

Abstract
Fibroblast growth factor (FGF) 9 is a secreted signaling molecule that is expressed in lung mesothelium and epithelium and is required for lung development. Embryos lacking FGF9 show mesenchymal hypoplasia, decreased epithelial branching and, by the end of gestation, hypoplastic lungs that cannot support life. Mesenchymal FGF signaling interacts with β-catenin-mediated WNT signaling in a feed-forward loop that functions to sustain mesenchymal FGF responsiveness and mesenchymal WNT/β-catenin signaling. During pseudoglandular stages of lung development, Wnt2a and Wnt7b are the canonical WNT ligands that activate mesenchymal WNT/β-catenin signaling, whereas FGF9 is the only known ligand that signals to mesenchymal FGF receptors (FGFRs). Here, we demonstrate that mesothelial- and epithelial-derived FGF9, mesenchymal Wnt2a and epithelial Wnt7b have unique functions in lung development in mouse. Mesothelial FGF9 and mesenchymal WNT2A are principally responsible for maintaining mesenchymal FGF-WNT/β-catenin signaling, whereas epithelial FGF9 primarily affects epithelial branching. We show that FGF signaling is primarily responsible for regulating mesenchymal proliferation, whereas β-catenin signaling is a required permissive factor for mesenchymal FGF signaling.
Keywords: Lung, Branching, FGF, FGF receptor, WNT, β-Catenin, BMP4, Noggin, Mouse
INTRODUCTION
Lung development is governed by complex interactions between embryonic lung endoderm, mesoderm and mesothelium. During the pseudoglandular stage of lung development [embryonic day (E) 9.5-16 in the mouse], fibroblast growth factor 9 (encoded by Fgf9) is expressed in the mesothelium, the single layer of cells that envelopes the lung, and in the pulmonary epithelium that will give rise to the proximal conducting airways and terminal epithelial buds (Colvin et al., 1999). In the absence of FGF9 (Fgf9–/–), lung development is severely impaired, resulting in perinatal death (Colvin et al., 2001a; White et al., 2006). Fgf9–/– embryos show decreased mesenchymal proliferation and impaired epithelial branching. FGF9 primarily signals to mesenchymal FGF receptors (FGFRs) 1 and 2, but also has the unique ability to activate epithelial FGFR signaling (del Moral et al., 2006; White et al., 2006; Yin et al., 2008).
Lung mesenchyme is not a homogeneous tissue, and using histological criteria and molecular markers it can be subdivided into sub-mesothelial (SMM) and sub-epithelial (SEM) domains (White et al., 2006). The pulmonary vasculature appears to form between these mesenchymal domains (White et al., 2006; White et al., 2007). SMM is marked by expression of Wnt2a, and SEM expresses Noggin (Nog) (Levay-Young and Navre, 1992; Bellusci et al., 1996; Weaver et al., 2003; Yin et al., 2008). Previous studies have identified interactions between FGF signaling and WNT/β-catenin signaling in lung mesenchyme that function to positively reinforce each other. Mesenchymal FGF signaling is required for expression of WNT2a and mesenchymal WNT/β-catenin signaling, and mesenchymal WNT/β-catenin signaling is required to sustain expression of FGFR1 and FGFR2 (De Langhe et al., 2008; Yin et al., 2008; Yi et al., 2009). In the absence of either FGF9 or mesenchymal WNT/β-catenin signaling, lung mesenchyme cannot be rescued by addition of exogenous FGF9 (Yin et al., 2008). This loss of responsiveness of mesenchyme is likely to be due to degeneration of this feed-forward loop and loss of FGFR expression in mesenchyme. In addition to mesothelial and epithelial Fgf9 and mesenchymal Wnt2a, Wnt7b (which is expressed in developing epithelium) is also required for lung development where it activates mesenchymal and, potentially, epithelial WNT/β-catenin signaling (Rajagopal et al., 2008). At early (lung bud) stages of development, Wnt2b is also expressed in foregut mesenchyme and signals redundantly with Wnt2a in endoderm to regulate lung bud formation (Goss et al., 2009). The mechanisms that sustain this feed-forward loop and the relative contributions of mesothelial and epithelial FGF9 and mesenchymal WNT2A and epithelial WNT7B are not known.
Here, we show that mesothelial and epithelial FGF9 have distinct functions during early pseudoglandular stage lung development. Mesothelial FGF9 is necessary for mesenchymal FGF signaling, WNT2A expression and mesenchymal growth, whereas epithelial FGF9 modulates epithelial branching. WNT2A, and to a much lesser extent WNT7B, functions to maintain mesenchymal growth through a required permissive action on mesenchymal FGFR signaling. Consistent with this, we show that exogenous activation of both FGF and WNT/β-catenin signaling are required to rescue growth of Fgf9–/– lung mesenchyme, and that mesenchymal WNT/β-catenin signaling is a required permissive factor for FGF signaling. Finally, we show that epithelial and mesothelial FGF9 and mesenchymal β-catenin function to suppress expression of Noggin in lung mesenchyme, providing a mechanism for coupling mesenchymal FGF-WNT/β-catenin signaling with epithelial proliferation.
MATERIALS AND METHODS
Mouse strains
All mouse strains, including Fgf9+/–, Fgf9f/f (f indicates a floxed allele), Dermo1-Cre and Shh-cre have been described previously (Colvin et al., 2001a; Colvin et al., 2001b; Yu et al., 2003; Harfe et al., 2004; Lin et al., 2006). For conditional inactivation of Fgf9 in lung mesenchyme, mice were generated with the genotype Dermo1Cre/+, Fgf9f/f (referred to as Fgf9Dermo1) by mating Dermo1Cre/+, Fgf9f/+ mice with Fgf9f/f mice. Control mice were of the genotype Dermo1Cre/+; Dermo1Cre/+, Fgf9f/+; Fgf9f/f; or Fgf9f/+, all of which showed no phenotypic differences from wild-type mice. For conditional inactivation of Fgf9 in lung epithelium, mice were generated with the genotype ShhCre/+, Fgf9f/f (referred to as Fgf9Shh) by mating ShhCre/+, Fgf9f/+ mice with Fgf9f/f mice. Control mice were of the genotype ShhCre/+; ShhCre/+, Fgf9f/+; Fgf9f/f; or Fgf9f/+, all of which showed no phenotypic differences from wild-type mice. All loss-of-function mice were maintained on a mixed 129SV/J-C57B6/J background.
Analyses of mouse embryos, histology and immunohistochemistry
Embryo tissues were collected in ice-cold PBS, fixed in 4% paraformaldehyde (PFA) overnight at 4°C, washed with PBS, photographed and embedded in paraffin prior to sectioning at 5 μm. For histology, slides were stained with Hematoxylin and Eosin (H&E). For immunohistochemistry, cryosections were rehydrated and treated with 0.3% hydrogen peroxide in methanol for 15 minutes to suppress the endogenous peroxidase activity. Antigen retrieval was achieved by microwaving the sections in 10 mM citrate buffer for 5 minutes followed by gradual cooling to room temperature. Sections were incubated overnight at 4°C with the primary antibodies against BrdU (347580, Becton Dickinson Immunocytometry Systems, 1:200) or FGF9 (AF-273-NA, R&D, 1:100) and visualized using Broad Spectrum (AEC) Kit (95-9743, Zymed Laboratories Inc.) or Vectastain Elite ABC (AEC) kit (PK-4005, Vector Laboratories). For 5-bromo-2′-deoxyuridine (BrdU) analysis, pregnant females were injected with BrdU at 0.1 mg/g body weight, 2 hours prior to harvest. Embryos were collected in ice-cold PBS, processed and sectioned as above. All staining patterns are representative of at least three embryos.
Cell death analysis
Cryosectioned slides, prepared as described above, were assayed by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) using the In Situ Cell Death Detection Kit (Roche Applied Science). Slides were mounted with DAPI containing Vectashield mounting medium (H-1200, Vector Laboratories) for fluorescent detection. All staining patterns are representative of at least three embryos.
Whole mount in situ hybridization
In situ hybridization probes were from the following sources: Lef1 (Kratochwil et al., 1996); Wnt2a (A. McMahon, Harvard University, Cambridge, MA, USA); Wnt7b (F. Long, Washington University, St Louis, MO, USA); Noggin (R. Harland, University of California, Berkeley, CA, USA); Fgf10 (Bellusci et al., 1997); Spry2 (Minowada et al., 1999); Fgfr2 (Yu et al., 2003), Evt4 (Chen et al., 2005); N-Myc (E. Morrisey, University of Pennsylvania, Philadelphia, PA, USA). Probes were synthesized and labeled with a kit from Roche Applied Science. Whole-mount in situ hybridization was performed as described (Colvin et al., 2001a). Following color reaction and methanol dehydration, tissues were photographed and then cryosectioned (6 μm), mounted on slides and re-photographed. In situ hybridizations of tissue sections were performed as previously described (Colvin et al., 1999). All staining patterns are representative of at least three embryos.
Whole-mount lacZ staining
Lungs were dissected in ice-cold PBS and fixed with 0.5% glutaraldehyde in PBT (PBS, 0.1% Tween-20) overnight at 4°C. Tissues were washed in PBT twice for 10 minutes prior to incubation with lacZ staining solution (2 mM MgCl2, 35 mM potassium ferrocyanide, 35 mM potassium ferricyanide, 1 mg/ml X-Gal in PBT) at room temperature in the dark. Following adequate color reaction, tissues were again washed twice in PBT for 10 minutes each to stop the reaction. Samples were then soaked in 30% sucrose overnight, photographed and then embedded and frozen in OCT for cryosectioning (6 μM). Sections were dried for ∼3 hours at room temperature, washed with PBS and mounted. All staining patterns are representative of at least three embryos.
Lung organ cultures
Lung explant cultures were performed as described (White et al., 2006). E11.5 control, Fgf9Dermo1, Fgf9–/– and wild-type embryonic lungs were dissected and cultured on Transwell filters (Costar, Corning) for 48 hours at 37°C, 5% CO2. Mouse FGF9 protein (PeproTech) was used at a concentration of 2.5 μg/ml in DMEM supplemented with 2 μg/ml heparin. LiCl and BIO were used at a concentration of 20 mM (Dean et al., 2005) and 1 μM (Meijer et al., 2003), respectively.
For morpholino knockdown experiments, E11.5 lung explant cultures were prepared as above. Explants were treated with control (CCTCTTACCTCAGTTACAATTTATA), Wnt2a (CCAGGAGAAGGAGACTCACTTCGGA), Wnt7b (AGGCCCAGAGACGCCCTTACCTACT), or Wnt2a and Wnt7b morpholino oligonucleotides (Gene Tools) at a final concentration of 10 μM in culture media. Morpholinos penetrated the epithelium and the mesenchyme of lung explant tissue and effectively blocked mRNA splicing as demonstrated by RT-PCR 48 hours post-explant (Fig. 5A). Primers were designed to detect the 5′ end of the Wnt2a transcript (forward primer: CGCACACGGAGTCTGACCTGATGTA, reverse primer: CTTTCTTCTTTGGATCACAGGAGCAG) and the Wnt2a splice site that is targeted by the morpholino (junction primers) (forward primer: GAACCAGGGTGGCACTGGCTTCAC, reverse primer: GATGTGATGCGTGCCATTGGCCTGC). Similar primers were designed to analyze the Wnt7b transcript: Wnt7b 5′ primers (forward primer: CCCTCCGGCCGCAGCTGTTGG, reverse primer: ACAATGATGGCATCGGGTCGGCTCT), Wnt7b splice junction primers (forward primer: TCCTCTACGTGAAGCTCGGAGCATT, reverse primer: CCGTGATGGCATAGGTGAAGGCAG).
Fig. 5.

Regulation of BMP pathways by mesothelial and epithelial FGF9. (A-C) Expression of Bmp4 in control (A), Fgf9Dermo1 (B) and Fgf9Shh (C) mouse lung mesenchyme at E12.5. (D-F) Expression of Nog in control (D), Fgf9Dermo1 (E) and Fgf9Shh (F) lung epithelium at E12.5. Bmp4 expression was similar but Nog expression was significantly increased in both Fgf9Dermo1 and Fgf9Shh lungs. (G-I) Frozen sections of the whole mounts shown in D-F. (J) Quantitative RT-PCR detection of Nog expression showing increased expression in Fgf9Dermo1 and Fgf9Shh E12.5 lung compared with control. Error bars represent s.d. *P<0.05, **P<0.01, Student's t-test. Scale bar: 200 μm for A-F; 25 μm for G-I.
To quantify mesenchymal thickness, explants were photographed and mesenchymal thickness was measured using Canvas X software. Data shown is representative of at least three independent experiments. P-values were calculated using the Student's t-test and mean ± s.d. were plotted.
Quantitative RT-PCR
Total RNA was extracted from individual control, Fgf9Dermo1 or Fgf9Shh E12.5 embryonic lungs (n=3 pooled samples for each genotype, each pooled sample contained four to six lungs) using the RNeasy kit (Qiagen) following manufacturer's instructions. Total RNA was reverse-transcribed using the iScript Select cDNA Synthesis Kit (BIO-RAD) following the manufacturer's recommendations. Quantitative RT-PCR was performed on an ABI 7500 machine using TaqMan probes for Gapdh (ABI Mm99999915_g1), Fgf10 (ABI Mn00433275_m1) and Noggin (ABI Mm00476456_s1). Amplification and analysis were performed according to the manufacturer's instructions. All reactions were normalized to Gapdh. Results were plotted as relative expression compared with control, where control was scaled to one. P-values were calculated using the Student's t-test and mean ± s.d. were plotted.
RESULTS
Mesothelial and epithelial FGF9 have unique functions in developing lung
During pseudoglandular lung development, Fgf9 is expressed exclusively in mesothelium and epithelium, but not in mesenchyme. To explore the role of mesothelial and epithelial FGF9, tissue-specific Cre recombinases were used to inactivate a floxed allele of Fgf9 (Fgf9f/f) (Lin et al., 2006) in mesothelial and epithelial tissues. To inactivate Fgf9 in mesothelium, embryos were generated with the genotype Dermo1Cre/+, Fgf9f/f (Fgf9Dermo1). The Dermo1Cre allele targets lung mesenchyme and mesothelium beginning before E9.5, but has no detectable activity in lung epithelium (Fig. 1A) (White et al., 2006). Thus, the Dermo1Cre allele can be used to specifically inactivate mesothelial Fgf9. Examination of FGF9 expression in Fgf9Dermo1 lungs by immunohistochemistry showed loss of expression in lung mesothelium, but retained expression in lung epithelium (Fig. 1D). Control (Fgf9f/f and Dermo1Cre/+) lung tissue showed FGF9 staining in both epithelium and mesothelium (Fig. 1C).
Fig. 1.
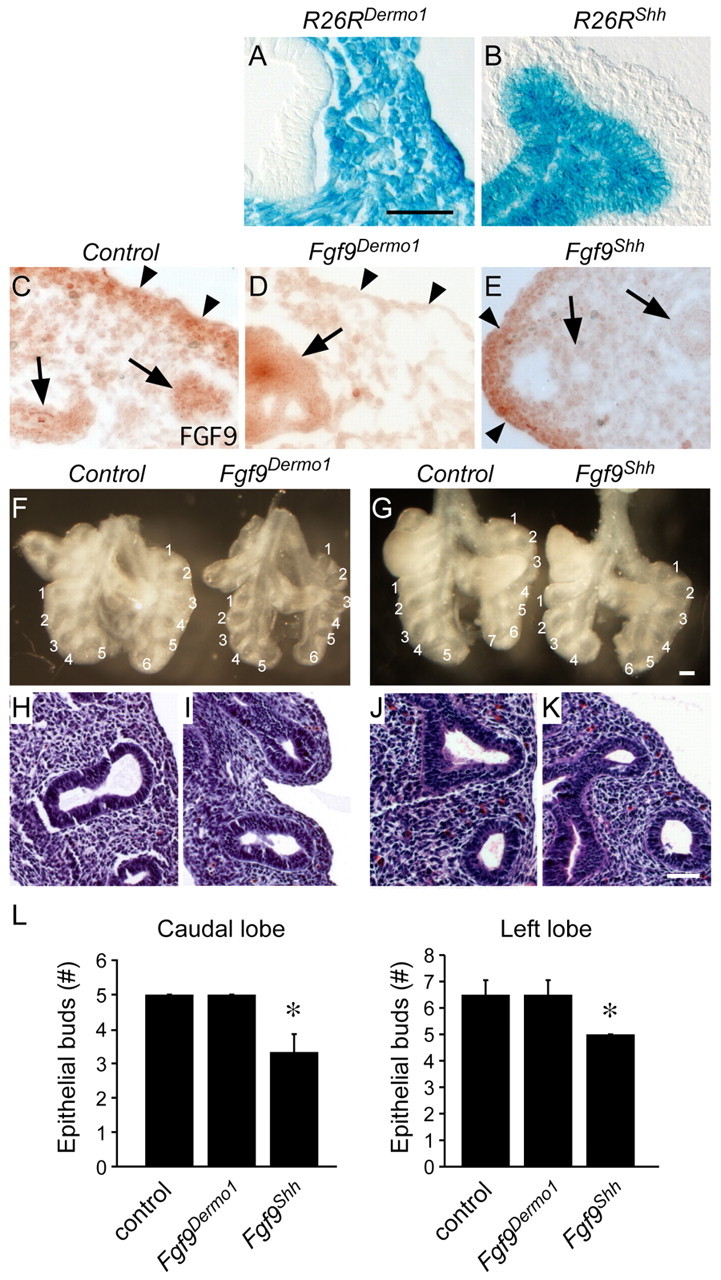
Mesothelial and epithelial FGF9 have unique functions in developing mouse lung. (A,B) lacZ staining for the Rosa26 reporter (R26R) targeted with Dermo1-Cre (A) and Shh-Cre (B) lungs at E10.5 showing mesenchymal/mesothelial and epithelial cell-specific β-gal staining, respectively. (C-E) Immunohistochemical detection of FGF9 in E12.5 lung showing expression in epithelium (arrows) and mesothelium and submesothelial mesenchyme (arrowheads) in control lungs (C). Fgf9Dermo1 lungs (D) have greatly reduced staining in submesothelial mesenchyme (arrowheads) and Fgf9Shh lungs (E) have reduced expression in epithelium (arrows). (F,G) Anterior views of gross dissections of control (Dermo1-Cre, Fgf9f/+) and Fgf9Dermo1 (F) and control (Shh-Cre, Fgf9f/+) and Fgf9Shh (G) lungs at E12.5. Note the decreased mesenchyme but normal epithelial branch number and orientation of lobes with the Fgf9Dermo1 lung (F) and decreased number of epithelial branches and normal mesenchyme in Fgf9Shh lung (G). (H-K) Hematoxylin and Eosin-stained histological sections of control (H,J), Fgf9Dermo1 (I) and Fgf9Shh (K) lungs at the same stages as shown in F,G. (L) Number of epithelial buds in the caudal and left lobes at E12.5. *P<0.001 (Student's t-test). Error bars represent s.d. Scale bars: 25 μm in A-E,H-L; 200 μm in F,G.
Fgf9Dermo1 embryos were born alive but only 67% of the expected number survived beyond weaning. Littermate control mice were phenotypically normal. At E12.5, Fgf9Dermo1 embryos had hypoplastic lungs with a normal number and orientation of lobes and a normal number of epithelial branches (Fig. 1F,L). Histological examination of Fgf9Dermo1 lungs showed hypoplastic mesenchyme (Fig. 1H,I). At E13.5 and E14.5, Fgf9Dermo1 lungs were smaller than control lungs, the edges were serrated and hypoplastic, and the histology indicated decreased mesenchyme and decreased epithelial branching (see Fig. S1A,C,E,F in the supplementary material).
To inactivate Fgf9 in epithelium, embryos were generated with the genotype ShhCre/+, Fgf9f/f (Fgf9Shh). The ShhCre allele targets lung epithelium beginning before E9.5, but has no detectable activity in lung mesenchyme or mesothelium (Fig. 1B) (Harfe et al., 2004; Harris et al., 2006). Thus, the ShhCre allele can be used to specifically inactivate epithelial Fgf9. Examination of FGF9 expression in Fgf9Shh lungs by immunohistochemistry showed loss of expression in lung epithelium, but retained expression in lung mesothelium (Fig. 1C,E). Fgf9Shh embryos had a normal number and orientation of primary lobes. However, at E12.5, Fgf9Shh lungs were slightly smaller than control (Fgf9f/f and ShhCre/+) lungs, and, in contrast to Fgf9Dermo1 lungs, Fgf9Shh lungs showed decreased epithelial branching (Fig. 1G,J-L). Histological examination of E12.5 Fgf9Shh lungs showed fewer epithelial ducts but normal appearing mesenchyme. At E13.5 and E14.5, Fgf9Shh lungs were noticeably smaller than control lungs, but showed smooth edges. Lung histology showed normal mesenchymal histology but decreased epithelial branching (see Fig. S1B,D,G,H in the supplementary material).
Mesothelial and epithelial FGF9 differentially affect mesenchymal and epithelial proliferation at early developmental stages
We hypothesized that changes in cell proliferation could account for the observed differences in lung phenotypes of Fgf9Dermo1 and Fgf9Shh embryos. At E12.5, Fgf9Dermo1 lungs showed significantly decreased mesenchymal proliferation but normal epithelial proliferation (Fig. 2A,B,G). At E14.5, both mesenchymal and epithelial proliferation was decreased (Fig. 2D,E,H). By contrast, at E12.5, Fgf9Shh lungs showed decreased epithelial proliferation but normal mesenchymal proliferation (Fig. 2A,C,G). At E14.5, Fgf9Shh lungs showed decreased proliferation in both epithelial and mesenchymal compartments (Fig. 2D,F,H), similar to that observed in Fgf9Dermo1 lungs at this stage. We also examined cell death by TUNEL labeling. At E12.5 and E14.5, no significant change in cell death was detected in Fgf9Dermo1 or Fgf9Shh lung tissues (Fig. 2I-K; data not shown). This is consistent with previous studies showing that FGF signaling does not regulate cell survival at these stages of lung development (Yin et al., 2008).
Fig. 2.
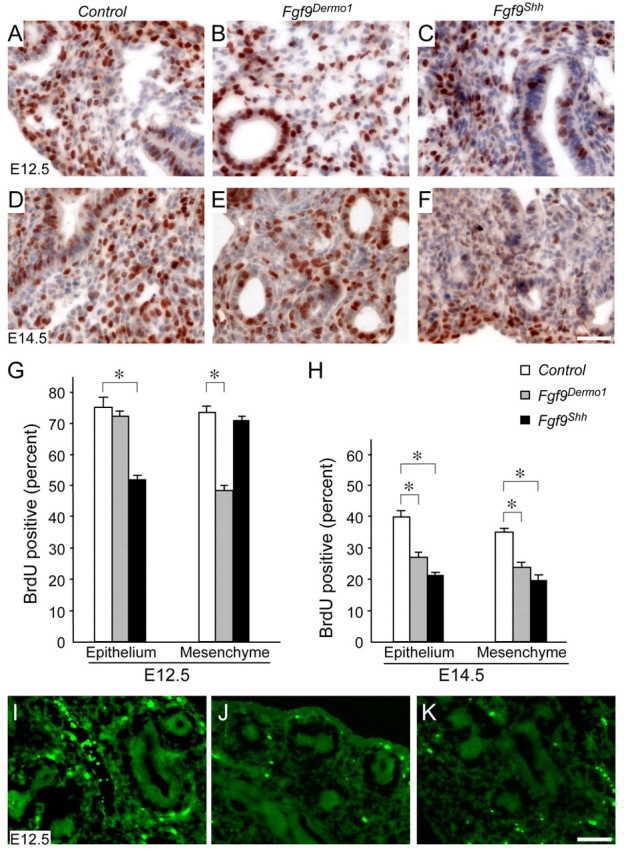
Mesothelial and epithelial FGF9 differentially affect mesenchymal and epithelial proliferation. (A-F) BrdU labeling of control (A,D), Fgf9Dermo1 (B,E) and Fgf9Shh (C,F) mouse lung showing decreased proliferation in the mesenchyme of the Fgf9Dermo1 lung and the epithelium of the Fgf9Shh lung at E12.5 and E14.5. Note that at E14.5 proliferation was decreased in both the epithelium and mesenchyme in Fgf9Dermo1 and Fgf9Shh lung. (G,H) Quantitation of BrdU labeling at E12.5 (G) and E14.5 (H). *P<0.01 (Student's t-test). Error bars represent s.d. (I-K) TUNEL labeling of control (I), Fgf9Dermo1 (J) and Fgf9Shh (K) lung at E12.5 showing no difference in rates of cell death. Scale bars: 50 μm.
Taken together, these data demonstrate that FGF9 derived from mesothelial and epithelial tissues have different functions during early stages (E12.5) of lung development: mesothelial FGF9 functions to maintain lung mesenchymal proliferation, whereas epithelial FGF9 primarily affects epithelial proliferation. At later stages (E14.5), both epithelial and mesenchymal compartments were affected in both conditional knockouts, suggesting that secondary interactions between tissue compartments might override these local signals to coordinate growth of lung mesenchyme and epithelium.
Mesothelial FGF9 regulates Wnt2a expression and mesenchymal WNT/β-catenin signaling but not Fgf10 expression or signaling
To examine the effects of mesothelial and epithelial FGF9 on mesenchymal and epithelial WNT/β-catenin signaling, we examined the expression of mesenchymal Wnt2a, epithelial Wnt7b and the WNT target gene Lef1 in E12.5 Fgf9Dermo1 and Fgf9Shh lungs. In the Fgf9Dermo1 lungs, Wnt2a expression was absent in lung mesenchyme (Fig. 3A,B), similar to what was observed in Fgf9–/– lungs (Yin et al., 2008). By contrast, in Fgf9Shh lungs, Wnt2a expression was reduced but still detectable in distal mesenchyme (Fig. 3A,C,A′,C′). In control lung, Lef1 was expressed in both the SMM and SEM domains (Fig. 3D,D′). In Fgf9Dermo1 lungs, Lef1 expression was reduced in the SMM but retained in the SEM (Fig. 3E,E′). By contrast, in Fgf9Shh lungs, Lef1 expression was reduced in the SEM but retained in the SMM (Fig. 3F,F′). Additionally, Wnt7b and N-Myc (Mycn – Mouse Genome Informatics) expression was not affected by inactivation of mesothelial or epithelial Fgf9 (Fig. 3G-I; data not shown). Taken together, these data are consistent with a model in which mesothelial FGF9 is the primary source of FGF9 that regulates mesenchymal FGF-WNT/β-catenin signaling.
Fig. 3.
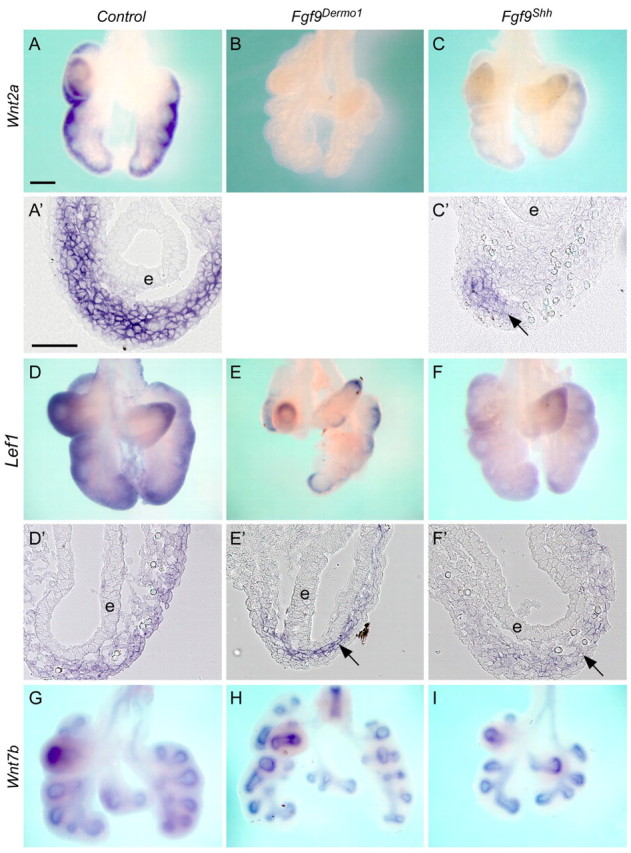
Mesothelial FGF9 regulates WNT2A expression and mesenchymal WNT/β-catenin signaling. (A-C′) Expression of Wnt2a in control (A,A′), Fgf9Dermo1 (B) and Fgf9Shh (C,C′) mouse lung mesenchyme at E12.5. A′ and C′ are cryosections of the corresponding whole mounts. Note that in Fgf9Dermo1 lung mesenchyme Wnt2a is not detected. In Fgf9Shh lung, Wnt2a is still expressed in the distal mesenchyme (arrow) but not in proximal mesenchyme of epithelium (e). (D-F′) Expression of Lef1 in control (D,D′), Fgf9Dermo1 (E,E′) and Fgf9Shh (F,F′) lung mesenchyme at E12.5. D′, E′ and F′ are cryosections of the corresponding whole mounts. Note that in Fgf9Dermo1 lung, Lef1 expression was detected in the proximal mesenchyme (arrow) and in Fgf9Shh lung Lef1 was detected in the distal mesenchyme (arrow). (G-I) Wnt7b expression in lung epithelium was not significantly changed in control (G), Fgf9Dermo1 (H) and Fgf9Shh (I) lung epithelium at E12.5. Scale bars: 200 μm in A-C,D-E,G-I; 50 μm in A′,C′,D′-F′.
FGF10 is essential for lung epithelial branching (Bellusci et al., 1997; Min et al., 1998; Sekine et al., 1999; Weaver et al., 2000). We hypothesized that the reduced branching observed in Fgf9Shh lungs could result from reduced Fgf10 expression or signaling. However, at E12.5, control and Fgf9Shh lungs exhibited comparable expression patterns and levels of Fgf10 in mesenchyme distal to budding airways (Fig. 4A-C). Quantitative RT-PCR of Fgf9Shh or Fgf9Dermo1 E12.5 whole lung, compared with control lung, showed no significant difference in Fgf10 expression (n=3; P=0.6 and 0.7, respectively). Furthermore, Sprouty2 (Spry2), one of the earliest targets induced in the lung epithelium in response to FGF10 and a negative regulator of the FGF signaling pathway (Minowada et al., 1999; Mailleux et al., 2001), showed no significant difference in expression in control and Fgf9Shh lungs (or Fgf9Dermo1 lungs) at E12.5 (Fig. 4D-F). Similarly to Spry2, other molecules in the FGF10 signaling pathway, such as epithelial Fgfr2 (Fig. 4G-I) and the downstream transcription factor Etv4 (Fig. 4J-L), showed no significant difference in expression in control and Fgf9Shh (or Fgf9Dermo1) lungs at E12.5. Note that Fgfr2 expression was absent in mesenchyme in the Fgf9Dermo1 lung at E12.5. These data suggest that Fgf10 and the FGF10 signaling pathway is not a primary target of mesenchymal FGF9 signaling. These data are consistent with our previous studies in which Fgf10 expression was not significantly affected in the mesenchyme of Fgf9–/– lungs or β-cateninDermo1 lungs at E12.5 (Colvin et al., 2001a; Yin et al., 2008).
Fig. 4.

FGF10 signaling pathways are not affected by mesothelial and epithelial FGF9. (A-C) Expression of Fgf10 in control (A), Fgf9Dermo1 (B) and Fgf9Shh (C) mouse lung mesenchyme at E12.5. (D-F) Expression of Spry2 in control (D), Fgf9Dermo1 (E) and Fgf9Shh (F) lung epithelium at E12.5. No significant differences in Fgf10 or Spry2 expression were observed. (G-I) Expression of Fgfr2 in control (G), Fgf9Dermo1 (H) and Fgf9Shh (I) lung epithelium at E12.5. (J-L) Expression of Etv4 in control (J), Fgf9Dermo1 (K) and Fgf9Shh (L) lung mesenchyme at E12.5. Scale bar: 200 μm.
Mesenchymal FGF and WNT/β-catenin signaling suppress Noggin expression
BMP signaling through epithelial ALK3 (BMPR1A – Mouse Genome Informatics) has been shown to regulate proliferation of distal lung epithelium (Eblaghie et al., 2006). To determine whether mesenchymal FGF or WNT/β-catenin signaling could affect the BMP pathway and indirectly epithelial proliferation, we examined expression of Bmp4 and its inhibitor Noggin. Bmp4 is expressed in distal epithelium and adjacent SEM and Noggin is expressed in SEM (Weaver et al., 1999; Weaver et al., 2003; White et al., 2006; Jang et al., 2010). Consistent with previous observations of E12.5 Fgf9–/– lungs (White et al., 2006), Bmp4 expression was not changed in Fgf9Dermo1 or Fgf9Shh lungs (Fig. 5A-C). However, examination of the BMP inhibitor Noggin showed significantly increased expression in Fgf9Dermo1 or Fgf9Shh lungs (Fig. 5D-E), as well as in Fgf9–/– lungs and β-cateninDermo1 conditional knockout lungs (see Fig. S2 in the supplementary material). This result was also confirmed with quantitative RT-PCR, which showed significantly increased expression of Noggin in Fgf9Dermo1 or Fgf9Shh lungs compared with control lungs (n=3; P<0.01, P<0.03, respectively; Fig. 5J). Interestingly, frozen sections of the whole mounts showed increased Noggin throughout the mesenchyme of Fgf9Dermo1 and Fgf9–/– lung, but only in SEM of Fgf9Shh lungs (Fig. 5G-I, see Fig. S2 in the supplementary material). De-repression of Noggin and suppression of BMP signaling provides a mechanism by which loss of mesenchymal FGF or WNT/β-catenin signaling could indirectly lead to decreased epithelial proliferation.
Wnt2a maintains mesenchymal FGF responsiveness and both Wnt2a and Wnt7b are required for mesenchymal growth
Wnt2a, Wnt2b and Wnt7b are the only canonical WNT ligands that are expressed in the developing lung (Shu et al., 2002; Goss et al., 2009). Wnt2b is expressed at highest levels at the lung bud stage of development (Lin et al., 2001). Wnt2b–/– mice do not exhibit any phenotype, but in combination with knockout of Wnt2a, lung buds fail to form (Goss et al., 2009). These results suggest that WNT2B primarily functions at the lung bud stage of development or earlier. To explore the relative contribution of Wnt2a and Wnt7b to lung pseudoglandular-stage mesenchyme growth and maintenance of mesenchymal FGF signaling, antisense morpholino oligonucleotides (MOs) were used to inhibit Wnt2a and Wnt7b mRNA splicing in explanted lung tissue. Treatment of E11.5 lung explants with antisense MOs for 48 hours did not affect mRNA expression but effectively blocked mRNA splicing of both the Wnt2a and Wnt7b transcripts (Fig. 6A). Treatment of lung explants with Wnt2a MOs resulted in significantly decreased mesenchymal growth (thickness), which was further enhanced by simultaneous treatment with Wnt7b MOs (Fig. 6B,D,F,H,J).
Fig. 6.
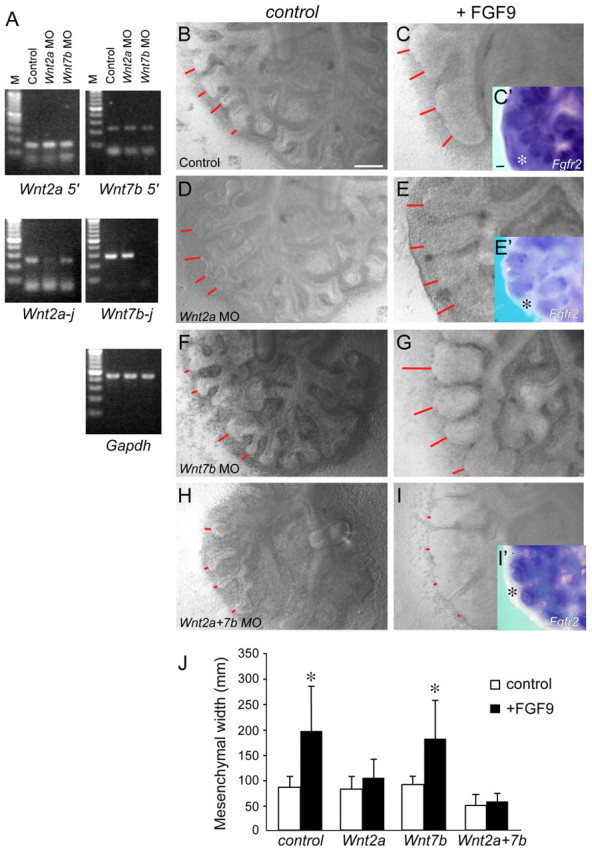
WNT2A and WNT7B are required for mesenchymal expression of Fgfr2 and response to FGF9. (A) PCR detection of Wnt2a and Wnt7b mRNA in mouse lung explants treated with control or splice site morpholinos. Top: amplification of the 5′ end of the transcripts showed normal expression in control, Wnt2a MO and Wnt7b MO treated explants. Middle: amplification of the splice site (j) of the transcripts showed reduced or absent mRNA expression in the corresponding treated explant. (B-I′) Wild-type lung explants were untreated (B,C) or exposed to morpholinos targeting a splice site in the Wnt2a (D,E), Wnt7b (F,G) or both Wnt2a and Wnt7b (H,I) transcripts for 48 hours. Explants were also treated with media (B,D,F,H) or FGF9 (C,E,G,I). Expression of Fgfr2 in control (C′), Wnt2a (E′) and Wnt2a and Wnt7b MO (I′) treated explants showed loss of mesenchymal (asterisks) Fgfr2 expression following knockdown of Wnt2a or Wnt2a and Wnt7b. Red lines indicate mesenchymal thickness. (J) Quantification of mesenchymal width. Explants treated with Wnt2a, or Wnt2a and Wnt7b MO showed no mesenchymal growth response to FGF9. (n=3, *P<0.01, Student's t-test). Error bars represent s.d. Scale bars: 200 μm.
Treatment of lung explants with FGF9 resulted in increased mesenchymal growth and epithelial dilation (del Moral et al., 2006; White et al., 2006; Yin et al., 2008). To assay mesenchymal response to FGF signaling following knockdown of Wnt2a and Wnt7b, explants were treated with MOs and with FGF9. After 48 hours in culture, mesenchymal thickness was measured (Fig. 6C,E,G,I,J). Consistent with previous observations, control lung explants showed an approximate twofold increase in mesenchymal thickness following culture with FGF9. Explants treated with a Wnt7b MO also showed a near-wild-type response to FGF9. However, explants treated with a Wnt2a MO, or with both Wnt2a and Wnt7b MOs, did not respond to FGF9. Consistent with loss of responsiveness to FGF9, mesenchymal Fgfr2 expression assayed by in situ hybridization was absent in mesenchyme from explants treated with Wnt2a or with Wnt2a and Wnt7b MOs, but not in control explants (Fig. 6C′,E′,I′). These data are consistent with a model in which Wnt2a functions primarily to regulate mesenchymal WNT/β-catenin signaling and expression of mesenchymal FGFRs. However, Wnt7b has either an indirect or redundant function with Wnt2a in regulating mesenchymal growth.
Mesenchymal FGF and WNT/β-catenin signaling requires both FGF9 and WNT inputs to maintain FGF9 responsiveness
Previous studies and data presented here showed that, during the early stages of mouse lung development, both FGF9 and WNT/β-catenin signaling are required to sustain mesenchymal FGFR expression and mesenchymal response to FGF9 (De Langhe et al., 2008; Yin et al., 2008). In the absence of either FGF9 or canonical WNT ligands, the mesenchymal FGF9-WNT/β-catenin feed-forward network degenerates, resulting in loss of mesenchymal FGFR expression and FGF responsiveness. A prediction of this model is that Fgf9–/– lung tissue could be rescued, in terms of responsiveness to FGF9 and mesenchymal growth, by simultaneous addition of FGF9 and activation of WNT/β-catenin signaling.
To test the mesenchymal FGF9-WNT/β-catenin network model, control and Fgf9–/– lung explants were treated with the GSK3β inhibitors LiCl or BIO [(2′Z,3′E)-6-bromoindirubin-3′-oxime], either alone or in combination with FGF9. Treatment of wild-type lung explants with LiCl or BIO did not affect mesenchymal growth and did not affect responsiveness to FGF9 (Fig. 7A-C,M). Fgf9–/– explants failed to respond to FGF9, as previously observed (Yin et al., 2008) (Fig. 7D,J). However, Fgf9–/– explants treated with LiCl and FGF9, or BIO and FGF9, showed a 2.6- and 1.8-fold increase, respectively, in mesenchymal thickness in response to FGF9 (Fig. 7E,K,F,L). Similarly, Fgf9Dermo1 lung explants did not respond to FGF9 unless simultaneously treated with BIO (see Fig. S3 in the supplementary material). These data indicate that WNT/β-catenin signaling must be maintained in order to induce or maintain FGFR expression, FGF9 responsiveness, and continuation of FGF-WNT/β-catenin feed-forward signaling.
Fig. 7.

Mesenchymal FGF and WNT/β-catenin signaling requires both FGF9 and WNT inputs to maintain FGF9 responsiveness. (A-L) Lung explant cultures from control (A-C,G-I) and Fgf9–/– (D-F,J-L) mouse embryos were untreated (A,D,G,J) or treated with LiCl (B,E,H,K) or BIO (C,F,I,L). Control and Fgf9–/– explants were also treated with FGF9 (G-L). Red lines indicate mesenchymal thickness. Scale bar: 200 μm. (M) Quantification of mesenchymal growth in response to LiCl, BIO and FGF9 (n=3 explants for each condition). Dashed red line shows mesenchymal thickness of wild-type lung explants. Error bars represent s.d. *P<0.05, **P<0.01, ***P<0.001, Student's t-test.
DISCUSSION
During lung development, lung mesenchymal cells not only serve as precursors to stromal fibroblasts and peribronchial and vascular smooth muscle cells, but they also have a crucial role in regulating epithelial growth and branching (Morrisey and Hogan, 2010; Warburton et al., 2010). Although many of the essential signals that regulate epithelial growth and branching arise from lung mesenchyme, the mechanisms that regulate and pattern these signals are poorly understood. Similarly, essential signals that regulate lung mesenchyme have been identified in mesothelium and epithelium. However, the mechanisms by which extrinsic signals derived from mesothelium and epithelium regulate proliferation, differentiation and molecular patterning of lung mesenchyme are poorly understood.
Mesothelial FGF9 signals to sub-mesothelial mesenchyme to regulate lung mesenchymal growth
Previous work from our laboratory and others identified FGF9 as an essential factor that functions to regulate lung mesenchyme during early stages of lung development (Colvin et al., 2001a; Hyatt et al., 2002; Weaver et al., 2003; del Moral et al., 2006; White et al., 2006; De Langhe et al., 2008; Yin et al., 2008; Yi et al., 2009). As Fgf9 is expressed in both the mesothelium and epithelium, we hypothesized that these sources of FGF9 might have unique and specific regulatory functions. To identify the relative contributions of Fgf9 derived from mesothelium and epithelium, we conditionally inactivated the Fgf9 gene in these tissue compartments. Targeting Fgf9 in mesothelium with Dermo1-cre demonstrated that mesothelial-derived FGF9 is primarily responsible for regulating mesenchymal proliferation and has little influence on epithelial branching. By contrast, epithelial-derived Fgf9 (targeted with Shh-Cre) influences epithelial branching. Thus, these two sources of FGF9 must be considered independently when modeling mechanisms that regulate lung development.
In studies aimed at understanding signaling mechanisms that operate within lung mesenchyme, we have identified a feed-forward signaling network in which FGF9 regulates Wnt2a expression and mesenchymal WNT/β-catenin signaling, and in which mesenchymal WNT/β-catenin signaling is required for mesenchymal FGFR expression and mesenchymal responsiveness to FGF9 (Yin et al., 2008). Deficiencies of either pathway results in decreased mesenchymal proliferation.
The question of whether mesenchymal WNT/β-catenin signaling or FGF signaling regulates mesenchymal proliferation directly is difficult to test owing to the feed-forward relationship of these two signaling pathways in lung mesenchyme. However, examination of our data and that from several other studies collectively suggests that FGF9, but not mesenchymal WNT/β-catenin signaling, is the primary pathway that regulates mesenchymal proliferation. In wild-type explant cultures, activation of β-catenin signaling with either LiCl or BIO did not affect growth or morphology of the explant (Fig. 7A-C, see Fig. S3 in the supplementary material). Consistent with these observations, Rajogopal et al. showed that activation of β-catenin signaling in lung explants (with LiCl) activated Axin2 and Lef1 expression, yet did not affect the size or the morphology of the explant (Rajagopal et al., 2008). However, in the absence of mesenchymal FGF signaling (Fgf9–/– explants), activation of WNT signaling did support mesenchymal growth, but not beyond that of untreated wild-type explants (Fig. 7D-F,M, see Fig. S3 in the supplementary material). In contrast to these gain-of-function studies, morpholino knockdown of Wnt2a alone and of Wnt2a plus Wnt7b (Fig. 6) and knockout of Wnt2a (Shu et al., 2002) or Wnt7b (Shu et al., 2002; Rajagopal et al., 2008) resulted in decreased growth of lung mesenchyme. Given that mesenchymal WNT/β-catenin signaling is required for expression of FGFR1 and FGFR2 (De Langhe et al., 2008; Yin et al., 2008) (Fig. 6), we posit that mesenchymal WNT/β-catenin signaling has a primary permissive function for FGFR signaling and that FGFR signaling directly regulates mesenchymal proliferation and differentiation (Fig. 8).
Fig. 8.
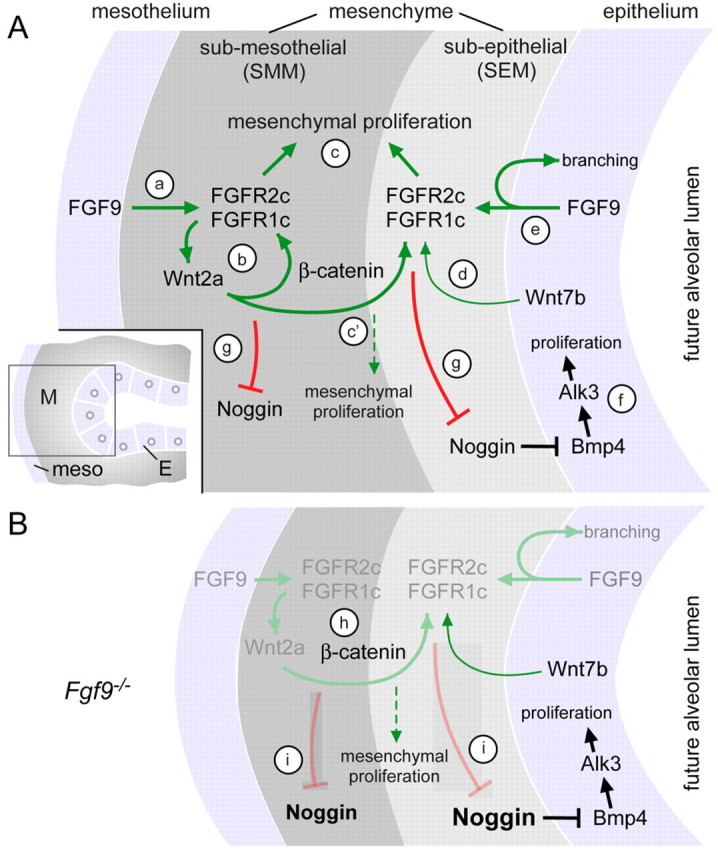
Regulatory mechanisms governing mesenchymal FGF-WNT/β-catenin signaling during early pseudoglandular stages of lung development. (A) (a) Mesothelial FGF9 signals to mesenchymal FGFR1C and FGFR2C to regulate mesenchymal proliferation and Wnt2a expression. (b) Mesenchymal β-catenin signaling is required for mesenchymal FGFR expression and mesenchymal response to FGF9. (c) FGFR signaling primarily regulates mesenchymal proliferation but requires WNT/β-catenin signaling to maintain FGFR expression. (c′) In the absence of mesenchymal FGFR signaling, WNT/β-catenin signaling has a limited capacity to induce mesenchymal expansion. (d) Wnt7b, expressed in airway epithelium, is partially redundant with Wnt2a for regulation of mesenchymal β-catenin signaling. (e) Epithelial FGF9 regulates branching either by direct autocrine activation of epithelial FGFRs or indirectly through regulation of mesenchymal FGF-WNT/β-catenin signaling and their downstream targets. (f) BMP4, primarily expressed in epithelium, acts as an autocrine factor to regulate epithelial proliferation and, secondarily, branching, through activation of BMPR1A (ALK3). (g) FGF-WNT/β-catenin signaling in the SEM and SMM negatively regulates Noggin expression. Inset: Diagram showing the region near the distal epithelial bud in which these signaling interactions occur. (B) When Fgf9 is absent, the FGF-WNT/β-catenin feed-forward signaling loop degenerates (gray) and only Wnt7b signaling remains. (h) In the absence of Fgf9, β-catenin is still present but Fgfr1 and Fgfr2 expression is lost. (i) Loss of mesenchymal FGF-WNT/β-catenin signaling results in increased Noggin expression in both the SEM and SMM. Noggin repression of epithelial BMP signaling is proposed to result in decreased epithelial proliferation. E, epithelium; M, mesenchyme.
This model posits that mesothelial FGF9 is a key factor for the regulation of lung growth during development and suggests that mechanisms that regulate the expression of mesothelial FGF9 will be crucial for lung development. Although the signals that regulate Fgf9 expression are not known, the possibility of feedback regulation by WNT2A signaling to the mesothelium is intriguing. Studies in endometrial carcinoma cells show that WNT signaling can regulate Fgf9 expression; however, this signal appears to be indirect (Hendrix et al., 2006). Other factors that could regulate mesothelial Fgf9 include biomechanical forces acting on mesothelial cells, other signaling molecules expressed in the SMM or signaling molecules expressed in the pleural fluid. Future studies will be required to determine the mechanisms that regulate Fgf9 expression in the mesothelium.
Epithelial FGF9 signals regulate lung epithelial branching
The primary consequence of inactivating Fgf9 in lung epithelium is decreased epithelial branching. The mechanism could include direct autocrine signaling of FGF9 to epithelium or indirect effects of epithelial FGF9 on adjacent mesenchyme. Accumulating evidence suggests that FGF9 can directly affect epithelial growth and branching. FGF9 can induce proliferation in isolated lung epithelium (del Moral et al., 2006) and FGF9 treatment of lung explants in which mesenchymal FGFRs have been inactivated (either directly with Dermo1-Cre-mediated targeting of Fgfr genes or indirectly, secondary to loss of mesenchymal β-catenin signaling) results in epithelial dilation in the absence of a mesenchymal response (Figs 6, 7) (White et al., 2006; Yin et al., 2008).
Indirect mechanisms by which epithelial FGF9 could regulate epithelial branching could be through the regulation of expression or activity of mesenchymal-expressed molecules that signal to epithelium. Candidate pathways include FGF10/FGFR2B and BMP4/ALK3. FGF10/FGFR2B signaling regulates epithelial branching (Morrisey and Hogan, 2010). However, the observation that Fgf10 expression in lung mesenchyme, and Spry2 and Etv4 expression in lung epithelium were not changed in Fgf9Shh lungs suggests that this pathway is not responsible for the observed decreased branching.
BMP4 regulates epithelial proliferation through signaling to BMPR1A (ALK3) (Eblaghie et al., 2006; Sun et al., 2008). The precise source of BMP4 is controversial. A recent study indicates that Bmp4 might only be expressed in lung epithelium (Jang et al., 2010) where it would function as an autocrine factor or as a paracrine signal to adjacent SEM, but other studies localize Bmp4 to both epithelium and SEM (Weaver et al., 2003). However, expression of the secreted BMP antagonist Noggin in the SEM provides a mechanism by which a mesenchymal-expressed factor can modulate epithelial BMP signaling. Our finding that mesenchymal FGF and WNT/β-catenin signaling functions to suppress Noggin and that Noggin is highly upregulated in mice lacking mesenchymal FGF or WNT/β-catenin signaling is consistent with previous studies in which FGF9 beads suppressed Noggin expression in explant cultures (Weaver et al., 2003) and in which NogginlacZ expression was increased in distal mesenchyme of Fgf9–/– lungs (Yi et al., 2009). Specific induction of Noggin in the SEM of Fgf9Shh lungs might explain the preferential effects on epithelium observed in this mutant. Future studies will be required to determine the extent to which mesenchymal FGF-WNT/β-catenin signaling suppression of Noggin affects epithelial development and which pathway(s) directly regulate Noggin.
Supplementary Material
Acknowledgments
We thank C. Smith for animal husbandry and genotyping. This work was funded by a grant from the March of Dimes Foundation, the Digestive Diseases Research Core Center Grant P30 DK052574 (transgenic mouse production), funds from the Department of Developmental Biology and a gift from Linda and Edward Ornitz.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.065110/-/DC1
References
- Bellusci S., Henderson R., Winnier G., Oikawa T., Hogan B. L. (1996). Evidence from normal expression and targeted misexpression that bone morphogenetic protein (Bmp-4) plays a role in mouse embryonic lung morphogenesis. Development 122, 1693-1702 [DOI] [PubMed] [Google Scholar]
- Bellusci S., Grindley J., Emoto H., Itoh N., Hogan B. L. (1997). Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 124, 4867-4878 [DOI] [PubMed] [Google Scholar]
- Chen C., Ouyang W., Grigura V., Zhou Q., Carnes K., Lim H., Zhao G. Q., Arber S., Kurpios N., Murphy T. L., et al. (2005). ERM is required for transcriptional control of the spermatogonial stem cell niche. Nature 436, 1030-1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin J. S., Feldman B., Nadeau J. H., Goldfarb M., Ornitz D. M. (1999). Genomic organization and embryonic expression of the mouse fibroblast growth factor 9 gene. Dev. Dyn. 216, 72-88 [DOI] [PubMed] [Google Scholar]
- Colvin J. S., White A., Pratt S. J., Ornitz D. M. (2001a). Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development 128, 2095-2106 [DOI] [PubMed] [Google Scholar]
- Colvin J. S., Green R. P., Schmahl J., Capel B., Ornitz D. M. (2001b). Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell 104, 875-889 [DOI] [PubMed] [Google Scholar]
- De Langhe S. P., Carraro G., Tefft D., Li C., Xu X., Chai Y., Minoo P., Hajihosseini M. K., Drouin J., Kaartinen V., et al. (2008). Formation and differentiation of multiple mesenchymal lineages during lung development is regulated by beta-catenin signaling. PLoS ONE 3, e1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C. H., Miller L. A., Smith A. N., Dufort D., Lang R. A., Niswander L. A. (2005). Canonical Wnt signaling negatively regulates branching morphogenesis of the lung and lacrimal gland. Dev. Biol. 286, 270-286 [DOI] [PubMed] [Google Scholar]
- del Moral P. M., De Langhe S. P., Sala F. G., Veltmaat J. M., Tefft D., Wang K., Warburton D., Bellusci S. (2006). Differential role of FGF9 on epithelium and mesenchyme in mouse embryonic lung. Dev. Biol. 293, 77-89 [DOI] [PubMed] [Google Scholar]
- Eblaghie M. C., Reedy M., Oliver T., Mishina Y., Hogan B. L. (2006). Evidence that autocrine signaling through Bmpr1a regulates the proliferation, survival and morphogenetic behavior of distal lung epithelial cells. Dev. Biol. 291, 67-82 [DOI] [PubMed] [Google Scholar]
- Goss A. M., Tian Y., Tsukiyama T., Cohen E. D., Zhou D., Lu M. M., Yamaguchi T. P., Morrisey E. E. (2009). Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev. Cell 17, 290-298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe B. D., Scherz P. J., Nissim S., Tian H., McMahon A. P., Tabin C. J. (2004). Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118, 517-528 [DOI] [PubMed] [Google Scholar]
- Harris K. S., Zhang Z., McManus M. T., Harfe B. D., Sun X. (2006). Dicer function is essential for lung epithelium morphogenesis. Proc. Natl. Acad. Sci. USA 103, 2208-2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix N. D., Wu R., Kuick R., Schwartz D. R., Fearon E. R., Cho K. R. (2006). Fibroblast growth factor 9 has oncogenic activity and is a downstream target of Wnt signaling in ovarian endometrioid adenocarcinomas. Cancer Res. 66, 1354-1362 [DOI] [PubMed] [Google Scholar]
- Hyatt B. A., Shangguan X., Shannon J. M. (2002). BMP4 modulates fibroblast growth factor-mediated induction of proximal and distal lung differentiation in mouse embryonic tracheal epithelium in mesenchyme-free culture. Dev. Dyn. 225, 153-165 [DOI] [PubMed] [Google Scholar]
- Jang C. W., Gao L., Dickinson M. E., Behringer R. R. (2010). Bmp4-directed nuclear cyan fluorescent protein provides a tool for live imaging and reveals cellular resolution of Bmp4 expression patterns during embryogenesis. Int. J. Dev. Biol. 54, 931-938 [DOI] [PubMed] [Google Scholar]
- Kratochwil K., Dull M., Fariñas I., Galceran J., Grosschedl R. (1996). Lef1 expression is activated by BMP-4 and regulates inductive tissue interactions in tooth and hair development. Genes Dev. 10, 1382-1394 [DOI] [PubMed] [Google Scholar]
- Levay-Young B. K., Navre M. (1992). Growth and developmental regulation of wnt-2 (irp) gene in mesenchymal cells of fetal lung. Am. J. Physiol. 262, L672-L683 [DOI] [PubMed] [Google Scholar]
- Lin Y., Liu A., Zhang S., Ruusunen T., Kreidberg J. A., Peltoketo H., Drummond I., Vainio S. (2001). Induction of ureter branching as a response to Wnt-2b signaling during early kidney organogenesis. Dev. Dyn. 222, 26-39 [DOI] [PubMed] [Google Scholar]
- Lin Y., Liu G., Wang F. (2006). Generation of an Fgf9 conditional null allele. Genesis 44, 150-154 [DOI] [PubMed] [Google Scholar]
- Mailleux A. A., Tefft D., Ndiaye D., Itoh N., Thiery J. P., Warburton D., Bellusci S. (2001). Evidence that SPROUTY2 functions as an inhibitor of mouse embryonic lung growth and morphogenesis. Mech. Dev. 102, 81-94 [DOI] [PubMed] [Google Scholar]
- Meijer L., Skaltsounis A. L., Magiatis P., Polychronopoulos P., Knockaert M., Leost M., Ryan X. P., Vonica C. A., Brivanlou A., Dajani R., et al. (2003). GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 10, 1255-1266 [DOI] [PubMed] [Google Scholar]
- Min H., Danilenko D. M., Scully S. A., Bolon B., Ring B. D., Tarpley J. E., DeRose M., Simonet W. S. (1998). Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev. 12, 3156-3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowada G., Jarvis L. A., Chi C. L., Neubuser A., Sun X., Hacohen N., Krasnow M. A., Martin G. R. (1999). Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development 126, 4465-4475 [DOI] [PubMed] [Google Scholar]
- Morrisey E. E., Hogan B. L. (2010). Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev. Cell 18, 8-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal J., Carroll T. J., Guseh J. S., Bores S. A., Blank L. J., Anderson W. J., Yu J., Zhou Q., McMahon A. P., Melton D. A. (2008). Wnt7b stimulates embryonic lung growth by coordinately increasing the replication of epithelium and mesenchyme. Development 135, 1625-1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine K., Ohuchi H., Fujiwara M., Yamasaki M., Yoshizawa T., Sato T., Yagishita N., Matsui D., Koga Y., Itoh N., et al. (1999). Fgf10 is essential for limb and lung formation. Nat. Genet. 21, 138-141 [DOI] [PubMed] [Google Scholar]
- Shu W., Jiang Y. Q., Lu M. M., Morrisey E. E. (2002). Wnt7b regulates mesenchymal proliferation and vascular development in the lung. Development 129, 4831-4842 [DOI] [PubMed] [Google Scholar]
- Sun J., Chen H., Chen C., Whitsett J. A., Mishina Y., Bringas P., Jr, Ma J. C., Warburton D., Shi W. (2008). Prenatal lung epithelial cell-specific abrogation of Alk3-bone morphogenetic protein signaling causes neonatal respiratory distress by disrupting distal airway formation. Am. J. Pathol. 172, 571-582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton D., El-Hashash A., Carraro G., Tiozzo C., Sala F., Rogers O., De Langhe S., Kemp P. J., Riccardi D., Torday J., et al. (2010). Lung organogenesis. Curr. Top. Dev. Biol. 90, 73-158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver M., Yingling J. M., Dunn N. R., Bellusci S., Hogan B. L. (1999). Bmp signaling regulates proximal-distal differentiation of endoderm in mouse lung development. Development 126, 4005-4015 [DOI] [PubMed] [Google Scholar]
- Weaver M., Dunn N. R., Hogan B. L. (2000). Bmp4 and Fgf10 play opposing roles during lung bud morphogenesis. Development 127, 2695-2704 [DOI] [PubMed] [Google Scholar]
- Weaver M., Batts L., Hogan B. L. (2003). Tissue interactions pattern the mesenchyme of the embryonic mouse lung. Dev. Biol. 258, 169-184 [DOI] [PubMed] [Google Scholar]
- White A. C., Xu J., Yin Y., Smith C., Schmid G., Ornitz D. M. (2006). FGF9 and SHH signaling coordinate lung growth and development through regulation of distinct mesenchymal domains. Development 133, 1507-1517 [DOI] [PubMed] [Google Scholar]
- White A. C., Lavine K. J., Ornitz D. M. (2007). FGF9 and SHH regulate mesenchymal Vegfa expression and development of the pulmonary capillary network. Development 134, 3743-3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi L., Domyan E. T., Lewandoski M., Sun X. (2009). Fibroblast growth factor 9 signaling inhibits airway smooth muscle differentiation in mouse lung. Dev. Dyn. 238, 123-137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y., White A. C., Huh S. H., Hilton M. J., Kanazawa H., Long F., Ornitz D. M. (2008). An FGF-WNT gene regulatory network controls lung mesenchyme development. Dev. Biol. 319, 426-436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K., Xu J., Liu Z., Sosic D., Shao J., Olson E. N., Towler D. A., Ornitz D. M. (2003). Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130, 3063-3074 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.