

Abstract
Dlx homeobox genes play a crucial role in the migration and differentiation of the subpallial precursor cells that give rise to various subtypes of γ-aminobutyric acid (GABA)-expressing neurons of the forebrain, including local-circuit cortical interneurons. Aberrant development of GABAergic interneurons has been linked to several neurodevelopmental disorders, including epilepsy, schizophrenia, Rett syndrome and autism. Here, we report in mice that a single-nucleotide polymorphism (SNP) found in an autistic proband falls within a functional protein binding site in an ultraconserved cis-regulatory element. This element, I56i, is involved in regulating Dlx5/Dlx6 homeobox gene expression in the developing forebrain. We show that the SNP results in reduced I56i activity, predominantly in the medial and caudal ganglionic eminences and in streams of neurons tangentially migrating to the cortex. Reduced activity is also observed in GABAergic interneurons of the adult somatosensory cortex. The SNP affects the affinity of Dlx proteins for their binding site in vitro and reduces the transcriptional activation of the enhancer by Dlx proteins. Affinity purification using I56i sequences led to the identification of a novel regulator of Dlx gene expression, general transcription factor 2 I (Gtf2i), which is among the genes most often deleted in Williams-Beuren syndrome, a neurodevelopmental disorder. This study illustrates the clear functional consequences of a single nucleotide variation in an ultraconserved non-coding sequence in the context of developmental abnormalities associated with disease.
Keywords: Autism, Enhancer, Polymorphism, Telencephalon, Transgenics, Mouse
INTRODUCTION
During embryonic development, two major types of neurons migrate and populate the cortex: the excitatory glutamate-releasing neurons and the inhibitory γ-aminobutyric acid (GABA)-releasing interneurons. It has been proposed that some forms of autism are the result of an imbalance in the excitation/inhibition ratio of the cortical neurons, leading to an increase in the excitatory state of the brain (Rubenstein and Merzenich, 2003). This excitatory state could be achieved by a reduction in the inhibitory signaling or an increase in the excitatory system.
The differentiation of multipotent neuronal progenitors into various neuronal cell types requires complex genetic regulatory mechanisms. For instance, the differentiation and proper migration of the GABAergic interneurons to the cortex are controlled to a large extent by homeodomain-containing transcription factors of the Dlx family (Anderson et al., 1997a; Anderson et al., 1997b). Four Dlx genes, Dlx1, Dlx2, Dlx5 and Dlx6, are expressed in two distinct forebrain domains: the ventral telencephalon and parts of the diencephalon (Bulfone et al., 1993; Robinson et al., 1991). Their expression in the ventral telencephalon is restricted to the differentiating GABAergic projection neurons and interneurons, which will later migrate to the cortex and olfactory bulb (Anderson et al., 1997a; Stuhmer et al., 2002a; Stuhmer et al., 2002b).
The Dlx genes are organized in convergently transcribed bigene clusters and have highly similar patterns of expression, possibly owing to shared cis-acting regulatory elements (CREs) (Nakamura et al., 1996; Porteus et al., 1991; Price et al., 1991; Robinson and Mahon, 1994; Robinson et al., 1991; Scherer et al., 1995; Simeone et al., 1994; Stock et al., 1996). Through phylogenetic footprinting and transgenic analysis, two CREs, called I56i and I56ii, have been discovered in the Dlx5/Dlx6 intergenic region (Zerucha et al., 2000). In reporter assays, both elements are active in the telencephalon and diencephalon. In-depth analysis of their enhancer activities has shown that these individual enhancers may participate in Dlx gene regulation during the development of specific populations of GABAergic interneurons (Ghanem et al., 2007; Ghanem et al., 2008).
The Dlx genes have been associated with human autism spectrum disorders (ASDs) (Hamilton et al., 2005). Two Dlx bigene clusters, DLX1/DLX2 and DLX5/DLX6, are found in autism susceptibility loci on chromosomes 2q31.1 and 7q21.3. Two recent linkage studies have provided evidence that these two loci can be linked to ASDs (Liu et al., 2009; Nakashima et al., 2009). Liu and collaborators have shown that two polymorphisms found in the DLX1/DLX2 loci could potentially increase susceptibility or cause autism in cohorts of multiplex families in which there is a greater genetic component for these conditions (Liu et al., 2009). In a search for autism genomic variants in the DLX1/2 and DLX5/6 loci, we identified 31 single-nucleotide polymorphisms (SNPs) and two insertion/deletion polymorphisms. One adenine-to-guanine SNP was found at position 182 of I56i (Hamilton et al., 2005). The location of the SNP coincides with one of the two putative Dlx binding sites (Feledy et al., 1999) within I56i (Zerucha et al., 2000). This region of I56i also encodes the second exon of the long polyadenylated non-coding RNA (lpncRNA) Evf2 (Dlx6os1 – Mouse Genome Informatics) (Feng et al., 2006), which was recently shown to contribute to the mechanisms by which the Dlx5/Dlx6 bigene cluster is regulated (Bond et al., 2009). Furthermore, the I56i CRE was identified as one of 481 ultraconserved elements of the mouse genome (exhibiting 100% identity, with no insertion or deletions, on DNA fragments longer than 200 bp between orthologous regions of the human, rat and mouse genomes) (Bejerano et al., 2004). The 182-SNP was found to be part of a completely conserved 8 bp motif based on the sequence conservation between 40 vertebrate genomes (Fig. 1). Therefore, the unexpected identification of this SNP in the I56i enhancer suggested that it could have functional consequences for Dlx5/Dlx6 regulation during development.
Fig. 1.

The SNP found in I56i is located in a highly conserved sequence. Forty vertebrate genome sequences in the vicinity of the targeted variant (bold) were aligned. Non-consensus bases are underlined and inserted bases in tenrec and medaka are indicated as gaps in all other species. This region corresponds to bases 96,641,417-96,641,443 of chromosome 7 from the February 2009 human genome assembly (GRCh37).
Here, we present evidence suggesting that the I56i SNP affects transcriptional regulation of the Dlx5/Dlx6 bigene cluster through the I56i CRE. Enhancer activity is impaired by the I56i SNP, leading to a decrease in the expression of a reporter gene in the developing telencephalon as well as in different subpopulations of GABAergic neurons in the adult cortex. Our in vivo and in vitro assays strongly suggest that Dlx proteins have a reduced affinity for the variant site. Affinity purification confirmed that Dlx proteins are bound at or near the SNP sequence and also led to the identification of Gtf2i as a new regulator of the Dlx5/Dlx6 bigene cluster. We propose that the impaired I56i enhancer activity resulting from the 182-SNP could impact on Dlx function and therefore cortical interneuron development and could constitute a contributing factor to developmental abnormalities underlying ASDs.
MATERIALS AND METHODS
Mutagenesis of mouse I56i
The 182-SNP-containing I56i enhancer was generated by PCR using the overlapping fragment technique. A first fragment was amplified from the mouse I56i enhancer using oligonucleotides I56i-1.for and I56i-316.rev (for oligonucleotide sequences, see Table S1 in the supplementary material). A second overlapping fragment was generated using an oligonucleotide containing the desired mutation I56i SNP in combination with the I56i-436 primer. After purification by agarose gel electrophoresis, the two fragments were used as templates for a final PCR reaction with I56i-1.for and I56i-436.rev. A mix (1:3) of Taq and Pfx DNA polymerase (Invitrogen) was used to avoid unwanted mutations. Finally, the mutant enhancer was purified on a gel, TA-cloned and sequenced.
Transgenic experiments
The I56i and vI56i enhancers were subcloned into the XhoI site of the p1230 vector (Yee and Rigby, 1993) that contains a human β-globin minimal promoter and a lacZ reporter gene. Transgenic animals were produced and analyzed as described (Zerucha et al., 2000). Two transgenic lines and three primary transgenic embryos were obtained for the vI56i-lacZ construct. These transgenic embryos were compared with two mouse transgenic lines obtained with the I56i-lacZ construct (Ghanem et al., 2003; Ghanem et al., 2007; Zerucha et al., 2000).
Histology and double immunohistochemistry
Fixation of E13.5 telencephalon was carried out overnight in 4% paraformaldehyde (PFA) in 1×PBS. For adult brains, mice (P30) were deeply anesthetized and subjected to intracardiac perfusion with 10% saline solution followed by 4% PFA in 1×PBS. The adult brains were removed and post-fixed in 4% PFA for 2 hours at room temperature. After fixation, all brains were cryoprotected by immersion in 30% sucrose, frozen in OCT (Tissue-Tek) on dry ice, and stored at −80°C until use. For single and double immunohistochemistry, E13.5 brains were cut at 60 μm and adult brains at 40-45 μm on a cryostat (Leica CM3050 S). Double immunostaining was performed as previously described (Ghanem et al., 2007). An antibody from Abnova was used for Gtf2i immunohistochemistry (IHC-00273).
Nuclear extract preparation
Nuclear extracts were prepared from the brains of E13.5 mouse embryos (1 ml of tissue) as described (Sambrook et al., 1989). In addition, the protein extract was dialyzed against the resuspension buffer using a Spectra/Por dialysis cuvette (VWR) overnight at 4°C. Aliquots of 50 μl were frozen in liquid nitrogen and stored at −80°C.
DNase I footprinting analysis
A portion of the I56i enhancer corresponding to nucleotides 187-316 was amplified by PCR using primers zI56i-187 and zI56i-316. PCR fragments were TA-cloned in the pDrive vector (Qiagen). This fragment was recovered by digestion with EcoRI and KpnI followed by gel purification (QIAquick, Qiagen). Directional labeling of each fragment was performed by 5′ end fill using the large fragment of DNA polymerase I (Invitrogen). Footprinting reactions were carried out as described (Poitras et al., 2007).
Protein-DNA complex purification
Nuclear protein extracts from E13.5 mouse embryos were incubated with 2 μg of a double-stranded biotinylated oligonucleotide encompassing the two Dlx binding sites (see mI56i FP3-FP5.For and .Rev in Table S1 in the supplementary material) in 1× EMSA binding buffer [7 mM Tris pH 7.5, 81 mM NaCl, 2.75 mM dithiothreitol, 5 mM MgCl2, 0.05% NP40, 1 mg/ml bovine serum albumin, 25 μg/ml poly(dI-dC), 10% glycerol] at room temperature for 60 minutes. After 30 minutes of incubation, streptavidin-coated agarose (Sigma, S1638) was added to the reaction. Protein-DNA complexes coupled to the streptavidin-sepharose beads were washed with 1× EMSA binding buffer. Elution of the protein components of the complexes was carried out by boiling the reaction mixture in Laemmli loading buffer (final concentration 2% SDS, 10% glycerol, 5% 2-mercaptoethanol, 0.002% Bromophenol Blue and 0.0625 M Tris-HCl). Removal of the sepharose beads was achieved by filtering the boiled reaction through a SPIN-X centrifuge tube filter (Corning).
Sample preparation for mass spectrometry analysis
Eluted proteins were resolved using a 4-12% Criterion XT Bis-Tris gradient gel (Bio-Rad) and stained with Sypro Ruby (Invitrogen) according to the manufacturer's instructions. Images were acquired on a Geliance CCD-based bioimaging system (PerkinElmer).
Liquid chromatography (LC)-tandem mass spectrometry (MS/MS) analysis
The entire protein profile on SDS-PAGE was sliced from the gel into 25 bands using a gel excision Lanepicker (The Gel Company). In-gel protein digestion was performed on a MassPrep liquid handling station (Micromass) according to the manufacturer's specifications and using sequencing grade modified trypsin (Promega). Peptide extracts were dried using a SpeedVac.
Peptide extracts were separated by online reversed-phase (RP) nanoscale capillary liquid chromatography (nanoLC) and analyzed by electrospray mass spectrometry (ES MS/MS). The experiments were performed on a Thermo Surveyor MS pump connected to an LTQ linear ion-trap mass spectrometer equipped with a nanoelectrospray ion source (Thermo Electron, San Jose, CA, USA). Peptide separation took place within a PicoFrit BioBasic C18 column of 10 cm × 0.075 mm internal diameter (New Objective, Woburn, MA, USA) with a linear gradient from 2 to 50% solvent B (acetonitrile, 0.1% formic acid) in 30 minutes at 200 nl/minute. Mass spectra were acquired using the data-dependent acquisition mode (Xcalibure Software, version 2.0). Each full-scan mass spectrum (400 to 2000 m/z) was followed by collision-induced dissociation of the seven most intense ions. The dynamic exclusion function was enabled (30 second exclusion) and the relative collisional fragmentation energy was set to 35%.
Interpretation of tandem mass spectrometry spectra
All MS/MS samples were analyzed using Mascot (version 2.2.0; Matrix Science, London, UK). Mascot was set up to search against the rodent Uniref_100 protein database (TaxID: 9989) assuming digestion with trypsin. Fragment and parent ion mass tolerance were 0.5 Da and 2.0 Da, respectively. Iodoacetamide derivative of cysteine was specified as a fixed modification and oxidation of methionine was specified as variable modification. Two missed cleavages were allowed.
Criteria for protein identification
Scaffold (version 02_02_03; Proteome Software, Portland, OR, USA) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 95.0% probability, as specified by the Peptide Prophet algorithm (Keller et al., 2002). Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al., 2003). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Using these stringent identification parameters, the rate of false positive identifications was less than 1%.
Co-transfection experiments
A zebrafish dlx2a cDNA (845 bp) encompassing the full coding sequence was amplified by PCR and cloned into the EcoRI site of the pcDNA3-FLAG expression vector. The construction of the DLX2A homeodomain mutant was described previously (Poitras et al., 2007). The zebrafish dlx5a cDNA (1.2 kb) was amplified by PCR and cloned into the BglII/XhoI sites of the pTL2 expression vector. The rat Gtf2i cDNA (accession BC085815) subcloned into the expression vector pExpress-1 was purchased from Open Biosystems.
The wild-type and 182-SNP variant of the mouse I56i enhancer were PCR amplified and inserted upstream of a minimal promoter driving expression of the synthetic firefly luciferase (luc2) gene (pGL4.23 vector from Promega). Cell culture and transfection were carried out as previously described (Zerucha et al., 2000). Luciferase assays were performed according to the manufacturer's protocol (Promega).
Electrophoretic mobility shift assay (EMSA)
EMSAs were performed using double-stranded oligonucleotides corresponding to the identified putative binding site. Complementary 21 nucleotide oligonucleotides (mI56i FP3.for and mI56i FP3.rev) were annealed and end-labeled using [32P]γ-ATP (Amersham) and T4 polynucleotide kinase (New England Biolabs). For each binding reaction, 1 ng of labeled DNA was incubated with recombinant Flag-Dlx2 (TNT Quick Coupled Transcription/Translation Kit, Promega), GST-Dlx proteins or with nuclear extracts in reaction buffer. EMSAs were carried out as described (Poitras et al., 2007).
In the competition assay using nuclear extract, increasing amounts of unlabeled competitor (0, 5, 20, 50 and 100 ng) were added to the binding reaction. As a competitor, an unlabeled double-stranded wild-type oligonucleotide competitor or a version of equal length containing the 182-SNP (see Fig. 1A) was used.
The competition assay with recombinant Dlx proteins was carried out using double-stranded 32P-labeled oligonucleotides corresponding to a triplicate of the core FP3 site (ATAATT), termed I56i FP3 X3, with fixed amounts of GST-Dlx5a protein. These binding reactions were competed with increasing amounts (1, 3 and 10 ng) of the unlabeled double-stranded wild-type oligonucleotide competitor (I56i FP3 X3) or the variant version, vI56i FP3 X3. Quantification of the percentage of the remaining complexes was achieved using ImageJ (http://rsbweb.nih.gov/ij/index.html).
For the supershift assay, the Flag-Dlx2 unpurified TNT reaction was preincubated overnight at 4°C with 1 μg of anti-Flag antibody (F7425, Sigma).
Western blots
Proteins from the previously described E13.5 nuclear extract were separated on an 8% SDS-PAGE gel and electrophoretically transferred to Hybond-C membrane (GE Healthcare) in 25 mM Tris, 192 mM glycine, 20% methanol. Membranes were blocked in PBS-T (145 mM NaCl, 10 mM Na-phosphate buffer pH 7.4, 0.2% Tween 20) containing 2.5% dried milk. The membranes were then incubated with an anti-Gtf2i antibody (Abnova, IHC-00273; 1/1000) in PBS-T containing 1% dried milk overnight at 4°C. After washing with PBS-T containing 1% dry milk, blots were incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (1/5000) in PBS-T containing 1% dry milk. The blots were developed using an HRP chemiluminescence substrate kit (Immun-Star).
Statistical analysis
Statistical significance (P<0.05), as presented in Fig. 6C,D, was determined using the paired Student's t-test. All quantitative data are presented as the mean ± s.e.m.
Fig. 6.

Transcriptional activation of I56i by Dlx and Gtf2i is impaired by the 182-SNP. (A) The molecular weight of the protein component(s) of the E4 complex is similar to that of Dlx2. EMSA using the I56i FP3 oligonucleotide with E13.5 mouse embryonic forebrain nuclear extract (Nuclear Extract) or increasing amounts of recombinant Flag-tagged Dlx2 (Flag-Dlx2). Preincubation of the recombinant Dlx2 with an anti-Flag antibody leads to the formation of a high molecular weight complex (asterisk, Flag-Ab lane). (B) Affinity of Dlx proteins for the FP3 site is affected by the 182-SNP. EMSA was performed using the I56i X3 oligonucleotide and a fixed amount of recombinant Dlx5. The resulting complexes (arrowhead) were competed more efficiently by the unlabeled wild-type I56i X3 (increasing amounts in lanes 3 to 5) than by the variant (vI56i X3, lanes 6 to 8) competitor. Competition (%) refers to the percentage of remaining complexes with respect to the control reaction (without competitor, 100%). (C) Replacement of I56i by the 182-SNP-containing variant (vI56i-pGL4.23) impaired transcriptional activation by Dlx2 or Dlx5. (D) Dlx2 and Dlx5 synergize with Gtf2i in activating the transcription of a luc2 reporter construct containing the I56i enhancer. Values shown in C and D represent the mean of relative luciferase activity obtained from five (C) or six (D) independent experiments ± s.e.m. Relative luciferase activity was normalized to Dlx2 plus mI56i-pGL4 (100%). *, P<0.05.
RESULTS
An SNP affects the activity of the I56i enhancer
We previously identified seven variants in CREs of Dlx genes from a collection of DNA samples from autistic probands (Hamilton et al., 2005). Using sequence conservation analysis in orthologous sequences (see Fig. S1 in the supplementary material) and DNaseI footprinting to localize binding of trans-acting (Poitras et al., 2007) (data not shown; see also Fig. 4A), we found that position 182 in I56i (base 96,641,429 on chromosome 7, see Fig. 2A), changing an adenine for a guanine, was the only sequence variant predicted to have a major impact on CRE function. By comparing the nucleotide sequence of the I56i enhancer from 40 vertebrate genomes, we found that this adenine (underlined) was part of a completely conserved 8 bp motif (ATAATTAG) (Fig. 1).
Fig. 4.

I56i enhancer activity is impaired by the 182-SNP in the adult cortex. Quantification of the percentage of lacZ-expressing cells that co-express various GABAergic interneuron subtypes (PV, parvalbumin; SOM, somatostatin; CB, calbindin; CR, calretinin) in the adult mouse somatosensory cortex at P35. The contribution of vI56i-lacZ-expressing precursors to adult cortical interneuron populations expressing PV, SOM, CB and CR was significantly reduced compared with that of I56i-lacZ-expressing cells. All data are presented as mean ± s.e.m. *, P<0.001.
Fig. 2.
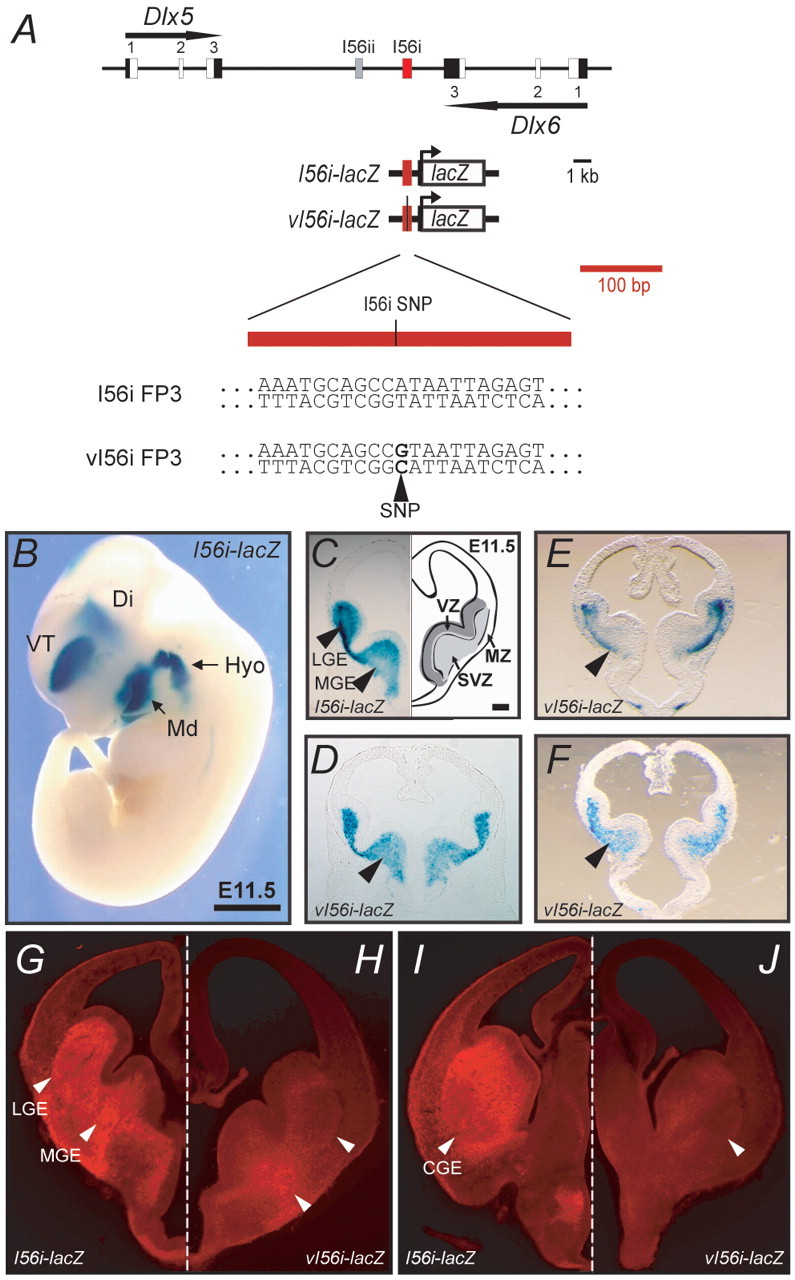
The 182-SNP affects I56i enhancer activity in the developing forebrain. (A) Dlx5/Dlx6 bigene cluster. Exons are shown in black and white (coding), the enhancers I56i in red and I56ii in gray. The I56i enhancer is enlarged to show the position of the 182-SNP variant identified by Hamilton and collaborators (Hamilton et al., 2005). The sequence of the 182-SNP variant (vI56i) and wild-type I56i are shown beneath. The remainder of the mouse and human I56i sequences are virtually identical (Zerucha et al., 2000). (B) At E11.5, the mouse I56i enhancer drives lacZ reporter expression to the ventral telencephalon (VT), diencephalon (Di) and to the hyoid (Hyo) and mandibular (Md) arches. (C-J) Transverse sections through the telencephalon of I56i-lacZ (C,G,I) and vI56i-lacZ (D-F,H,J) mouse embryos at E11.5 (C-F) and E13.5 (G-J). (A-F) β-galactosidase assay staining. (G-J) Immunohistochemistry for β-galactosidase. (E,F) Sections from primary transgenic embryos. Black arrowheads (C-F) and white arrowheads (I,J) indicate reduced vI56i enhancer activity in the MGE at E11.5 and in the MGE, LGE and CGE at E13.5, respectively. The asymmetric staining observed in E,F could be attributed to tilted sectioning and is not the result of asymmetric expression of the lacZ transgene; it is similar to the results of transgenic experiments reported by Lettice and collaborators concerning a point mutation in the ZRS enhancer of the Shh gene (Lettice et al., 2008). Scale bars: 1 mm in B; 500 μm in C-J.
To evaluate potential effects of the 182-SNP on the in vivo enhancer activity of I56i, we produced transgenic mouse embryos with reporter constructs that contained either the wild-type I56i or its corresponding 182-SNP-containing variant (hereafter referred to as vI56i) in combination with a β-globin minimal promoter-lacZ cassette (Yee and Rigby, 1993). As previously reported, the I56i enhancer targets the expression of the reporter cassette to the developing forebrain and branchial arches (Ghanem et al., 2003; Ghanem et al., 2007; Zerucha et al., 2000) (Fig. 2B). In the telencephalon of E11.5 mouse embryos, I56i targets lacZ expression to the subventricular zone (SVZ) and mantle zone (MZ) of the medial and lateral ganglionic eminences (MGE and LGE) (Fig. 2C). By contrast, lacZ expression driven by vI56i-lacZ was reduced in all the aforementioned regions of the forebrain. A more pronounced effect was observed in the MGE (Fig. 2D-F). The reductions in transgene expression varied when comparing embryos from two independent transgenic lines and three primary transgenic animals, ranging from slight reductions to an almost total absence of transgene expression (Fig. 2C-F; data not shown). As we never observed such variations in activity in at least nine primary transgenic embryos and four established lines expressing I56i-lacZ (Zerucha et al., 2000) (data not shown), it is most unlikely that the decreases we observed with vI56i-lacZ are attributable to integration or to copy number effects.
The activity of the variant enhancer was more severely affected at E13.5 than at E11.5 (Fig. 2G,H). Moreover, almost no vI56i-lacZ-expressing cells could be detected in the SVZ and MZ of the caudal ganglionic eminence (CGE) (Fig. 2I,J). Reductions in enhancer activity were also observed in more rostral sections of the telencephalon (data not shown).
We have previously shown that the transcriptional activity of the I56i enhancer can be observed in GABAergic interneurons that are migrating tangentially from the ventral telencephalon to the cortex (Ghanem et al., 2007). Hence, we evaluated the activity of the vI56i-lacZ transgene in migrating interneurons at E13.5. In the brains of I56i-lacZ embryos, numerous lacZ-expressing neurons, organized in streams, were observed migrating towards the pallium at the levels of the mid-telencephalon (LGE/MGE) and caudal telencephalon (CGE) (Fig. 3A-D). By contrast, only a few scattered migrating cells expressed the vI56i-lacZ transgene at both levels of the embryonic telencephalon (Fig. 3E-H).
Fig. 3.

I56i enhancer activity is impaired by the 182-SNP in migrating neuronal precursors at E13.5. (A-H) Immunolocalization of β-galactosidase (red) in I56i-lacZ (A-D) and vI56i-lacZ (E-H) transgenic mouse brains at the level of the LGE/MGE (A,B,E,F) and CGE (C,D,G,H). The boxed regions in A,C,E,G are shown at higher magnification in B,D,F,H. Numerous lacZ-expressing neurons were observed migrating from the MGE and CGE towards the developing cortex in I56i-lacZ brains (A-D), whereas only a few scattered lacZ-expressing cells were detected in the migrating streams leaving both eminences from the vI56i-lacZ brains (E-H). Scale bars: 500 μm in A,C,E,G; 250 μm in B,D,F,H.
To determine whether the effect of the 182-SNP on I56i enhancer activity persists after birth, we performed double immunohistochemistry on the somatosensory cortex of P35 transgenic mice using cortical interneuron markers. We co-labeled lacZ-expressing cells with markers for four major subtypes of interneurons: the calcium-binding proteins calbindin, calretinin (calbindin 2) and parvalbumin (CB, CR and PV) and the neuropeptide somatostatin (SOM). These four markers were specifically chosen on the basis of previous findings showing that most CB+, PV+ and SOM+ interneurons originate from the MGE, whereas the CGE is one major origin of CR+ interneurons (Fogarty et al., 2007; Wonders and Anderson, 2006; Wonders et al., 2008). We observed a significant decrease in co-labeling in the somatosensory cortex of the vI56i-lacZ mice as compared with the I56i-lacZ mice (Fig. 4). The mutant vI56i-lacZ transgene was expressed in 32.4% of CB+, 22.8% of CR+, 31.2% of PV+ and 46.5% of SOM+ interneurons. By contrast, the wild-type I56i-lacZ was found in 89.5% of CB+, 75.9% of CR+, 75.8% of PV+ and 76.4% of SOM+ interneurons (Ghanem et al., 2007). Taken together, these results suggest that the 182-SNP has important functional consequences for I56i enhancer activity at both embryonic and postnatal stages of brain development. Therefore, we propose that a mouse homozygous for the 182-SNP mutation may present reduced levels of Dlx5/Dlx6 expression, and that this in turn may affect the development of cortical interneurons
Binding of a forebrain transcription factor is affected by the presence of the 182-SNP
To characterize the DNA-protein interactions taking place on the I56i enhancer, we performed a DNaseI footprinting analysis using mouse E13.5 telencephalon nuclear protein extract. Seven DNA-protein complexes were observed, four of which are shown in Fig. 5A. The 182-SNP is located within the boundary of DNA-protein complex 3 (FP3). To examine the effects of the 182-SNP on the binding of forebrain transcription factors to the I56i enhancer, a competitive electrophoretic mobility shift assay (EMSA) was performed. When a 21 bp double-stranded oligonucleotide corresponding to the FP3 binding site (Fig. 2A) was incubated with the E13.5 telencephalon nuclear extract, four DNA-protein complexes were observed (Fig. 5B, E1-E4). As increasing amounts of the unlabeled I56i-FP3 oligonucleotide were added to the reaction, all four complexes were abolished. When the unlabeled vI56i-FP3 oligonucleotide (Fig. 2A) was used as a competitor, three DNA-protein complexes were efficiently competed. However, the E4 complex was unaffected by the vI56i-FP3 oligonucleotide. Interestingly, the molecular weights of the E3 and E4 complexes were comparable to those of complexes produced by a recombinant Dlx2 (Fig. 6A). This suggests that the 182-SNP is interfering, in vitro, with the formation of a DNA-protein complex, which either includes Dlx protein(s) or proteins of similar size.
Fig. 5.

Binding of a forebrain transcription factor to I56i is affected by the 182-SNP. (A) The 182-SNP is located within a nuclear protein binding site (FP3) in the I56i enhancer as shown by DNaseI footprinting analysis. A DNA fragment corresponding to the I56i enhancer (nucleotides 1-276) was incubated with a nuclear protein extract from E13.5 mouse forebrain (+ Extract). Four regions of the enhancer (FP1 to FP4) were bound by nuclear proteins. Protected areas (footprints) are represented by thick black bars. Protein-DNA interactions were mapped on the mouse I56i enhancer sequence using a Maxam-Gilbert guanine/adenine chemical sequencing reaction (GA) as a standard. The 182-SNP was pinpointed within the protected area FP3. The position on the I56i sequence is shown to the left (Hamilton et al., 2005) (see Fig. S1 in the supplementary material). (B) A competitive electrophoretic mobility shift assay (EMSA) on I56i FP3 was performed using E13.5 mouse embryonic forebrain nuclear extract. In the presence of nuclear extract, four protein-DNA complexes are observed (lanes 1 and 6, complexes E1 to E4). These four complexes were competed by increasing amounts of the unlabeled I56i FP3 oligonucleotide (lanes 2 to 5, I56i FP3). In competition using the SNP-containing vI56i FP3 oligonucleotide (see Fig. 1A for sequence), protein-DNA complexes E1 to E3 were competed, whereas the lower-mobility complex E4 was unaffected (lanes 7 to 10, vI56i FP3).
To identify factors that bind to the I56i at and near the FP3 binding site, we carried out an affinity purification assay using a DNA fragment that encompasses the two Dlx binding sites of the I56i enhancer and nuclear proteins prepared from E13.5 mouse embryonic telencephalon. Nuclear proteins that were bound to the affinity matrix were identified by mass spectrometry (see Table S2 in the supplementary material).
Consistent with previous observations that Dlx2 is able to bind and activate transcription through the I56i enhancer (Zerucha et al., 2000; Zhou et al., 2004), Dlx proteins were identified among the purified proteins. However, the most abundant protein isolated from the purification was general transcription factor 2 I (Gtf2i), with 49 distinct peptides identified by mass spectrometry (see Table S2 in the supplementary material). Interestingly, a potential Gtf2i binding site (Chimge et al., 2008) is located halfway between the two Dlx binding sites of the sequence used for affinity purification (see Fig. S1 in the supplementary material).
Transcriptional activity of I56i is affected by the presence of the 182-SNP
We evaluated the affinity of the Dlx proteins for the FP3 binding site in its wild-type or 182-SNP-containing versions by a competitive EMSA. The Dlx5-I56i FP3 X3 complexes were competed more efficiently by the unlabeled I56i FP3 X3 than by the variant competitor (vI56i FP3 X3), suggesting that the Dlx proteins possess more affinity for the wild-type FP3 binding site than for the variant sequence (Fig. 6B). Next, we examined the effects of the 182-SNP on the transcriptional activity of the I56i enhancer in response to Dlx2 or Dlx5 using luciferase reporter assays following co-transfections in murine fibroblast NIH3T3 cells. In the presence of the 182-SNP, the response of the I56i enhancer to Dlx2 and Dlx5 decreased by 39% and 24%, respectively (Fig. 6C).
To test a possible involvement of Gtf2i in Dlx5/Dlx6 regulation via the I56i enhancer, additional co-transfections were performed in NIH3T3 cells. Gtf2i and the Dlx proteins were shown to synergize in activating the expression of the reporter luc2 (Fig. 6D). The transfection of a Gtf2i-expressing plasmid per se did not stimulate transcriptional activity (0.04%, relative to Dlx2 activation taken as a 100% reference). However, when co-transfected with the Dlx2 or Dlx5 expression plasmids, the transcriptional activity increased to 138.2% and 60.8%, respectively. These activations represent an increase of 38% (Dlx2) and 24% (Dlx5) when compared with the transfection reactions in which only Dlx2 or Dlx5 plasmids were transfected (100% and 49.2%, respectively).
These results indicate that the 182-SNP interferes with the activation of the I56i enhancer by Dlx proteins and suggest a role for interactions between Dlx and Gtf2i proteins in Dlx gene regulation via the I56i enhancer.
DISCUSSION
In this report, we demonstrated that an SNP, originally isolated in a screen of autistic probands and located in an ultraconserved Dlx5/Dlx6 CRE (I56i), has functional consequences for the activity of the enhancer throughout forebrain development, continuing into adulthood. The identification of this functionally important SNP in the I56i enhancer provides an entry point to address the relationship between Dlx gene regulation in the telencephalon, GABAergic interneuron development and, potentially, the etiology of ASD.
Since this element was identified as one of 481 ultraconserved DNA segments among orthologous regions of the human, rat and mouse genomes, the I56i enhancer is considered as one region of the genome that is not susceptible to variation (Bejerano et al., 2004). Furthermore, the SNP was found in a region of I56i that is identical in 40 vertebrates species surveyed (Fig. 1). Such sequence conservation made this SNP an unexpected finding and thus suggestive of a causal relationship. DNaseI footprinting analysis showed that the 182-SNP is located within the FP3 protein binding site. This site was previously identified as one of two Dlx binding sites in the I56i enhancer. These Dlx binding sites play crucial roles in the auto- or cross-regulation mechanisms of the Dlx5/6 bigene (Zerucha et al., 2000). Thus, the presence of the 182-SNP within one of these important binding sites might, by itself, have functional consequences for the regulatory cascades controlling Dlx expression.
The hypothesis that the 182-SNP would affect auto- or cross-regulatory mechanisms at I56i is supported by our EMSA and co-transfection experiments. Dlx affinity for the FP3 sequence was impaired by the presence of the 182-SNP as was the transcriptional activity of I56i in response to Dlx2 or Dlx5. Co-transfection assays provided further evidence that the 182-SNP affected the I56i enhancer activity induced by the Dlx proteins.
Our EMSA analysis also showed that binding of one factor to I56i was specifically affected by the SNP. We have shown that this factor was similar in size to the Dlx proteins. Complexes formed by the FP3 oligonucleotide with the recombinant Dlx2 migrated in a similar manner to the E3 and E4 nuclear complexes. A potential explanation for the presence of two complexes is that Dlx2 binds to the FP3 site as a monomer and/or as a homodimer. It has been demonstrated that Dlx proteins can bind DNA as hetero- and homodimers (Zhang et al., 1997). Supershift experiments on these complexes with a number of anti-Dlx antibodies were inconclusive (data not shown). However, our affinity purification experiment enabled us to identify a new regulator of the Dlx genes: Gtf2i. In co-transfection assays, this factor was shown to synergize with the Dlx proteins in activating transcription through I56i and the overall activity of I56i in the presence of a Dlx plus Gtf2i combination was affected by the SNP. The mechanism by which Gtf2i interacts with Dlx proteins on the I56i enhancer is still unknown, but possibilities include: (1) Gtf2i interacts directly, or through an intermediate factor, with Dlx proteins independently of the ability of Gtf2i to bind to DNA [attempts at co-immunoprecipitating Gtf2i and Dlx2 in vitro have not been successful (data not shown)]; and (2) Gtf2i binds the I56i sequence, possibly at the putative binding site located between the two Dlx binding sites.
The identification of Gtf2i as a potential regulator of Dlx gene expression is particularly relevant because GTF2I is a candidate gene for Williams-Beuren syndrome (WBS), a neurodevelopmental disorder. A hemizygous deletion of a cluster of 26 genes including three members of the GTF2 family on chromosome 7 (7q11.23) has been observed in a majority of the WBS cases. Children with WBS display distinctive facial features, mental disability and growth retardation. They also share a unique set of personality traits, which include over-friendliness, abnormal social behavior (outwardly social) and an increased trust in strangers (reviewed by Francke, 1999). Although WBS children show high sociability, almost half of them display socio-communicative deficits characteristic of ASD (Klein-Tasman et al., 2007; Lincoln et al., 2007). Moreover, the genetic analysis of an autistic child exhibiting the cognitive-behavioral profile (CBP) associated with WBS revealed an atypical deletion of the WBS critical region (Edelmann et al., 2007). Although the authors of this study proposed that the WBS CBP was due to the deletion of the member of the GTF2 family and that the symptoms of autism were due to the deletion of another gene outside the typical WBS deletion, one cannot exclude the possibility that the mental retardation and autistic traits observed in this patient were due to a dosage effect of GTF2I gene expression. Finally, a case of an autistic individual with a 7q11.23 duplication was recently reported (Van der Aa et al., 2009). Altogether, there is accumulating evidence suggesting that members of the GTF2I gene family play an important role in WBS, even though it remains unclear whether the GTF2I genes can account for the socio-communicative deficits (characteristic of ASD) seen in WBS patients.
Dlx genes are key members of genetic cascades responsible for the establishment of a diversity of neuronal types in the central nervous system, such as the GABAergic interneurons. In transgenic animals, the 182-SNP affects I56i enhancer activity in vivo at three key time points during GABAergic interneuron development: their early differentiation, their migration to the pallium and their differentiated phase in the somatosensory cortex. From these results, we can extrapolate that the presence of the 182-SNP in I56i may lead to a decrease in Dlx5/Dlx6 expression, which in turn might affect the early process of GABAergic interneuron differentiation, as Dlx proteins have been shown to regulate Gad1 and Gad2 expression (Long et al., 2009a; Long et al., 2009b). As for the impact of the SNP on precursor migration, it has been shown that neuropilin 2 (Nrp2) is ectopically expressed when Dlx1 and Dlx2 are inactivated (Le et al., 2007). Nrp2 is a transmembrane protein that is implicated in repulsive axon guidance and, therefore, it was proposed that downregulation of Nrp2 by Dlx proteins could be a mechanism by which the migration of GABAergic interneurons was facilitated. We predict that reduced levels of Dlx5/Dlx6 proteins caused by the SNP could interfere with Nrp2 regulation, potentially leading to the impaired migration of interneuron progenitors to the cortex.
The impact of altered Dlx gene regulation in GABAergic interneurons of newborn or adult mice remains to be established. In mice, the brain of Dlx1-null mutants displays no overt phenotype at birth, but later in life they develop epileptic seizures (Cobos et al., 2005). Dlx5/6+/− mice, the cortex of which appears normal, have spontaneous electrographic seizures and reduced power of gamma oscillations suggesting defects in cortical interneurons; in addition, there is evidence for defective development of PV-expressing interneurons in Dlx5/6−/− and Dlx5−/− mutants (Wang et al., 2010). Thus, Dlx proteins could adopt a specific role in the adult brain or subtle alterations that occur during development could have delayed consequences. In either case, a decrease in I56i activity in mature neurons could potentially lead to dysfunctions of neural networks.
The 182-SNP is a rare polymorphism with a frequency of 0.3% amongst the autistic probands (one affected individual from 161 probands screened) (Hamilton et al., 2005). This SNP was paternally transmitted and was absent from an affected sibling (Richler et al., 2006). This situation is not exceptional considering that siblings, who share half the genetic risk, often display a different autistic phenotype (Skuse, 2007). It was proposed that one or several independent risk factors can elevate the chance of displaying the full autistic phenotype. Furthermore, using a broader concept of autism, two independent studies have reported that an increased proportion of unaffected siblings has subsyndromal autistic impairments (Bishop et al., 2006; Constantino et al., 2006), supporting the notion of independent risk factors or genetic susceptibility factors.
Paternal imprinting of the Dlx5 locus was recently documented (Horike et al., 2005; Okita et al., 2003) and would have phenotypic implications for an SNP in I56i, depending on the parent of origin. Horike and colleagues demonstrated that the loss of Dlx5 imprinting, through Mecp2 inactivation, leads to biallelic expression of Dlx5, resulting in increased Dlx5 expression. Since Mecp2 mutations are associated with Rett syndrome, it was suggested that the misregulation of several genes, including Dlx5, resulting from the loss of their imprinting, might contribute to the etiology of this syndrome. Imprinting of Dlx5 has also been observed in human and swine loci (Cheng et al., 2008; Okita et al., 2003). However, paternal imprinting of Dlx5 has been challenged by other studies reporting biallelic expression of the gene (Bischoff et al., 2009; Kimura et al., 2004; Schule et al., 2007). When one copy of the Dlx5/Dlx6 locus is silenced by imprinting, the presence of the SNP on the remaining allele will result in a decrease in Dlx5/Dlx6 expression that is much more substantial than in the absence of imprinting.
The reason for the exceptional sequence conservation of some non-coding regulatory elements in vertebrate genomes is still puzzling. Although it is unlikely to be due to chance that these regions have been maintained for hundreds of millions of years, it is also unclear what their function is, given the lack of a phenotype when some of the ultraconserved regions are deleted (Ahituv et al., 2007). Our study provides an example of the consequences that a single nucleotide change could have on the function of an ultraconserved sequence.
Supplementary Material
Acknowledgements
We thank Mélanie Debiais-Thibaud and Fabien Avaron for helpful discussions and Adriana Gambarotta for technical assistance in the production of transgenic mice. This work was supported by grants MOP14460 and MOP-74564 from the Canadian Institutes of Health Research (CIHR). Work in the laboratory of J.L.R.R. was supported by research grants from: Nina Ireland, the Althea Foundation, Cure Autism Now and R37 MH049428. Work in the laboratory of G.G.P. was supported by grants from Genome Canada and Génome Québec. G.G.P. is a recipient of a Canadian Research Chair in Proteomics.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Author contributions
L.P. performed experiments in Figs 2, 3, 4, 5 and 6 and contributed to the writing of the manuscript. M.Y. carried out the immunolocalization experiments presented in Figs 2, 3 and 4. C.L.-P. carried out transfections and EMSA experiments shown in Fig. 6. R.B.M. participated in the EMSA experiments shown in Fig. 6. J.-P.G., I.K. and G.G.P. were involved in the purification and identification of candidate proteins by mass spectrometry. G.H. participated in the design of the in vivo experiments. S.P.H. and J.L.R.R. identified the SNP presented in this study and contributed the writing of this manuscript. M.E. participated in the design of the experiments and contributed to the writing of the manuscript.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.051052/-/DC1
References
- Ahituv N., Zhu Y., Visel A., Holt A., Afzal V., Pennacchio L. A., Rubin E. M. (2007). Deletion of ultraconserved elements yields viable mice. PLoS Biol. 5, e234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S. A., Eisenstat D. D., Shi L., Rubenstein J. L. R. (1997a). Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 278, 474-476 [DOI] [PubMed] [Google Scholar]
- Anderson S. A., Qiu M., Bulfone A., Eisenstat D. D., Meneses J., Pedersen R., Rubenstein J. L. R. (1997b). Mutations of the homeobox genes Dlx-1 and Dlx-2 disrupt the striatal subventricular zone and differentiation of late-born striatal neurons. Neuron 19, 27-37 [DOI] [PubMed] [Google Scholar]
- Bejerano G., Pheasant M., Makunin I., Stephen S., Kent W. J., Mattick J. S., Haussler D. (2004). Ultraconserved elements in the human genome. Science 304, 1321-1325 [DOI] [PubMed] [Google Scholar]
- Bischoff S., Tsai S., Hardison N., Motsinger-Reif A., Freking B., Nonneman D., Rohrer G., Piedrahita J. (2009). Characterization of conserved and nonconserved imprinted genes in swine. Biol. Reprod. 81, 906-920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop D. V., Maybery M., Wong D., Maley A., Hallmayer J. (2006). Characteristics of the broader phenotype in autism: a study of siblings using the children's communication checklist-2. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141, 117-122 [DOI] [PubMed] [Google Scholar]
- Bond A., Vangompel M., Sametsky E., Clark M., Savage J., Disterhoft J., Kohtz J. (2009). Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat. Neurosci. 12, 1020-1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulfone A., Puelles L., Porteus M. H., Frohman M. A., Martin G. R., Rubenstein J. L. R. (1993). Spatially restricted expression of Dlx-1, Dlx-2 (Tes-1), Gbx-2, and Wnt-3 in the embryonic day 12.5 mouse forebrain defines potential transverse and longitudinal segmental boundaries. J. Neurosci. 13, 3155-3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H., Zhang F., Jiang C., Li F., Xiong Y., Deng C. (2008). Isolation and imprinting analysis of the porcine DLX5 gene and its association with carcass traits. Anim. Genet. 39, 395-399 [DOI] [PubMed] [Google Scholar]
- Chimge N., Makeyev A., Ruddle F., Bayarsaihan D. (2008). Identification of the TFII-I family target genes in the vertebrate genome. Proc. Natl. Acad. Sci. USA 105, 9006-9010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos I., Calcagnotto M. E., Vilaythong A. J., Thwin M. T., Noebels J. L., Baraban S. C., Rubenstein J. L. (2005). Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat. Neurosci. 8, 1059-1068 [DOI] [PubMed] [Google Scholar]
- Constantino J. N., Lajonchere C., Lutz M., Gray T., Abbacchi A., McKenna K., Singh D., Todd R. D. (2006). Autistic social impairment in the siblings of children with pervasive developmental disorders. Am. J. Psychiatry 163, 294-296 [DOI] [PubMed] [Google Scholar]
- Edelmann L., Prosnitz A., Pardo S., Bhatt J., Cohen N., Lauriat T., Ouchanov L., González P., Manghi E., Bondy P., et al. (2007). An atypical deletion of the Williams-Beuren syndrome interval implicates genes associated with defective visuospatial processing and autism. J. Med. Genet. 44, 136-143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feledy J. A., Morasso M. I., Jang S.-I., Sargent T. D. (1999). Transcriptional activation by the homeodomain protein Distal-less 3. Nucl Acids Res. 27, 764-770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J., Bi C., Clark B. S., Mady R., Shah P., Kohtz J. D. (2006). The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 20, 1470-1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty M., Grist M., Gelman D., Marín O., Pachnis V., Kessaris N. (2007). Spatial genetic patterning of the embryonic neuroepithelium generates GABAergic interneuron diversity in the adult cortex. J. Neurosci. 27, 10935-10946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francke U. (1999). Williams-Beuren syndrome: genes and mechanisms. Hum. Mol. Genet. 8, 1947-1954 [DOI] [PubMed] [Google Scholar]
- Ghanem N., Jarinova O., Amores A., Hatch G., Park B. K., Rubenstein J. L. R., Ekker M. (2003). Regulatory roles of conserved intergenic domains in vertebrate Dlx bigene clusters. Genome Res. 13, 533-543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem N., Yu M., Long J., Hatch G., Rubenstein J. L., Ekker M. (2007). Distinct cis-regulatory elements from the Dlx1/Dlx2 locus mark different progenitor cell populations in the ganglionic eminences and different subtypes of adult cortical interneurons. J. Neurosci. 27, 5012-5022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem N., Yu M., Poitras L., Rubenstein J., Ekker M. (2008). Characterization of a distinct subpopulation of striatal projection neurons expressing the Dlx genes in the basal ganglia through the activity of the I56ii enhancer. Dev. Biol. 322, 415-424 [DOI] [PubMed] [Google Scholar]
- Hamilton S. P., Woo J. M., Carlson E. J., Ghanem N., Ekker M., Rubenstein J. L. (2005). Analysis of four DLX homeobox genes in autistic probands. BMC Genet. 6, 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horike S., Cai S., Miyano M., Cheng J. F., Kohwi-Shigematsu T. (2005). Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 37, 31-40 [DOI] [PubMed] [Google Scholar]
- Keller A., Nesvizhskii A., Kolker E., Aebersold R. (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383-5392 [DOI] [PubMed] [Google Scholar]
- Kimura M., Kazuki Y., Kashiwagi A., Kai Y., Abe S., Barbieri O., Levi G., Oshimura M. (2004). Dlx5, the mouse homologue of the human-imprinted DLX5 gene, is biallelically expressed in the mouse brain. J. Hum. Genet. 49, 273-277 [DOI] [PubMed] [Google Scholar]
- Klein-Tasman B., Mervis C., Lord C., Phillips K. (2007). Socio-communicative deficits in young children with Williams syndrome: performance on the Autism Diagnostic Observation Schedule. Child Neuropsychol. 13, 444-467 [DOI] [PubMed] [Google Scholar]
- Le T., Du G., Fonseca M., Zhou Q., Wigle J., Eisenstat D. (2007). Dlx homeobox genes promote cortical interneuron migration from the basal forebrain by direct repression of the semaphorin receptor neuropilin-2. J. Biol. Chem. 282, 19071-19081 [DOI] [PubMed] [Google Scholar]
- Lettice L., Hill A., Devenney P., Hill R. (2008). Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum. Mol. Genet. 17, 978-985 [DOI] [PubMed] [Google Scholar]
- Lincoln A., Searcy Y., Jones W., Lord C. (2007). Social interaction behaviors discriminate young children with autism and Williams syndrome. J. Am. Acad. Child Adolesc. Psychiatry 46, 323-331 [DOI] [PubMed] [Google Scholar]
- Liu X., Novosedlik N., Wang A., Hudson M., Cohen I., Chudley A., Forster-Gibson C., Lewis S., Holden J. (2009). The DLX1and DLX2 genes and susceptibility to autism spectrum disorders. Eur. J. Hum. Genet. 17, 228-235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J., Cobos I., Potter G., Rubenstein J. (2009a). Dlx1&2 and Mash1 transcription factors control MGE and CGE patterning and differentiation through parallel and overlapping pathways. Cereb. Cortex 19, i96-i106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J., Swan C., Liang W., Cobos I., Potter G., Rubenstein J. (2009b). Dlx1&2 and Mash1 transcription factors control striatal patterning and differentiation through parallel and overlapping pathways. J. Comp. Neurol. 512, 556-572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S., Stock D. W., Wynder K. L., Bollekens J. A., Takeshita K., Nagai B. M., Chiba S., Kitamura T., Freeland T. M., Zhao Z., et al. (1996). Genomic analysis of a new mammalian distal-less gene: Dlx7. Genomics 38, 314-324 [DOI] [PubMed] [Google Scholar]
- Nakashima N., Yamagata T., Mori M., Kuwajima M., Suwa K., Momoi M. (2009). Expression analysis and mutation detection of DLX5 and DLX6 in autism. Brain Dev. 32, 98-104 [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A., Keller A., Kolker E., Aebersold R. (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646-4658 [DOI] [PubMed] [Google Scholar]
- Okita C., Meguro M., Hoshiya H., Haruta M., Sakamoto Y. K., Oshimura M. (2003). A new imprinted cluster on the human chromosome 7q21-q31, identified by human-mouse monochromosomal hybrids. Genomics 81, 556-559 [DOI] [PubMed] [Google Scholar]
- Poitras L., Ghanem N., Hatch G., Ekker M. (2007). The proneural determinant MASH1 regulates forebrain Dlx1/2 expression through the I12b intergenic enhancer. Development 134, 1755-1765 [DOI] [PubMed] [Google Scholar]
- Porteus M. H., Bulfone A., Ciaranello R. D., Rubenstein J. L. R. (1991). Isolation and characterization of a novel cDNA clone encoding a homeodomain that is developmentally regulated in the ventral forebrain. Neuron 7, 221-229 [DOI] [PubMed] [Google Scholar]
- Price M., Lemaistre M., Pischetola M., Di Lauro R., Duboule D. (1991). A mouse gene related to distal-less shows a restricted expression in the developing forebrain. Nature 351, 748-751 [DOI] [PubMed] [Google Scholar]
- Richler E., Reichert J. G., Buxbaum J. D., McInnes L. A. (2006). Autism and ultraconserved non-coding sequence on chromosome 7q. Psychiatr. Genet. 16, 19-23 [DOI] [PubMed] [Google Scholar]
- Robinson G. W., Mahon K. A. (1994). Differential and overlapping expression domains of Dlx-2 and Dlx-3 suggest distinct roles for Distal-less homeobox genes in craniofacial development. Mech. Dev. 48, 199-215 [DOI] [PubMed] [Google Scholar]
- Robinson G. W., Wray S., Mahon K. A. (1991). Spatially restricted expression of a member of a new family of murine distal-less homeobox genes in the developing forebrain. New Biol. 3, 1183-1194 [PubMed] [Google Scholar]
- Rubenstein J. L., Merzenich M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255-267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E. F., Maniatis T. (1989). Molecular Cloning, a Laboratory Manual, 2nd edn. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; [Google Scholar]
- Scherer S. W., Heng H. H. Q., Robinson G. W., Mahon K. A., Evans J. P., Tsui L.-C. (1995). Assignment of the human homolog of mouse Dlx3 to chromosome 17q21.3-q22 by analysis of somatic cell hybrids and fluorescence in situ hybridization. Mamm. Genome 6, 310-311 [DOI] [PubMed] [Google Scholar]
- Schule B., Li H. H., Fisch-Kohl C., Purmann C., Francke U. (2007). DLX5 and DLX6 expression is biallelic and not modulated by MeCP2 deficiency. Am. J. Hum. Genet. 81, 492-506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone A., Acampora D., Pannese M., Desposito M., Stornaiuolo A., Gulisano M., Mallamaci A., Kastury K., Druck T., Huebner K., et al. (1994). Cloning and characterization of two members of the vertebrate Dlx gene family. Proc. Natl. Acad. Sci. USA 91, 2250-2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuse D. H. (2007). Rethinking the nature of genetic vulnerability to autistic spectrum disorders. Trends Genet. 23, 387-395 [DOI] [PubMed] [Google Scholar]
- Stock D. W., Ellies D. L., Zhao Z., Ekker M., Ruddle F. H., Weiss K. M. (1996). The evolution of the vertebrate Dlx gene family. Proc. Natl. Acad. Sci. USA 93, 10858-10863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhmer T., Anderson S. A., Ekker M., Rubenstein J. L. R. (2002a). Ectopic expression of the Dlx genes induces glutamic acid decarboxylase and Dlx expression. Development 129, 245-252 [DOI] [PubMed] [Google Scholar]
- Stuhmer T., Puelles L., Ekker M., Rubenstein J. L. (2002b). Expression from a Dlx gene enhancer marks adult mouse cortical GABAergic neurons. Cereb. Cortex 12, 75-85 [DOI] [PubMed] [Google Scholar]
- Van der Aa N., Rooms L., Vandeweyer G., van den Ende J., Reyniers E., Fichera M., Romano C., Delle Chiaie B., Mortier G., Menten B., et al. (2009). Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur. J. Med. Genet. 52, 94-100 [DOI] [PubMed] [Google Scholar]
- Wang Y., Dye C., Sohal V., Long J., Estrada R., Roztocil T., Lufkin T., Deisseroth K., Baraban S., Rubenstein J. (2010). Dlx5 and Dlx6 regulate the development of parvalbumin-expressing cortical interneurons. J. Neurosci. 30, 5334-5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonders C., Anderson S. (2006). The origin and specification of cortical interneurons. Nat. Rev. Neurosci. 7, 687-696 [DOI] [PubMed] [Google Scholar]
- Wonders C., Taylor L., Welagen J., Mbata I., Xiang J., Anderson S. (2008). A spatial bias for the origins of interneuron subgroups within the medial ganglionic eminence. Dev. Biol. 314, 127-136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee S.-P., Rigby P. W. J. (1993). The regulation of myogenin gene expression during the embryonic development of the mouse. Genes Dev. 7, 1277-1289 [DOI] [PubMed] [Google Scholar]
- Zerucha T., Stuhmer T., Hatch G., Park B. K., Long Q., Yu G., Gambarotta A., Schultz J. R., Rubenstein J. L. R., Ekker M. (2000). A highly conserved enhancer in the Dlx5/Dlx6 intergenic region is the site of cross-regulatory interactions between Dlx genes in the embryonic forebrain. J. Neurosci. 20, 709-721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Hu G., Wang H., Sciavolino P., Iler N., Shen M. M., Abate-Shen C. (1997). Heterodimerization of Msx and Dlx homeoproteins results in functional antagonism. Mol. Cell. Biol. 17, 2920-2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q. P., Le T. N., Qiu X., Spencer V., de Melo J., Du G., Plews M., Fonseca M., Sun J. M., Davie J. R., et al. (2004). Identification of a direct Dlx homeodomain target in the developing mouse forebrain and retina by optimization of chromatin immunoprecipitation. Nucleic Acids Res. 32, 884-892 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}