protease | 1514 | 1514 |
| Solvent | 351 | 142 |
| Average B factor (Å2) | ||
| Ligand | 10.91 | 35.67 |
| Protease | 13.38 | 18.58 |
| Solvent | 37.16 | 27.95 |
| RMSD bond length (Å) | 0.010 | 0.009 |
| RMSD bond angle (°) | 1.37 | 1.03 |
| Ramachandran plot | ||
| Allowed/generous/disallowed (%) | 94.9/5.1/0 | 92.9/7.1/0 |
Rmerge= Σhkl Σi |Ii (hkl) - <I(hkl)>| / Σhkl Σi Ii(hkl), where Ii(hkl) is the intensity of an observation and I(hkl) is the mean value for its unique reflection.
Rwork =Σhkl ∥Fo|−|Fc∥ / Σhkl |Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes. Rfree is calculated exactly as Rwork using a random 5% of the reflections omitted from refinement.
Fig. 1. Co-crystal structures of darunavir and tipranavir with the multi-drug resistant protease.

(A) The tipranavir-MDR 769 82T structure (magenta) was superimposed on the tipranavir-wild-type (WT) HIV-1 protease structure (green). The I54V mutation is displayed in stick model. The magenta and green dashed lines indicate the distance (Å) between the inhibitor and the MDR HIV-1 protease or the WT HIV-1 protease, respectively. (B) The conformational changes of darunavir binding to MDR 769 82T (magenta) in comparison with the darunavir-WT HIV-1 protease complex (green). (C) The electron density of tipranavir bound to MDR 769 82T protease is shown as an Fo–Fo OMIT map contoured at 2σ. Two tipranavir molecules (yellow and magenta) binding in different orientation are presented. (D) The electron density of darunavir bound to MDR 769 82T protease is shown as an Fo–Fc OMIT map contoured at 2σ.
Asymmetric movement of the 80s loop regions was observed in the darunavir-MDR complex. The root-mean-square deviation (RMSD) of Thr82 of superimposed protease monomers is 1.318 Å. The asymmetric P1 and P1' group of darunavir induced the asymmetric protease dimer. The smaller isobutyl P1' group of darunavir allowed the 80s loop of the MDR protease to move closer to the inhibitor. According to the superposition of MDR 769 82T and WT HIV-1 protease structures, Pro81 in one monomer of the MDR protease was 0.7 Å closer to the isobutyl P1' group of darunavir. Thr82 was 1.6 Å further from the isobutyl P1' group of darunavir compared to Val82 in WT HIV-1 protease (Fig. 1B). The V82T mutation changed the hydrophobicity of the binding pocket in the MDR protease. Therefore, the hydrophilic Thr82 pointed toward the solvent rather than the hydrophobic binding pocket. The 80s loop on the other monomer of the MDR protease showed an asymmetric pattern. Both Pro81 and Thr82 moved away from the benzyl P1 group of darunavir.
3.3 Specific contacts were lost between darunavir or tipranavir and multi-drug resistant HIV-1 protease
Hydrophobic interactions or hydrogen bonds were lost between the inhibitors and the MDR protease. According to the LIGPLOT analysis, the number of residues in MDR 769 82T involved in hydrophobic interactions with darunavir or tipranavir decreased by 1~2 residues compared to that of WT HIV-1 protease. Tipranavir lost hydrophobic interactions with Gly49, Val32, and Val84 of the MDR protease but gained additional hydrophobic interactions with Thr82. Similarly, darunavir failed to maintain hydrophobic interactions with Pro81 and Ile84 of the MDR protease but gained hydrophobic contacts with Leu23. The hydrogen bonding network of tipranavir was generally preserved (Fig. 2). Tipranavir formed hydrogen bonds with Asn25, Asp29, Asp30, Gly48, and Ile50. Except for the hydrogen bonds formed between the hydroxyl group of tipranavir and the catalytic residues of the protease, all other hydrogen bonds were formed on the main chain of the protease. Darunavir lost one hydrogen bond and altered the bonding of another hydrogen bond in the darunavir-MDR 769 82T complex structure. The bicyclic tetrahydrofuran P2 group of darunavir formed a pair of hydrogen bonds with Asp29 of WT HIV-1 protease. In the MDR protease complex, the dislocation of the P2 group of darunavir causes it to lose one hydrogen bond with Asp29 (Fig. 2). The dislocation of the sulfonamide P2' group lost the hydrogen bond with the carboxyl group of the Asp29 side chain, but the P2' group formed a new hydrogen bond with the carbonyl group of the Asp29 main chain (Fig. 2).
Fig. 2. Hydrogen bonds and hydrophobic interactions of tipranavir and darunavir with MDR 769 82T protease.
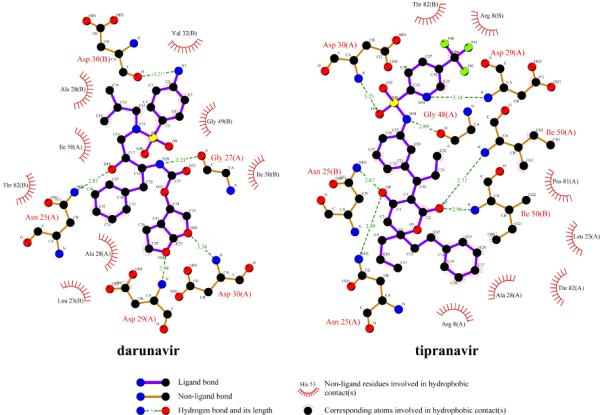
Hydrogen bonds are shown in green dashed line. The residues that form hydrophobic interactions with tipranavir are illustrated by spokes. The interaction map was produced using LIGPLOT V4.5.3 [19].
3.4 Tipranavir elevated the melting temperature of the protease-inhibitor complex suggesting enhanced stability
The melting temperatures of the apo-MDR 769 82T and the MDR 769 82T in complex with inhibitors were determined using differential scanning fluorimetry. While eight protease-inhibitor complexes displayed a similar melting temperature to that of the apo-MDR 769 82T (50.8 ± 0.6°C), the tipranavir-MDR 769 82T complex shifted the melting temperature to a higher value (62.0 ± 2.6°C). The melting temperature of the darunavir-protease complex was 52.6 ± 0.8°C. The difference between the melting temperatures of the two complexes was 9.4°C suggesting that tipranavir stabilized the protease-inhibitor complex (Fig. 3).
Fig. 3. The tipranavir-MDR 768 82T complex showed higher denaturation temperature compared to the darunavir-MDR 769 82T complex.
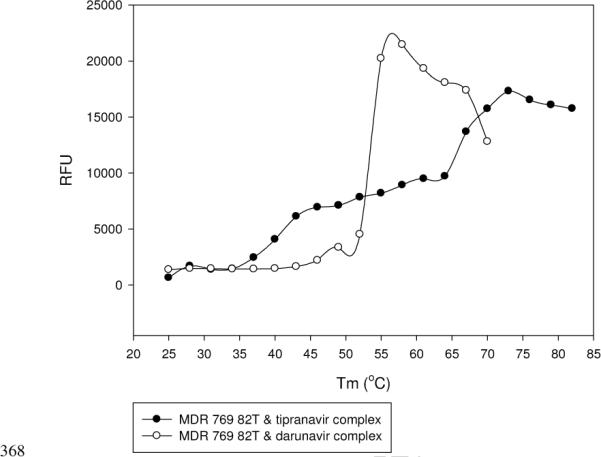
The X-axis is temperature in Celsius degree, and the Y-axis is fluorescence change in Relative Fluorescence Units (RFU).
4. Discussion
The objective of this study is to investigate how darunavir and tipranavir maintain their inhibitory efficacy against MDR HIV-1 protease. Darunavir preserves its residual inhibitory efficacy by forming altered hydrogen bonds while tipranavir overcomes drug resistance by stabilizing the protease-inhibitor complex through direct interactions with the protease flaps.
The valine to threonine mutation at residue 82 is an isosteric switch that alters the hydrophilic environment of the S1 and S1' binding pocket in the protease and greatly enhances the fold resistance to inhibitors. The valine to alanine mutation in MDR 769 is a change from a longer side chain to a shorter side chain, but the hydrophobicity of this mutation is preserved. In both darunavir and tipranavir MDR 769 82T complex structures, the Thr82 residue moves away from the binding pocket towards the hydrophilic solvent environment exhibiting greater fold resistance than the Ala82 mutation. Darunavir, tipranavir, and lopinavir inhibit the MDR 769 at sub-nanomolar concentration; however, they only inhibit MDR 769 82T at approximately 3 nM. Therefore, the hydrophobicity change in the binding pocket is more severe than contact loss in the binding pocket.
Darunavir or tipranavir binding to MDR 769 82T effectively triggers the protease flap closure. MDR 769 82T tends to maintain the wide-open form even in the presence of other inhibitors [14]. The wide-open form of MDR 769 82T is possibly due to the fact that M46L and I54V disrupt the rigid β-sheet of the protease flaps and therefore increase the flexibility of the flaps.
Tipranavir, the only FDA-approved non-peptidomimetic HIV-1 protease inhibitor, does not form the same hydrogen bonds with the flaps through a bridging water molecule as peptidomimetic inhibitors do. Instead, tipranavir forms a hydrogen bond directly between the carbonyl group of tipranavir and the Ile50 residue on each MDR protease flap. The DSF results indicate that tipranavir greatly stabilizes the inhibitor-protease complex. Therefore, tipranavir may partially overcome drug resistance of the MDR protease by tightly closing the protease flaps.
Two possible drug design strategies could be considered to improve the potency of inhibitors against MDR proteases based on the advantageous characteristics of darunavir and tipranavir that elevate the resistance barrier to MDR protease. The first strategy is to design chemical groups that make direct contact with the flap residues of MDR proteases rather than mimicking substrates to interact with flaps via a bridging water molecule. Stronger interactions between the inhibitor and protease flaps decrease the probability of the protease-inhibitor complex dissociation. The hydrogen bonds formed directly between the flap residue Ile50 and tipranavir reduce the protease flap flexibility caused by the mutations, demonstrating the stability of the tipranavir-MDR protease complex.
The second strategy to improve the potency of inhibitors against MDR proteases is to design inhibitor interactions with the main chain of the HIV-1 protease by flexible pharmacophores. When the carboxyl group of Asp29 is far away from the sulfonamide P2' group of darunavir, the P2' group preserves a hydrogen bond with the main chain carbonyl group of Asp29 (Fig. 2). The main chain hydrogen bonds contribute to the ability of darunavir to overcome drug resistance conferred by protease active site mutations [21]. Other than the P2 group, the darunavir structure is identical to amprenavir. The flexible bicyclic tetrahydrofuran P2 group of darunavir enhances the hydrogen bond forming propensity relative to amprenavir [22]. According to the darunavir-MDR 769 82T complex, the P2 group of darunavir adopts a different conformation relative to the darunavir-WT HIV-1 protease complex but preserves hydrogen bonds with the main chain of the MDR protease, suggesting that flexible drugs favorably form contacts in the HIV-1 protease active site cavity and maintain potency.
In summary, the inhibitor-MDR HIV-1 protease complex structures could serve as models for drug optimization. Rather than the de novo design of new inhibitors, optimization of existing inhibitors may have a higher chance of success against MDR variants. Minor modifications on the protease inhibitors may restore the contacts without affecting the bioavailability and toxicity of the molecule.
Highlights
-
>
Darunavir and tipranavir show higher drug-resistance barrier for HIV-1 protease.
-
>
Darunavir preserves its potency by forming protease backbone hydrogen bonds.
-
>
Tipranavir maintains its potency by stabilizing the protease flaps.
Acknowledgments
This work was supported by the National Institutes of Health (R21AI65294). Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Shafer RW. Rationale and uses of a public HIV drug-resistance database. J Infect Dis. 2006;194(Suppl 1):S51–58. doi: 10.1086/505356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003;31:298–303. doi: 10.1093/nar/gkg100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rhee SY, Taylor J, Fessel WJ, Kaufman D, Towner W, Troia P, Ruane P, Hellinger J, Shirvani V, Zolopa A, Shafer RW. HIV-1 Protease Mutations and Protease Inhibitor Cross Resistance. Antimicrob Agents Chemother. 2010 doi: 10.1128/AAC.00574-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Palmer S, Shafer RW, Merigan TC. Highly drug-resistant HIV-1 clinical isolates are cross-resistant to many antiretroviral compounds in current clinical development. AIDS. 1999;13:661–667. doi: 10.1097/00002030-199904160-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Logsdon BC, Vickrey JF, Martin P, Proteasa G, Koepke JI, Terlecky SR, Wawrzak Z, Winters MA, Merigan TC, Kovari LC. Crystal structures of a multidrug-resistant human immunodeficiency virus type 1 protease reveal an expanded active-site cavity. J Virol. 2004;78:3123–3132. doi: 10.1128/JVI.78.6.3123-3132.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yedidi RS, Proteasa G, Martinez JL, Vickrey JF, Martin PD, Wawrzak Z, Liu Z, Kovari IA, Kovari LC. Contribution of the 80s loop of HIV-1 protease to the multidrug-resistance mechanism: crystallographic study of MDR769 HIV-1 protease variants, Acta crystallographica. Section D. Biological crystallography. 2011;67:524–532. doi: 10.1107/S0907444911011541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tenore SB, Ferreira PR. The Place of protease inhibitors in antiretroviral treatment. Braz J Infect Dis. 2009;13:371–374. doi: 10.1590/S1413-86702009000500012. [DOI] [PubMed] [Google Scholar]
- [8].Temesgen Z, Feinberg J. Tipranavir: a new option for the treatment of drug-resistant HIV infection. Clin Infect Dis. 2007;45:761–769. doi: 10.1086/520847. [DOI] [PubMed] [Google Scholar]
- [9].Ghosh AK, Dawson ZL, Mitsuya H. Darunavir, a conceptually new HIV-1 protease inhibitor for the treatment of drug-resistant HIV. Bioorg Med Chem. 2007;15:7576–7580. doi: 10.1016/j.bmc.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Streeck H, Rockstroh JK. Review of tipranavir in the treatment of drug-resistant HIV. Ther Clin Risk Manag. 2007;3:641–651. [PMC free article] [PubMed] [Google Scholar]
- [11].Welch M, Govindarajan S, Ness JE, Villalobos A, Gurney A, Minshull J, Gustafsson C. Design parameters to control synthetic gene expression in Escherichia coli. PLoS One. 2009;4:7002. doi: 10.1371/journal.pone.0007002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Vickrey JF, Logsdon BC, Proteasa G, Palmer S, Winters MA, Merigan TC, Kovari LC. HIV-1 protease variants from 100-fold drug resistant clinical isolates: expression, purification, and crystallization. Protein Expr Purif. 2003;28:165–172. doi: 10.1016/s1046-5928(02)00650-2. [DOI] [PubMed] [Google Scholar]
- [13].Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- [14].Liu Z, Wang Y, Brunzelle J, Kovari IA, Kovari LC. Nine crystal structures determine the substrate envelope of the MDR HIV-1 protease. The protein journal. 2011;30:173–183. doi: 10.1007/s10930-011-9316-2. [DOI] [PubMed] [Google Scholar]
- [15].Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW, Sweet RM, editors. Methods in Enzymology. Academic Press, Inc.; New York, NY: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- [16].The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- [17].Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- [18].Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- [19].Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- [20].Liu TF, Shafer RW. Web resources for HIV type 1 genotypic-resistance test interpretation. Clin Infect Dis. 2006;42:1608–1618. doi: 10.1086/503914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Accounts of chemical research. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- [22].Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J Mol Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]