

Abstract
Many epidemiological studies show the benefit of fruits and vegetables on reducing risk of lung cancer, the leading cause of cancer death in the United States. Previously, we demonstrated that cigarette smoke exposure (SM)-induced lung lesions in ferrets were prevented by a combination of low dose of β-carotene, α-tocopherol (AT), and ascorbic acid (AA). However, the role of a combination of AT and AA alone in the protective effect on lung carcinogenesis remains to be examined. In the present study, we investigated whether the combined AT (equivalent to ∼100 mg/day in the human) and AA (equivalent to ∼210 mg/day) supplementation prevents against SM (equivalent to 1.5 packs of cigarettes/day) induced lung squamous metaplasia in ferrets. Ferrets were treated for 6 weeks in the following three groups (9 ferrets/group): (i) Control (no SM, no AT+AA), (ii) SM alone, and (iii) SM+AT+AA. Results showed that SM significantly decreased concentrations of retinoic acid, AT, and reduced form of AA, not total AA, retinol and retinyl palmitate, in the lungs of ferrets. Combined AT+AA treatment partially restored the lowered concentrations of AT, reduced AA and retinoic acid in the lungs of SM-exposed ferrets to the levels in the control group. Furthermore, the combined AT+AA supplementation prevented SM-induced squamous metaplasia [0 positive /9 total ferrets (0%) vs. 5/8 (62%); p <0.05] and cyclin D1 expression (p < 0.05) in the ferret lungs, in which both were positively correlated with expression of c-Jun expression. Although there were no significant differences in lung microsomal malondialdehyde (MDA) levels among the three groups, we found a positive correlation between MDA levels and cyclin D1, as well as c-Jun expressions in the lungs of ferrets. These data indicate that the combination of antioxidant AT+AA alone exerts protective effects against SM-induced lung lesions through inhibiting cyclin D1 expression and partially restoring retinoic acid levels to normal.
Keywords: Antioxidants, retinoic acid, cigarette smoke, lung carcinogenesis, cyclin D1
1. Introduction
Despite the fact that clinical intervention trials conducted to determine the chemoprotective effect of β-carotene as a potential chemopreventive agent on the incidence of lung cancer in smokers found either no protective effect or a harmful effect, supporting evidence for a protective role of fruits and vegetables rich in antioxidants in the prevention of lung cancer continues to be reported in human epidemiological studies, as well as in mechanistic studies using cell culture and animal models [1]. Within the past years, we have gained greater knowledge of the biological effects of carotenoids, particularly the impact of oxidation on these carotenoids and the potential for beneficial effects of small quantities or harmful effects of large quantities of the resulting metabolic products. The dosages of β-carotene used in the ATBC (Alpha-Tocophoral, Beta-Carotene Cancer Prevention Study) [2, 3] and CARET (Beta-Carotene and Retinol Efficacy Trial) [4, 5] studies were 20 to 30 mg per day for 2–8 years, and these doses are 10- to 15- fold higher than the average intake of β-carotene in a typical American diet (2 mg per day). Such a pharmacological dose of β-carotene in humans could result in the accumulation of relatively high levels of β-carotene and its oxidative metabolites in lung tissue, especially after long periods of supplementation, which could lead to a decrease in lung retinoic acid concentration via induction of cytochrome p450 (CYP) enzymes [6, 7].
The low levels of retinoic acid could interfere with retinoid signal transduction and result in enhanced cell proliferation and potentially malignant transformation [8]. Our previous studies in ferrets (Mustela putorius furo), an excellent model for studying carotenoid absorption and metabolism, showed that cigarette smoke exposure decreased levels of retinoic acid and retinoic acid receptor β (RARβ) protein, but increased levels of c-Jun and cyclin D1 proteins, and induced precancerous lesions in lung tissue [6, 9]. Recently, we reported that a physiologic dose of β-carotene (equivalent to 6.7 mg/day in humans) in the presence of α-tocopherol (AT) and ascorbic acid (AA) protected against smoke-induced lung squamous metaplasia [10], but a physiologic dose of β-carotene alone provided a weak protection [9]. These results revealed that low dose of β-carotene in the presence of AT and AA prevented lung tissues from detrimental effects of heavy smoking. In addition, we demonstrated that AT and AA supplementation increased lung concentrations of retinoic acid in the presence of β-carotene supplementation [10]. It has been seen in the cases of cigarette smoking and excessive alcohol drinking, resulting in higher cytochrome P450 enzymes and breakdown of retinoic acid [11, 12]. Therefore, these studies raised two important questions: 1) whether a combination of AT and AA alone has the protective effect against smoke-induced lung lesions; and 2) whether the increased levels of lung retinoic acid with AT and AA supplementation could be due to their own action in the absence of β-carotene as a precursor. In the present study, we explored the protective effect of a combination of AT and AA on smoke-induced lung squamous metaplasia. We examined the expressions of several biomarkers that are involved in lung carcinogenesis such as proliferating cellular nuclear antigen (PCNA), cyclin D1, c-Jun, and c-Fos. In addition, we measured lung levels of retinoids and malondialdehyde (MDA) as a marker for lipid peroxidation to determine whether or not supplementation with combined antioxidant vitamins can modify both retinoid and MDA levels in smoke-exposed lungs of ferrets.
2. Materials and Methods
2.1. Animals, diet and study groups
The animals and diet used for this study have been described elsewhere [10]. Briefly, twenty-seven male adult ferrets from Marshall Farms (North Rose, NY) were randomly assigned to 3 groups (n =9 in each group) for a 6-week experiment as follows: 1) control; 2) smoke exposure (SM); and 3) smoke exposure plus AT and AA (SM+AT+AA). The ferrets were fed a semi-purified ferret diet (Research Diets, Inc. New Brunswick, NJ) and had access to food and water ad libitum. Body weights were recorded weekly.
2.2. AT and AA supplementation and cigarette smoke exposure
AT and AA supplementation and smoke exposure procedure have been described elsewhere [10]. All-rac-α-tocopherol acetate (AT) (F. Hoffman-La Roche Ltd, Basel, Switzerland) was dissolved in corn oil and fed orally every morning to the supplemented ferrets at a dose of 22 mg AT/kg body wt/day, which is equivalent to 100 mg AT/day in a 70-kg man based on the bioavailability of AT in a ferret being about 6% of that in a human [13]. Ascorbic acid (AA) (Roche Vitamins, Inc., NJ) was freshly prepared by dissolving it in distilled water and fed orally to the supplemented ferrets every morning at a dose of 3 mg AA/kg body wt/day. This amount of AA in the ferret is equivalent to 210 mg AA in a 70-kg man. The rationale for using these doses has been described elsewhere [10]. The animals without vitamin supplements received an equivalent amount of corn oil. The amount of cigarettes exposure in the ferret gave a similar concentration of urinary cotinine (∼12 μg/mL) equivalents as that found in humans smoking one and a half packs of cigarettes per day [6]. Control ferrets were housed in a separate room and underwent the same procedures as the smoke-exposed animals, except for smoke exposure.
2.3. Plasma and lung tissue extraction and HPLC analysis
Plasma and lung tissue extraction was conducted as described previously [10]. AT, total AA, reduced AA and retinoids (retinyl palmitate, retinol and retinoic acid) in lung tissue homogenates were measured by HPLC and UV detection, as described before [10].
2.4. Histopathology and immunohistochemistry
The histopathology examination procedure has been described elsewhere [10]. Both single- and double-labeling immunoperoxidase staining techniques were performed according to the manufacturer’s instructions (Vectastain® ABC Kit; Vector Laboratories, Inc., Burlingame, CA). Lung squamous metaplasia lesions were confirmed by immunoperoxidase staining with an antibody for cytokeratin polypeptide 19 (mouse monoclonal anti-cytokeratin 4.62, Sigma-Aldrich, Inc.). The anti-cytokeratin 4.62 antibody was used at a dilution of 1:20 at 37ºC for 45 minutes. For PCNA and RARβ staining, the anti-PCNA antibody and anti-RARβ antibody (PC-10 and C-19, respectively, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) were used at dilutions of 1:100 and 1:50, respectively, at 37ºC for 45 minutes. The secondary biotinylated antibody, species-correspondent to each primary antibody, was used at a dilution of 1:100 at room temperature for 15 minutes. The Vector® NovaRED™, giving the red color, was the substrate for cytokeratin polypeptide 19 staining. For PCNA and RARβ staining, 3,3′-diaminobenzidine (DAB) plus nickel was the substrate, giving a black color. Slides were counterstained with methyl green unless described otherwise. Sections were also performed as negative controls with corresponding non-immune serum.
2.5. Nuclear and microsomal fractions extraction and western blot analysis
Lung nuclear and microsomal extraction and western blot analysis were prepared as described previously [9, 14]. All primary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The dilutions of each antibody were as follows: PCNA (PC-10) 1:200; RARβ (C-19) 1:5000; c-Jun (D-11) 1:100; Cyclin D1 (HD 11) 1:100; c-Fos (4) 1:1000.
2.6. Lung malondialdehyde (MDA) analysis
A microsomal aliquot from lungs was suspended in 0.1 M PBS, pH 7.4 at a concentration of 1 mg protein/mL of suspension. An aliquot of 100 μL of microsomal suspension was mixed with 10 μL of 5% ethanolic butylated hydroxytoluene (BHT) and 1 mL thiobarbituric acid solution [0.4% thiobarbituric acid (TBA) in 0.2 M sodium acetate buffer, pH 3.5]. The mixture was heated at 95ºC for 45 minutes, then cooled down on ice for 5 minutes and centrifuged at 14,000 rpm for 10 minutes. The supernatant was passed through a 0.45 μm filter using a syringe, and 50 μL of filtered sample was injected into the HPLC. The thiobarbituric acid-malondialdehyde (TBA-MDA) adduct was separated by a isocratic reverse-phase HPLC system, consisting a Waters 600 S controller, a Waters 717 plus autosampler (Waters Corporate, Milford, MA), and a 0.46- × 8.3-cm Pecosphere-3 C18 cartridge column (Perkin-Elmer Analytical Instruments, Shelton, CT), and a fluorescence spectrophotometer (Hitachi F-1050). The excitation and emission wavelengths were 515 and 553 nm, respectively. The HPLC mobile phase consisted of 20% acetonitrile and 80% sodium phosphate buffer (20 mM, pH 7.0) at a flow rate of 0.8 mL/min, and running time was 7 minutes. This procedure gave a good precision (coefficient of variation within assay was 2.2% and that between assay was 8%), a good linearity (regression coefficient was 0.998), a high sensitivity (limit of detection and limit of quantitation were 0.1 and 0.25 pmol/injection, respectively); and the accuracy of plasma and tissue recovery was 101 ± 2.3% and 98 ± 2.4%, respectively.
2.7. Statistical analysis
Results are expressed as means ± SE unless described otherwise. One-way ANOVA and Dunnett t-test were used to compare control groups with vitamin groups (with or without a combination of AT and AA). Student’s t-test was performed to compare variables in the presence and absence of AT plus AA, respectively. The Pearson and Spearman correlations were used to analyze the correlations among parametric and nonparametric variables, respectively. A statistically significant difference was determined when the two-sided p-value was less than 0.05.
3. Results
3.1. Plasma and lung concentration of AT, AA, and retinoids in the ferrets after 6 weeks of tobacco smoke exposure
Plasma concentration of AT was not changed under smoke-exposure, but significantly increased by AT+AA supplementation, as compared with the control. Total AA concentration in plasma was not significantly different among the treatments groups (Table 1). In the lung, AT concentration was significantly decreased by smoke-exposure, but it was restored totally by AT+AA supplementation to normal level as the control group. The total AA concentration was not changed by either smoke exposure or AT+AA supplementation, however, the reduced AA was significantly decreased by smoke-exposure and restored partially to control level by AT+AA supplementation (Table 1). Compared to the control group, lung retinoic acid concentration was significantly lower in the smoke exposed alone group, but not in the smoke exposed group with AT+AA (Table 1). The levels of other retinoids including retinol and retinyl palmitate were not changed by treatments.
Table 1.
Plasma and lung concentrations of α-tocopherol (AT), ascorbic acid (AA), retinoic acid, retinol, and retinyl palmitate in ferrets from various treatment groups after 6 weeks of tobacco smoke exposure (SM) and with or without AT+AA supplementation.
| Control (n=9) | SM (n=9) | SM+AT+AA (n=9) | |
|---|---|---|---|
| Plasma | |||
| AT (μmol/L) | 17.3 ± 1.2 | 16.9 ± 1.5 | 50.0 ± 3.2* |
| Total AA (μmol/L) | 97.7 ± 6.4 | 99.8 ± 3.8 | 102.4 ± 7.0 |
| Retinoic acid (nmol/L) | 11.23 ± 0.81 | 9.60 ± 0.64 | 11.05 ± 1.11 |
| Retinol (μmol/L) | 1.69 ± 0.15 | 1.69 ± 0.09 | 1.78 ± 0.12 |
| Retinyl palmitate (μmol/L) | 4.32 ± 0.79 | 4.09 ± 0.58 | 4.52 ± 0.65 |
|
| |||
| Lung tissue | |||
| AT (nmol/g tissue) | 67.1 ± 17.2 | 43.5 ± 9.2* | 64.1 ± 4.7 |
| AA (μmol/g tissue) | |||
| Total | 1.84 ± 0.07 | 1.61 ± 0.07 | 1.71 ± 0.07 |
| Reduced | 1.34 ± 0.07 | 1.07 ± 0.08* | 1.19 ± 0.06 |
| Retinoic acid (pmol/g tissue) | 24.17 ± 3.89 | 10.52 ± 0.83* | 13.80 ± 1.80 |
| Retinol (nmol/g tissue) | 0.64 ± 0.03 | 0.56 ± 0.03 | 0.58 ± 0.10 |
| Retinyl palmitate (nmol/g tissue) | 12.21 ± 2.23 | 10.01 ± 1.86 | 8.06 ± 1.15 |
Values are mean ± SE. One-way ANOVA and Dunnett t-test were used to compare groups with the control group. Significantly different at * P < 0. 05. Abbreviations: SM; smoke-exposure; AA, ascorbic acid; AT, α-tocopherol.
3.2. Immunoperoxidase staining with antibodies for cytokeratin polypeptide 19, proliferating cell nuclear antigen (PCNA), and RARβ in the lungs of ferrets
There were no any squamous metaplasia detected in the lungs of control ferrets or combined AT+AA supplemented ferrets. However, smoke-exposure induced squamous metaplasia in the lungs of five out of eight ferrets, which reached a significant difference among three groups (0/9 (Control) vs. 5/8 (SM) vs. 0/9 (SM+AT+AA), P<0.05). Squamous metaplastic lesions showed more intense PCNA staining compared to normal tissue (Figs. 1A–1D). Staining the same lung section with anti-PCNA and anti-cytokeratin polypeptide 19 antibodies, both proteins were more expressed in the squamous metaplastic lesions than in either the adjacent or the normal regions (Figs. 1E, 1F). To the contrary, the expression of RARβ in the squamous metaplastic lesions was weaker than that in the normal lung section; almost entire areas of squamous metaplasia had no DAB-nickel stain for RARβ (Figs. 2C, 2D). In addition, bronchiolar epithelial cells adjacent to areas having squamous metaplasia had weaker expression of RARβ than those in the normal lung sections (Figs. 2E, 2F).
Fig. 1.
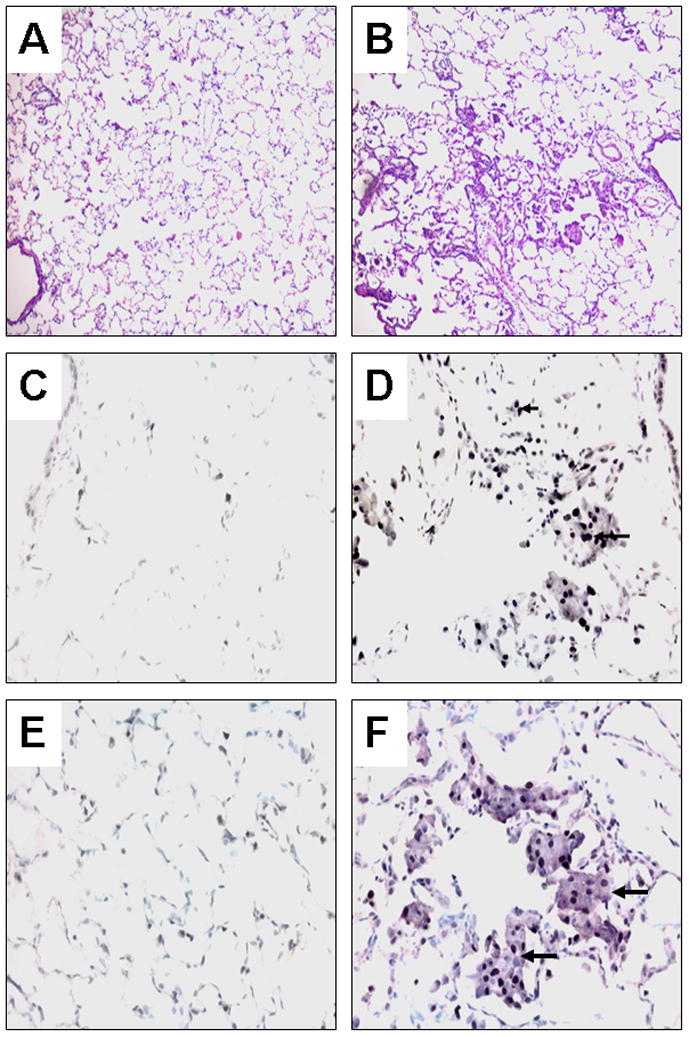
Histological examination and immunoperoxidase staining of ferret lung sections. Left panel, normal lung; Right panel, squamous metaplasia induced by tobacco smoke after 6 weeks of exposure; Hematoxylin-eosin staining at low magnification x 100 (A, B), Immunoperoxidase staining with anti-proliferating cell nuclear antigen (PCNA) antibody using 3, 3′-diaminobenzidine (DAB) plus nickel as the substrate (giving the black color) and a methyl green counterstain at high magnification x 400 (C, D), Double labeling immunoperoxidase staining with anti-cytokeratin polypeptide 19 (Vector®NovaRed as the substrate, giving the reddish-purple color) and anti-PCNA antibodies (DAB plus nickel as the substrate) and a methyl green counterstain (E, F). Arrowheads depict nuclei stained with anti-PCNA antibody. Arrows depict the cytoplasm of a cluster of cells stained with anti-cytokeratin polypeptide 19 antibody.
Fig. 2. Double-labeling immunoperoxidase staining of ferret lung sections with antibodies for cytokeratin polypeptide 19 (Vector®NovaRed as the substrate) and retinoic acid receptor (RAR)β (DAB plus nickel as the substrate) with methyl green counterstain.
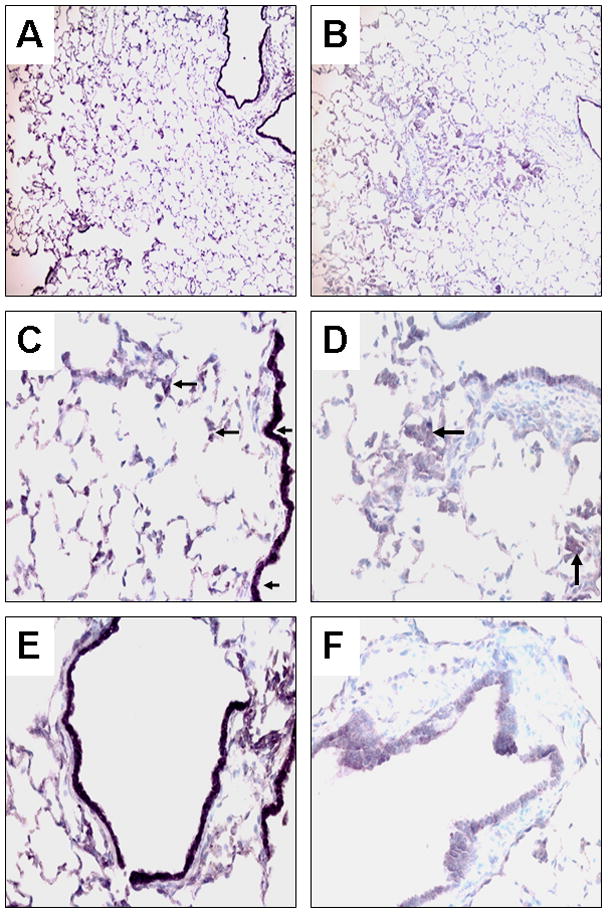
The left panel represents normal lung sections and the right panel represents lung squamous metaplasia and adjacent bronchioles after 6 weeks of smoke exposure at low magnification x 100 (A, B) and high magnification x 400 (C –F). Arrowheads point to representative nuclei expressing RARβ (stained in black). Arrows point representative areas expressing cytokeratin polypeptide 19 (stained in reddish-purple).
3.3 Western blot of nuclear protein for Cyclin D1, c-Jun, PCNA, and RARβ expressions in the lung of ferrets
PCNA expression at the protein level showed no significant difference among treatment groups (Figs. 3A, 3B); however, AT and AA supplementation slightly (but not significantly) decreased PCNA levels compared with corresponding groups without AT and AA supplementation (p = 0.053). We were unable to confirm the difference of PCNA expression shown in the squamous metaplastic lesions by Western blot, because the squamous metaplastic lesions are localized and spotty; thus PCNA protein levels may be diluted when the nuclear fractions of the tissue are used for the Western blot. Comparison of PCNA protein levels between individual ferrets having lung squamous metaplastic lesions and those not having lesions also showed no significant difference (data not shown). RARβ protein expression in lung nuclear fractions revealed no significant difference among treatment groups (Figs. 3A, 3C).
Fig. 3. The expression of cyclin D1, c-Jun, PCNA, and RARβ in lung nuclear fractions of ferrets after 6 weeks of smoke exposure and with or without AT+AA supplementation.
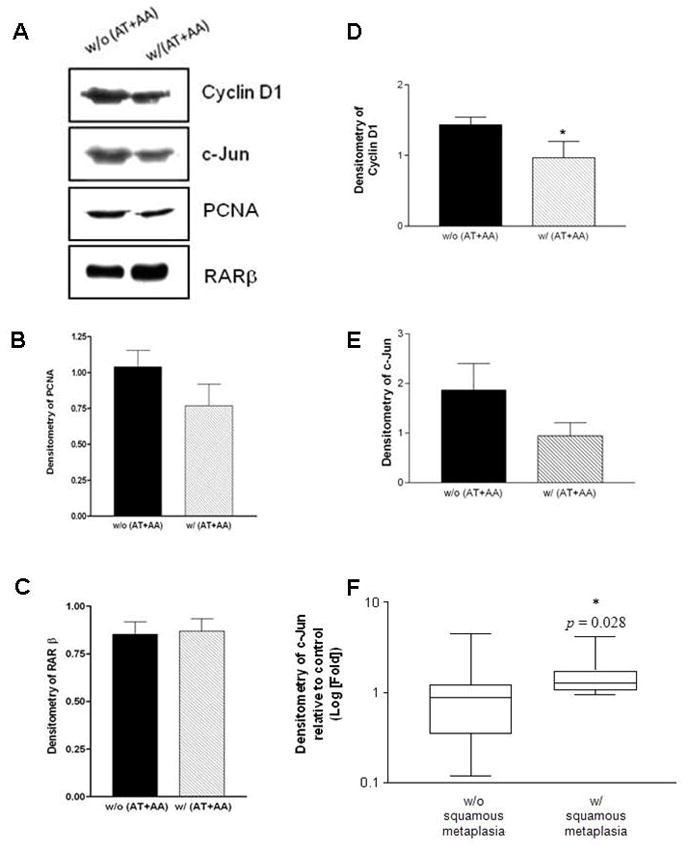
(A) Representative Western blot of cyclin D1, c-Jun, PCNA, and RARβ expression in nuclear fractions of ferret lungs after 6 weeks of smoke exposure and with or without AT+AA supplementation. (B-E) The densitometry of PCNA, RARβ, cyclin D1, and c-Jun. The asterisk (*) denotes a significant difference in cyclin D1 expression between the groups with AT and AA supplement compared to those without AT and AA supplementation (p < 0.05, Student’s t test); (F) The association of lung squamous metaplasia and c-Jun protein expression. The original unit of densitometry is transformed into the logarithm to the base 10 to obtain a normal distribution. The asterisk (*) denotes a significant difference in c-Jun expression between ferrets having squamous metaplasia and those not having squamous metaplasia (p < 0.05, Student’s t test). Abbreviations: AA, ascorbic acid; AT, α-tocopherol.
The expression of cyclin D1 in ferret lung nuclear fractions was significantly reduced by the AT and AA supplementation (p < 0.05), as compared with that of the smoke-exposed ferrets (Figs. 3A, 3D). There was a trend of a decreased c-Jun expression in the presence of AT and AA supplementation, but it was not statistically significant (p = 0.07) due to between animal variation (Figs. 3A, 3E). However, correlating lung squamous metaplasia with c-Jun expression, ferrets having the lesions had higher c-Jun levels than those not having the lesions (p < 0.05, Fig. 3F). Along with c-Jun expression, the c-Fos expression was not different among treatment groups (data not shown).
3.4. Associations among levels of AT, retinoic acid, squamous metaplasia, and c-Jun and cyclin D1 expression in smoke-exposed lungs
The concentration of AT in smoke-exposed lungs having squamous metaplasia was significantly lower than that in smoke-exposed lungs not having squamous metaplasia (Fig. 4A, p < 0.05 after transformation). In addition to a trend of decreased c-Jun expression in ferrets supplemented with AT and AA (Fig. 3E), there was a significantly inverse correlation between c-Jun expression and AT concentrations in the smoke-exposed lungs (correlation coefficient = −0.385, p-value = 0.019, Fig. 4B), but not between c-Jun expression and lung reduced ascorbic acid concentrations (data not shown). Furthermore, there was a strong correlation between c-Jun and cyclin D1 expressions in the smoke-exposed lungs (correlation coefficient = 0.886, p-value < 0.001, Fig. 4C). As shown in Fig. 4D, c-Jun expression was lower in the groups that had higher level of retinoic acid and/or AT although no statistical evidence of a correlation between the retinoic acid levels and c-Jun expression was found.
Fig. 4. Associations between retinoic acid and α-tocopherol (AT) levels, squamous metaplasia, and c-Jun and cyclin D1 expressions in the lungs of ferrets after 6 weeks of smoke exposure and with or without AT+AA supplementation.
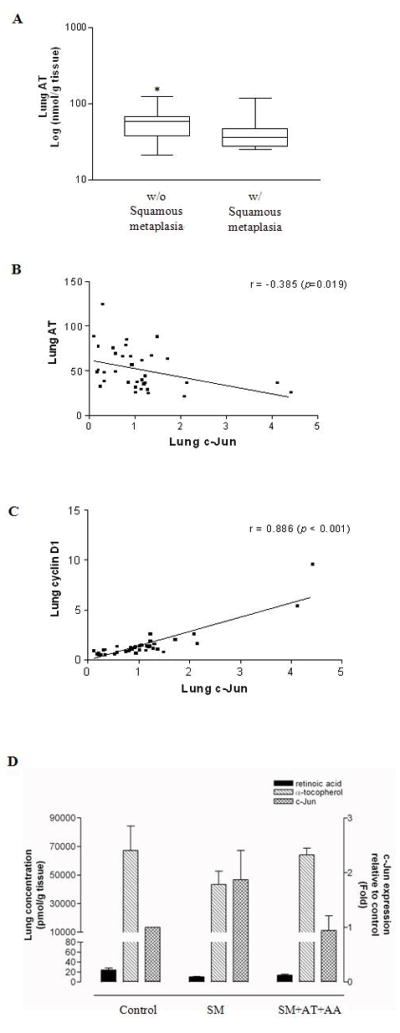
(A) α-Tocopherol levels by squamous metaplasia, *Significant difference between smoke-exposed lungs without vs. those with squamous metaplasia (p = 0.036 after transformation, Student’s t test); (B) Pair-wise scatter plot for the correlation between c-Jun expression and α-tocopherol levels in smoke-exposed lungs; (C) Pair-wise scatter plot for the correlation between c-Jun and cyclin D1 expressions in smoke-exposed lungs; (D) Levels of retinoic acid, α-tocopherol, and c-Jun protein expression in the lungs of ferrets from the various treatment groups. Abbreviations: same as in Figure 3, r = correlation coefficient.
3.5. MDA levels in lung microsomal fractions and association between MDA levels and c-Jun as well as cyclin D1 expression
In the lung of ferrets after 6-week smoke exposure, no significant differences in MDA levels were found between with and without AT+AA group (Fig. 5A). Nevertheless, there was a significant correlation between lung MDA and c-Jun (correlation coefficient = 0.466, p-value = 0.004) as well as cyclin D1 expression (correlation coefficient = 0.422, p-value = 0.006) (Figs. 5B, 5C, respectively).
Fig. 5.
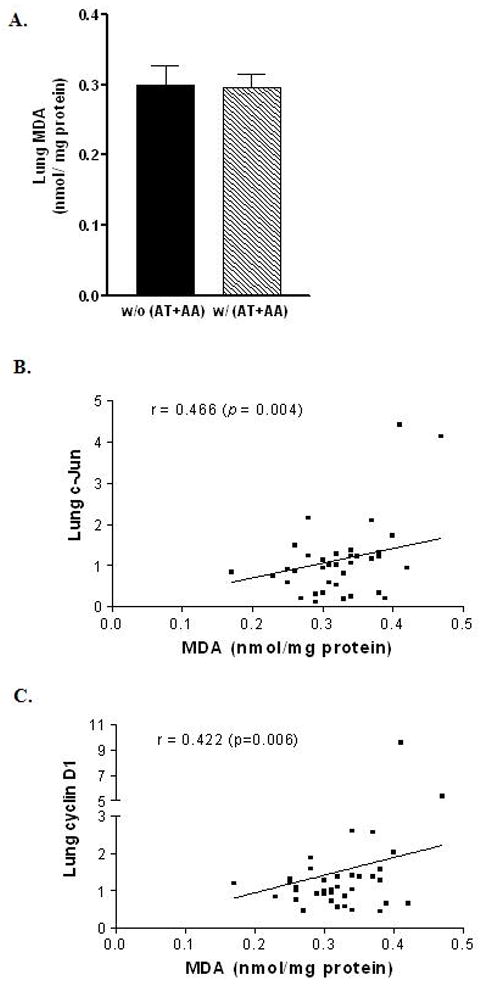
Malondialdehyde (MDA) levels in the lungs of ferrets after 6 weeks of smoke exposure and with or without AT+AA supplementation (A) Among treatment groups; (B) Pair-wise scatter plot for the correlation between lung MDA and c-Jun expression in smoke-exposed ferrets; (C) Pairwise scatter plot for the correlation between lung MDA and cyclin D1 expression in smoke-exposed ferrets; Abbreviations: same as in Figure 3. r = correlation coefficient.
4. Discussion
The present study demonstrates that a combination of antioxidant AT (at a dose equivalent to 100 mg/day in a human) and AA (at a dose equivalent to 210 mg/day in a human) protects against smoke-induced lung squamous metaplasia. In particular, the lowering protein expression of cyclin D1 by AT+AA supplementation was consistent with lung squamous metaplasia, suggesting that the mechanism could be mainly through inhibiting cyclin D1 expression in this study model. The presence of AT and AA had the effect on cyclin D1, which serves as a key sensor and integrator of extracellular signals of cells in G0 and early G1 phases and mediates its function through binding with Cdk4/6. Aberrant cyclin D1 expression is a critical event in lung cancer development, and it can be induced by many oncogenic signals [15]. Using immunohistochemistry, Caputi et al. [16] showed that cyclin D1 expression increased in human lung cancer tissues and that PCNA expression was positively associated with cyclin D1 protein expression. Emergence of cyclin D1 as a molecular pharmacologic target and biological marker for clinical response is based on experience of the proof of principle trials [17]. In our study, there was a strong correlation between c-Jun and cyclin D1 expression in the lung. Since c-Jun is involved in cell cycle progression and carcinogenesis via transcriptional activation of cyclin D1, the AT and AA treatment may inhibit smoke-induced abnormal cell proliferation by blocking c-Jun as well as cyclin D1 overexpression.
Interestingly, we found that levels of lung retinoic acid in ferrets exposed to smoke in the presence of AT and AA were slightly higher than the smoke-exposed alone group and did not differ with that of the control ferrets. It has been shown that retinoic acid regulates the growth and differentiation of bronchial epithelial cells in vitro [18–20] and suppresses lung carcinogenesis in animal studies [21, 22] and human trials [23]. Lee and colleagues demonstrated in vitro that retinoic acid inhibited Jun N-terminal kinase (JNK), which phosphorylates and thus activates c-Jun, and that the inhibitory effect of retinoic acid on JNK activity was through the induction of mitogen-activated protein (MAP) kinase phosphatase 1 (MKP-1) [24, 25]. Moreover, we found that the induction of MAP-1 was associated with the concentration of retinoic acid in the lungs of ferrets [7]. Our study indicates that the restoration of normal levels of retinoic acid by AT and AA supplementation in smoke-exposed lung tissue may contribute to the protective effects of AT and AA against smoke-induced cyclin D1 expression and lung squamous metaplasia. This notion was supported by our previous studies showing that the combined β-carotene, α-tocopherol, and ascorbic acid supplementation [10], not βcarotene alone [9] protected against various preneoplastic lesions, including squamous metaplasia, dysplasia, and atypical adenomatous hyperplasia, in the lungs of ferrets exposed to smoke-exposure. Particularly, loss of RARβ expression by epigenetic silencing due to genomic methylation or histone deacetylation can result in clinical resistance to classical retinoids in lung cancer [26, 27]. Hypermethylation of RARβ was found in heavy smokers [28], and the aberrant methylation status at the promoter of RARβ can be reversed by retinoic acids [29]. Therefore, it is possible that the combination of antioxidants (α-tocopherol and ascorbic acid) partially recover cigarette smoke-reduced level of retinoic acid, thereby regulating epigenetic of RARβ through the maintenance of retinoic acid. Clearly, this hypothesis warrants further investigation.
In the present study, the levels of AT in ferret lungs were increased greatly by supplementation of combined antioxidant vitamins, although the levels of reduced form of ascorbic acid was only slightly increased. Therefore, we cannot exclude the possibility that AT and AA inhibit cyclin D1 expression and smoke-induced metaplasia in the lungs of ferrets. This notion was supported by that lung AT levels in smoke-exposed lungs having squamous metaplasia were much lower than those not having squamous metaplasia. In addition, there was a significant inverse correlation between c-Jun expression and lung AT concentration. Chow et al. [30] measured levels of mitochondrial hydrogen peroxide in female B6C3F1 hybrid mice fed a basal AT-deficient diet with and without AT supplementation for 15 weeks, and found that AT decreased mitochondrial hydrogen peroxide in a dose-dependent manner. This effect of AT may explain the finding of our present study that a trend of decreased c-Jun expression in the presence of AT and AA, since hydrogen peroxide can induce c-Jun expression probably through the generation of hydroxyl radical [31, 32].
By double-labeling immunoperoxidase staining in lung sections, we found that PCNA protein was expressed more intensely in lung squamous metaplastic lesions than in normal areas (Figs. 1C–1D), indicating that most of the cells in the squamous metaplastic lesions were proliferating. PCNA is a nonhistone nuclear protein essential for DNA replication as well as being required for DNA recombination and repair, and the peak expression is found in the S-phase of cell cycle; therefore it is one of the most widely used markers for cell proliferation [33, 34].
Unlike our previous study demonstrating an increase of PCNA expression using Western blot analysis in lung nuclear fractions of ferrets exposed to smoke for 6 months, no evidence of a difference in PCNA expression among treatment groups was found in this study of smoke exposure for 6 weeks (Fig. 3A, 3B). Since the smoke exposure period in this study was much shorter than that in our earlier studies [6, 9], PCNA protein expression induced after 6 weeks of smoke exposure might not be strong enough to manifest a statistical difference by Western blot, suggesting a temporal association between the development of squamous metaplasia and the change of molecular markers. In addition, the squamous metaplastic lesions were spotty and localized, thus the PCNA protein levels may be diluted since normal lung areas and squamous metaplasia lesions were mixed during the preparation of nuclear fractions.
In the present study, although we found a correlation between MDA levels and c-Jun as well as cyclin D1 expression in lungs of ferrets, we did not find a difference in lung microsomal MDA levels among treatment groups of ferrets. Our study suggests that AT and AA inhibit the expressions of c-Jun and cyclin D1, independent of its antioxidant effect. This notion was supported by a study of Yano et al [35]. Those investigators reported a study in A/J mice with a single intraperitoneal injection of urethane, which promotes lung tumorigenesis, followed by a single oral administration of either corn oil, AT, or α-tocopheryloxybutyric acid (TSE, an ether derivative of AT that has no in vivo antioxidant effect). They demonstrated that both AT and TSE decreased the PCNA labeling index, and more interestingly, that both AT and TSE diminished the activity of urethane-induced extracellular signal-regulated kinase (ERK) and other members of the ERK cascade including MAPK/ERK kinase (MEK). MEK is an intermediate in the MAP kinase cascades and can activate JNK [36, 37]. Therefore, the expression of c-Jun was associated with the occurrence of lung squamous metaplasia, and was associated with levels of lung AT and retinoic acid. Clearly, the action of AT, independent of its antioxidant effect, needs further investigation.
Acknowledgments
The study was supported by the NIH grant R01CA104932, and U.S. Department of Agriculture, under agreement NO. 1950-51000-064. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the authors and do not necessarily reflect the views of the NIH and the US Department of Agriculture.
The abbreviations used are
- AP-1
activator protein-1
- AT
αtocopherol
- AA
ascorbic acid
- ERK
extracellular signal-regulated kinase
- JNK
Jun N-terminal kinase
- MAP
mitogen-activated protein
- PCNA
proliferating cellular nuclear antigen
- MDA
malondialdehyde
- MEK
mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- MAPK/ERK
mitogen-activated kinases/extracellular signal-regulated kinases
- MKP-1
MAP kinase phosphatase-1
- RAR
retinoic acid receptor
- ROS
reactive oxygen species
- Rb
retinoblastoma protein
- TGF
tumor growth factor
- TSE
α-tocopheryloxybutyric acid
Footnotes
Conflict of Interest Statement
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Veeramachaneni S, Wang XD. Carotenoids and lung cancer prevention. Front Biosci (Schol Ed) 2009;1:258–74. doi: 10.2741/S25. [DOI] [PubMed] [Google Scholar]
- 2.The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. The Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. N Engl J Med. 1994;330:1029–35. doi: 10.1056/NEJM199404143301501. [DOI] [PubMed] [Google Scholar]
- 3.Albanes D, Heinonen OP, Taylor PR, Virtamo J, Edwards BK, Rautalahti M, Hartman AM, Palmgren J, Freedman LS, Haapakoski J, Barrett MJ, Pietinen P, Malila N, Tala E, Liippo K, Salomaa ER, Tangrea JA, Teppo L, Askin FB, Taskinen E, Erozan Y, Greenwald P, Huttunen JK. Alpha-Tocopherol and beta-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: effects of base-line characteristics and study compliance. J Natl Cancer Inst. 1996;88:1560–70. doi: 10.1093/jnci/88.21.1560. [DOI] [PubMed] [Google Scholar]
- 4.Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, Keogh JP, Meyskens FL, Jr, Valanis B, Williams JH, Jr, Barnhart S, Cherniack MG, Brodkin CA, Hammar S. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. J Natl Cancer Inst. 1996;88:1550–9. doi: 10.1093/jnci/88.21.1550. [DOI] [PubMed] [Google Scholar]
- 5.Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, Keogh JP, Meyskens FL, Valanis B, Williams JH, Barnhart S, Hammar S. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334:1150–5. doi: 10.1056/NEJM199605023341802. [DOI] [PubMed] [Google Scholar]
- 6.Wang XD, Liu C, Bronson RT, Smith DE, Krinsky NI, Russell M. Retinoid signaling and activator protein-1 expression in ferrets given beta-carotene supplements and exposed to tobacco smoke. J Natl Cancer Inst. 1999;91:60–6. doi: 10.1093/jnci/91.1.60. [DOI] [PubMed] [Google Scholar]
- 7.Liu C, Russell RM, Wang XD. Low dose beta-carotene supplementation of ferrets attenuates smoke-induced lung phosphorylation of JNK, p38 MAPK, and p53 proteins. J Nutr. 2004;134:2705–10. doi: 10.1093/jn/134.10.2705. [DOI] [PubMed] [Google Scholar]
- 8.Bogos K, Renyi-Vamos F, Kovacs G, Tovari J, Dome B. Role of retinoic receptors in lung carcinogenesis. J Exp Clin Cancer Res. 2008;27:18. doi: 10.1186/1756-9966-27-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu C, Wang XD, Bronson RT, Smith DE, Krinsky NI, Russell RM. Effects of physiological versus pharmacological beta-carotene supplementation on cell proliferation and histopathological changes in the lungs of cigarette smoke-exposed ferrets. Carcinogenesis. 2000;21:2245–53. doi: 10.1093/carcin/21.12.2245. [DOI] [PubMed] [Google Scholar]
- 10.Kim Y, Chongviriyaphan N, Liu C, Russell RM, Wang XD. Combined antioxidant (beta-carotene, alpha-tocopherol and ascorbic acid) supplementation increases the levels of lung retinoic acid and inhibits the activation of mitogen-activated protein kinase in the ferret lung cancer model. Carcinogenesis. 2006;27:1410–9. doi: 10.1093/carcin/bgi340. [DOI] [PubMed] [Google Scholar]
- 11.Liu C, Russell RM, Seitz HK, Wang XD. Ethanol enhances retinoic acid metabolism into polar metabolites in rat liver via induction of cytochrome P4502E1. Gastroenterology. 2001;120:179–89. doi: 10.1053/gast.2001.20877. [DOI] [PubMed] [Google Scholar]
- 12.Liu C, Russell RM, Wang XD. Exposing ferrets to cigarette smoke and a pharmacological dose of beta-carotene supplementation enhance in vitro retinoic acid catabolism in lungs via induction of cytochrome P450 enzymes. J Nutr. 2003;133:173–9. doi: 10.1093/jn/133.1.173. [DOI] [PubMed] [Google Scholar]
- 13.Meydani SN, Meydani M, Blumberg JB, Leka LS, Siber G, Loszewski R, Thompson C, Pedrosa MC, Diamond RD, Stollar BD. Vitamin E supplementation and in vivo immune response in healthy elderly subjects. A randomized controlled trial. Jama. 1997;277:1380–6. doi: 10.1001/jama.1997.03540410058031. [DOI] [PubMed] [Google Scholar]
- 14.Mukhopadhyay CK, Chatterjee IB. NADPH-initiated cytochrome P450-mediated free metal ion-independent oxidative damage of microsomal proteins. Exclusive prevention by ascorbic acid. J Biol Chem. 1994;269:13390–7. [PubMed] [Google Scholar]
- 15.Gautschi O, Ratschiller D, Gugger M, Betticher DC, Heighway J. Cyclin D1 in non-small cell lung cancer: a key driver of malignant transformation. Lung Cancer. 2007;55:1–14. doi: 10.1016/j.lungcan.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 16.Caputi M, Groeger AM, Esposito V, Dean C, De Luca A, Pacilio C, Muller MR, Giordano GG, Baldi F, Wolner E, Giordano A. Prognostic role of cyclin D1 in lung cancer. Relationship to proliferating cell nuclear antigen. Am J Respir Cell Mol Biol. 1999;20:746–50. doi: 10.1165/ajrcmb.20.4.3366. [DOI] [PubMed] [Google Scholar]
- 17.Freemantle SJ, Liu X, Feng Q, Galimberti F, Blumen S, Sekula D, Kitareewan S, Dragnev KH, Dmitrovsky E. Cyclin degradation for cancer therapy and chemoprevention. J Cell Biochem. 2007;102:869–77. doi: 10.1002/jcb.21519. [DOI] [PubMed] [Google Scholar]
- 18.Jetten AM, Rearick JI, Smits HL. Regulation of differentiation of airway epithelial cells by retinoids. Biochem Soc Trans. 1986;14:930–3. doi: 10.1042/bst0140930. [DOI] [PubMed] [Google Scholar]
- 19.Jetten AM, George MA, Pettit GR, Rearick JI. Effects of bryostatins and retinoic acid on phorbol ester- and diacylglycerol-induced squamous differentiation in human tracheobronchial epithelial cells. Cancer Res. 1989;49:3990–5. [PubMed] [Google Scholar]
- 20.Saunders NA, Bernacki SH, Vollberg TM, Jetten AM. Regulation of transglutaminase type I expression in squamous differentiating rabbit tracheal epithelial cells and human epidermal keratinocytes: effects of retinoic acid and phorbol esters. Mol Endocrinol. 1993;7:387–98. doi: 10.1210/mend.7.3.8097865. [DOI] [PubMed] [Google Scholar]
- 21.Mernitz H, Smith DE, Wood RJ, Russell RM, Wang XD. Inhibition of lung carcinogenesis by 1alpha,25-dihydroxyvitamin D3 and 9-cis retinoic acid in the A/J mouse model: evidence of retinoid mitigation of vitamin D toxicity. Int J Cancer. 2007;120:1402–9. doi: 10.1002/ijc.22462. [DOI] [PubMed] [Google Scholar]
- 22.Mernitz H, Smith DE, Zhu AX, Wang XD. 9-cis-Retinoic acid inhibition of lung carcinogenesis in the A/J mouse model is accompanied by increased expression of RAR-beta but no change in cyclooxygenase-2. Cancer Lett. 2006;244:101–8. doi: 10.1016/j.canlet.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Khuri FR, Lee JS, Lippman SM, Lee JJ, Kalapurakal S, Yu R, Ro JY, Morice RC, Hong WK, Hittelman WN. Modulation of proliferating cell nuclear antigen in the bronchial epithelium of smokers. Cancer Epidemiol Biomarkers Prev. 2001;10:311–8. [PubMed] [Google Scholar]
- 24.Lee HY, Walsh GL, Dawson MI, Hong WK, Kurie JM. All-trans-retinoic acid inhibits Jun N-terminal kinase-dependent signaling pathways. J Biol Chem. 1998;273:7066–71. doi: 10.1074/jbc.273.12.7066. [DOI] [PubMed] [Google Scholar]
- 25.Lee HY, Sueoka N, Hong WK, Mangelsdorf DJ, Claret FX, Kurie JM. All-trans-retinoic acid inhibits Jun N-terminal kinase by increasing dual-specificity phosphatase activity. Mol Cell Biol. 1999;19:1973–80. doi: 10.1128/mcb.19.3.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Virmani AK, Rathi A, Zochbauer-Muller S, Sacchi N, Fukuyama Y, Bryant D, Maitra A, Heda S, Fong KM, Thunnissen F, Minna JD, Gazdar AF. Promoter methylation and silencing of the retinoic acid receptor-beta gene in lung carcinomas. J Natl Cancer Inst. 2000;92:1303–7. doi: 10.1093/jnci/92.16.1303. [DOI] [PubMed] [Google Scholar]
- 27.Suh YA, Lee HY, Virmani A, Wong J, Mann KK, Miller WH, Jr, Gazdar A, Kurie JM. Loss of retinoic acid receptor beta gene expression is linked to aberrant histone H3 acetylation in lung cancer cell lines. Cancer Res. 2002;62:3945–9. [PubMed] [Google Scholar]
- 28.Zochbauer-Muller S, Lam S, Toyooka S, Virmani AK, Toyooka KO, Seidl S, Minna JD, Gazdar AF. Aberrant methylation of multiple genes in the upper aerodigestive tract epithelium of heavy smokers. Int J Cancer. 2003;107:612–6. doi: 10.1002/ijc.11458. [DOI] [PubMed] [Google Scholar]
- 29.Fazi F, Travaglini L, Carotti D, Palitti F, Diverio D, Alcalay M, McNamara S, Miller WH, Jr, Lo Coco F, Pelicci PG, Nervi C. Retinoic acid targets DNA-methyltransferases and histone deacetylases during APL blast differentiation in vitro and in vivo. Oncogene. 2005;24:1820–30. doi: 10.1038/sj.onc.1208286. [DOI] [PubMed] [Google Scholar]
- 30.Chow CK, Ibrahim W, Wei Z, Chan AC. Vitamin E regulates mitochondrial hydrogen peroxide generation. Free Radic Biol Med. 1999;27:580–7. doi: 10.1016/s0891-5849(99)00121-5. [DOI] [PubMed] [Google Scholar]
- 31.Lee SF, Huang YT, Wu WS, Lin JK. Induction of c-jun protooncogene expression by hydrogen peroxide through hydroxyl radical generation and p60SRC tyrosine kinase activation. Free Radic Biol Med. 1996;21:437–48. doi: 10.1016/0891-5849(96)00040-8. [DOI] [PubMed] [Google Scholar]
- 32.Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. Faseb J. 1996;10:709–20. doi: 10.1096/fasebj.10.7.8635688. [DOI] [PubMed] [Google Scholar]
- 33.Kelman Z, Hurwitz J. Protein-PCNA interactions: a DNA-scanning mechanism? Trends Biochem Sci. 1998;23:236–8. doi: 10.1016/s0968-0004(98)01223-7. [DOI] [PubMed] [Google Scholar]
- 34.Iatropoulos MJ, Williams GM. Proliferation markers. Exp Toxicol Pathol. 1996;48:175–81. doi: 10.1016/S0940-2993(96)80039-X. [DOI] [PubMed] [Google Scholar]
- 35.Yano T, Yajima S, Hagiwara K, Kumadaki I, Yano Y, Otani S, Uchida M, Ichikawa T. Vitamin E inhibits cell proliferation and the activation of extracellular signal-regulated kinase during the promotion phase of lung tumorigenesis irrespective of antioxidative effect. Carcinogenesis. 2000;21:2129–33. doi: 10.1093/carcin/21.11.2129. [DOI] [PubMed] [Google Scholar]
- 36.Fanger GR, Gerwins P, Widmann C, Jarpe MB, Johnson GL. MEKKs, GCKs, MLKs, PAKs, TAKs, and tpls: upstream regulators of the c-Jun amino-terminal kinases? Curr Opin Genet Dev. 1997;7:67–74. doi: 10.1016/s0959-437x(97)80111-6. [DOI] [PubMed] [Google Scholar]
- 37.Force T, Bonventre JV. Growth factors and mitogen-activated protein kinases. Hypertension. 1998;31:152–61. doi: 10.1161/01.hyp.31.1.152. [DOI] [PubMed] [Google Scholar]