

Abstract
Hepatitis C virus (HCV) infection is a leading cause of chronic liver disease worldwide. Since several aspects of the infection remain unresolved, there is a pressing need for a convenient animal model that can mimic the clinical disease and aid the evaluation of treatment strategies. Although some success has been achieved in transgenic approaches for development of rodent models of HCV, transgenic expression of the complete HCV polyprotein or an entire set of the viral non-structural (NS) proteins continues to be a serious challenge. Using northern blot and 5′ rapid amplification of cDNA ends (RACE), we unraveled two possible mechanisms that can impede HCV NS transgene expression in the mouse liver. Several truncated transcripts are produced from alternate transcription start sites along the HCV NS sequence within the murine environment, in vivo. Translation of these shorter transcripts is blocked either by the positioning of a contextual stop codon or through a shift in the reading frame. In addition, the complete NS transcript undergoes trans-splicing through 5′ recombination with a non-transgene-derived, spliced leader sequence that appends a potential stop codon upstream of the translation start. These findings thus demonstrate that HCV NS-derived transgenes are subject to aberrant transcriptional initiation and post-transcriptional processing in the nucleus of a mouse host. Strategies to prevent such aberrant transcription start/RNA processing might be key to the development of a successful HCV transgenic mouse model.
Keywords: Hepatitis C virus, Non-structural proteins, Transgenic mouse, Aberrant transcription, Trans-splicing
Introduction
Hepatitis C is a major public health problem, affecting nearly 200 million people worldwide. While some acute hepatitis C patients are capable of eliminating the virus, about 70% of those infected develop chronic liver disease that may progress to liver cirrhosis and hepatocellular carcinoma. The hepatitis C virus (HCV) is an enveloped, positive, single-stranded RNA virus belonging to the Flaviviridae family. The HCV genome (9.6 kb) encodes a poly-protein precursor of about 3,000 amino acids, which is cleaved into the structural proteins (core, E1, E2, and p7) and the non-structural proteins (NS2, NS3, NS4A, NS4B, NS5A and NS5B) by host signal peptidases and the virally encoded proteases (Tang and Grise 2009).
The combined action of various host and viral factors is required for HCV replication and pathogenesis (Weber 2007; McGivern and Lemon 2009; Tang and Grise 2009). The successful establishment of permissible cell culture systems that can support replication of subgenomic and genome-length HCV RNA replicons has exponentially propelled research in the field (Blight et al. 2002; Bartenschlager 2006; Duverlie and Wychowski 2007). Yet, the complexity of HCV-host interactions underscores the need for practical animal models where the viral as well as host proteins are expressed at more biologically relevant levels. In this context, the limited availability of permissive hosts for HCV has been a challenge for the comprehensive, in vivo evaluation of the direct effects of viral proteins on hepatocytes and their role in HCV-associated liver injury. In the absence of a convenient small animal model, transgenic (tg) technology offers the opportunity to develop valuable rodent models of human diseases. Thus far, different tg mice carrying the gene or genes encoding HCV core and envelope (E)1 and E2, NS5A, NS3-4A and the full-length (fl) HCV polyprotein have been generated (Pasquinelli et al. 1997; Wakita et al. 1998; Sun et al. 2001; Lerat et al. 2002; Majumder et al. 2002; Alonzi et al. 2004; Sun et al. 2005; Frelin et al. 2006; Koike et al. 2010). Among these, tg mice specifically carrying an entire set of the viral NS genes have failed to definitively show a satisfactory expression of the relevant proteins. Earlier, Lerat et al. (2002) generated a tg mouse lineage carrying the complete HCV open reading frame (ORF) (FL-N/35) under the control of a mouse albumin enhancer/promoter. Although transgene-specific RNA was demonstrated in the liver tissues by RT–PCR with nine different primer sets amplifying consecutive regions spanning the transgene ORF, neither the related transcript could be detected by northern blot nor was any HCV protein expression shown in these mice. Subsequently, one group of investigators (Alonzi et al. 2004) produced constitutive HCV tg mice carrying the entire HCV ORF inserted in the α1 antitrypsin (A1AT) gene, with detectable HCV RNA in the liver. They also demonstrated HCV core, NS3 and NS5A plus NS5B proteins by immunohistochemical staining, but not by western blot. Another group (Koike et al. 2010) generated tg mice carrying the HCV NS genes, but failed to show expression of these proteins in the tg livers. It appeared that a considerable selection pressure due to the effect of NS proteins on the developing liver could be instrumental in the suppression of these genes within the murine system. Alternatively, certain part(s) of HCV NS sequence could negatively impact the transcriptional and/or post-transcriptional processing of these genes in the tg milieu.
In this report, we generated tg mice with inducible, liver-specific expression of the HCV NS (NS2 through 5B) genes by using the Cre/loxP DNA recombination system. To generate double tg mice that mediate NS transgene recombination constitutively, we bred these tg founders with an albumin-Cre (Alb-Cre) tg partner that has liver-specific expression of Cre-recombinase (Postic et al. 1999). Various aberrant transgene-specific RNAs were detected by northern blot in the liver tissues of these mice. We applied the 5′ and 3′ rapid amplification of cDNA ends (RACE) PCR procedure to sequence these RNA transcripts (Scotto-Lavino et al. 2006a, b). Our data provide insight into the possible molecular mechanisms responsible for the lack of HCV NS gene expression in tg mouse models.
Materials and methods
Plasmid construction
To establish tg mice with liver-specific inducible expression of HCV NS genes, we generated a transgene construct (Alb-NS) (Fig. 1a) in which HCV genotype 1b NS gene expression was inducible through the Cre/loxP DNA recombination system. The vector used for the construction of the transgene was pGEMAlbSVPA, which contains the murine albumin enhancer/promoter with a downstream SV40 intron and polyadenylation signal (McPherson et al. 1993). The sequence of the genotype 1b NS ORF was derived from the insert in the infectious plasmid pHCV-N (Beard et al. 1999) and contains a start codon and a typical Kozak sequence. It was sub-cloned into the XbaI site of pGEMAlbSVPA upstream of the SV40 intron/polyadenylation cassette. A 1.4-kb spacer, containing stop codons in all three reading frames and flanked by loxP sequences at both ends, was inserted between the albumin promoter and the NS transgene by using a XhoI site. Thus, the HCV NS ORF in the construct was transcribed, but not translated, unless the spacer was removed by Cre-mediated DNA recombination. Each cloning step was confirmed by restriction enzyme digestion and sequence analysis of the junctions. Also, the entire NS ORF was sequenced.
Fig. 1.
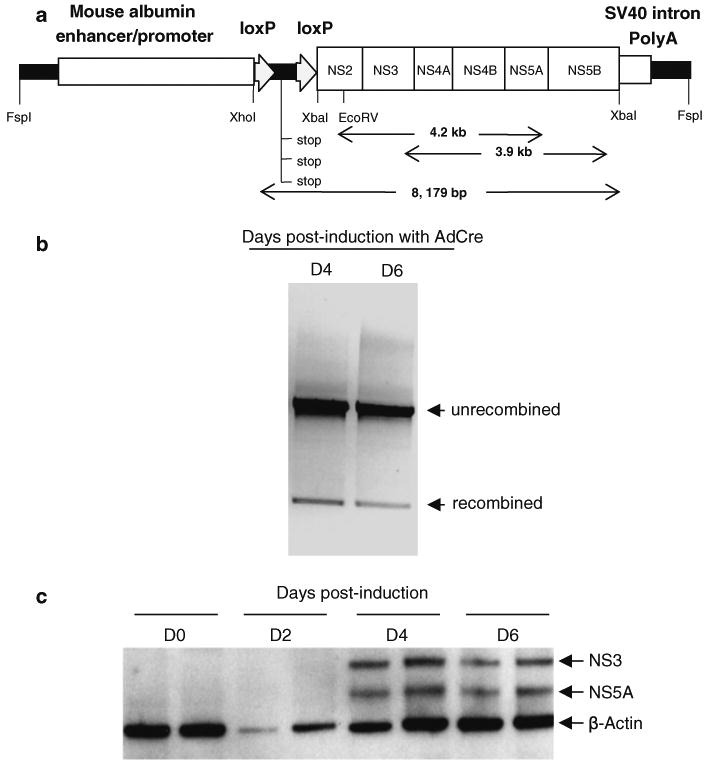
a Schematic representation of the Alb-NS construct. The complete HCV genotype 1b non-structural genomic region (NS2, NS3, NS4A, NS4B, NS5A and NS5B) was cloned under the control of the mouse minimal albumin enhancer/promoter and followed by a SV40 intron and a polyadenylation signal. RT–PCR products of 4.2 and 3.9 kb are shown. b Gel picture showing PCR bands representing recombined and unrecombined transgene. Huh-7 cells were transfected with the Alb-NS construct. At 24 h post-transfection, transgene recombination was induced by infecting the cells with a replication-deficient adenovirus expressing Cre DNA recombinase. c Following transgene induction in the Huh-7 cells, 72 and 58 kDa proteins were detected by western blot analysis in the Alb-NS transfectant cell lysates by using anti-NS3 and NS5A-specific monoclonal antibodies, respectively
Analysis of transgene inducibility and expression
Prior to microinjection, the plasmid was tested for its inducibility and expression of the HCV NS proteins by transient transfection of the human hepatoma cell line Huh-7. Transgene recombination was induced by infecting the cells at 24 h post-transfection with a replication-deficient adenovirus vector expressing Cre recombinase under the control of a cytomegalo-virus immediate-early promoter (AdCre) (Kanegae et al. 1995). Genomic DNA isolated from the transfected cells was extracted before and post-AdCre infection and analyzed for transgene recombination by using the primer pair ALB5 (5′-GGAACCAATGAAATGCGAGG-3′) and RevNS1 (5′-CGCCATAGAAAGCATATGCC-3′), which amplified both unrecombined (1,636 bp) and recombined (370 bp) transgene fragments, distinguishable on the basis of their molecular size. To assess transgene expression, cells were harvested in a cell culture lysis reagent (Promega, Madison, WI) and 10 μg of the extracted protein was resolved by SDS–PAGE. The proteins were electrotransferred to a polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA) and probed with monoclonal antibodies to HCV NS3 (NovoCastra, Newcastle, UK) and NS5A (Biodesign, Saco, ME), followed by horseradish peroxidase-conjugated antimouse immunoglobulin G (IgG) (Southern Biotechnology Associates, Birmingham, AL) and ECL detection reagents (Amersham Pharmacia, Buckinghamshire, UK).
Generation and characterization of transgenic mice
The transcriptional unit of the Alb-NS plasmid was excised with FspI and electrophoretically purified before microinjection into fertilized egg pronuclei of C57BL/6 × SJL hybrids at the University of Michigan Transgenic Facility (Ann Arbor, MI). Potential founders were screened for the presence of transgene by PCR of mouse genomic DNA isolated from tail biopsies by using two independent primer pairs located within the HCV NS4B and NS5B genomic regions. The nucleotide sequences of the sense and antisense primers derived from the NS4B gene were NSP8 5′-(AAGGTGCTAGTGGACATTCTGG)-3′ and RNS8 5′-(AGTATTGCTGCACACACGAC)-3′, respectively, and those of the NS5B derived primers were S8 5′-(TGCTGGAGGACACTGTGACACCAAT)-3′ and NSR 5′-(AGGAGTTAACTGGAGTGTGTCTAGC)-3′, respectively. Further, the number of integrated transgene copies in each founder line was determined by slot blot hybridization as follows: One μg of individual founder DNA was blotted onto a positively charged nylon membrane (S & S, Keene, NH) along with 1 μg of mouse genomic DNA mixed with serial copies of the Alb-NS plasmid as a copy number control. The membranes were probed independently with NS3-4A and NS5B-specific 32P-labeled probes. A graph was plotted based on the band densities of the transgene copy standards and the number of transgene copies per founder line determined from the standard graph. Additionally, the presence of the cognate transgene was confirmed by Southern hybridization with P32-labeled NS3-4A and NS5B-specific probes, following digestion with restriction endonucleases, XbaI (which cuts out the complete NS sequence from the transgene) and XhoI-EcoRV (which cut the 5′ end of the loxP spacer and about 345 bp from the 5′ end of the NS, respectively) (Fig. 1a).
The tg founders were first backcrossed to C57BL/6 mice for the N1 generation to evaluate breeding potential and transgene transmissibility. Subsequent generations of Alb-NS founder lines were crossed with a line of albumin-Cre (Alb-Cre) tg mice {C57BL/6-TgN(AlbCre)21Mgn}, in which the promoter/enhancer of the rat albumin gene was used to direct the liver-specific expression of Cre (Postic et al. 1999), to generate mice with constitutive NS transgene recombination. The Alb-NS × Alb-Cre double tg mice were screened by using the above-mentioned NS-specific primers and Alb-Cre-specific primers Alb-Cre/F 5′-(ACCGTCAGTACGTGAGATATCTT)-3′ and Alb-Cre/R 5′-(ACCTGAAGATGTTCGCGATTATCT)-3′. Transgene recombination was detected as above. All animal care and experimentation were performed according to the National Institutes of Health Guidelines and with the approval of the Institutional Animal Care and Use Committee.
Transgene mRNA detection
Double and single tg mice from different founder lines were euthanized at 8 weeks, and their liver tissue was snap frozen in RNAlater (Ambion, Austin, TX) for future analysis. Total RNA was extracted from the liver by using an RNAqueous® kit (Ambion, Austin, TX). After clearing contaminating DNA with DNase I, the RNA was reverse transcribed with NS5A-specific primer GSP3 5′-(CAATTCGGAGCTGCCGAAGG)-3′ and NS5B-specific primer NSR by using OmniScript reverse transcriptase (Qiagen, Valencia, CA). PCR was carried out for 40 cycles with AccuTaq LA DNA polymerase (Sigma–Aldrich, St Louis, MO) by using two primer sets. The first set comprising sense primer NSP2 5′-(TCTTCTCTGACATGGAGACC)-3′ and antisense primer GSP3 amplified a 4.2-kb fragment spanning the NS2 to NS5A genomic region, while the other set, comprising sense primer NS3-5′ 5′-(CGGGCTTACCTAAATACACC)-3′ and antisense primer NSR amplified a 3.9-kb fragment spanning the NS3 to NS5B genomic region (Fig. 1a).
For northern blotting, RNA was isolated from frozen tissue by using Trizol reagent (Life Technologies, Gaithersburg, MD). Total RNA (20 μg) was separated on a 1% formaldehyde denaturing agarose gel, and then transferred onto a nitrocellulose membrane (S & S). RNA was cross-linked to the membrane by ultraviolet irradiation at 254 nm (UV Stratalinker; Stratagene, La Jolla, CA) and hybridized to NS5B-, NS4B-5A- and GAPDH-specific 32P-labeled probes. GAPDH (1.27 kb) and in vitro transcribed HCV RNA (9.6 kb) were used as molecular size controls, while RNA isolated from the FL-N/35 line (Lerat et al. 2002) was used for comparison. The analysis was repeated with RNA extracted from three independent animals of each founder line.
Sequencing of the 5′ and 3′ terminal ends of transgene-specific mRNA
Isolation of the 5′ and 3′ terminal fragments of the Alb-NS and FL-N/35 mRNA transcripts was carried out by using classic 5′ and 3′ rapid amplification of cDNA ends (RACE) procedures (Scotto-Lavino et al. 2006a, b). In brief, for the 5′ RACE the RNA was reverse transcribed to cDNA by using a random hexamer. Following this, a 5′ poly(A) tail was appended by employing terminal deoxynucleotidyl transferase (Tdt) (New England BioLabs, Ipswich, MA) and dATP. Amplification was then carried out with JumpStart™ REDTaq® DNA Polymerase (Sigma-Aldrich, St Louis, MO), for which we used the hybrid primer QT to form the second strand of cDNA, and the forward primer QO and three separate reverse primers NS2Rv, RevNSP8 and NSR-3′ specific for the NS2, NS5A and NS5B sequences, respectively, in the case of the Alb-NS × Alb-Cre double tg cDNA, and reverse primers Seq1R and NS2-Rv3, specific for the core and NS2 segments, respectively, in the case of the FL-N/35 tg cDNA. Subsequently, a second set of PCR cycles was carried out in which we used the nested primers QI and RevNS1, NSR2 and NSR, respectively, in the case of Alb-NS × Alb-Cre cDNA, and QI and HCV3A and NS2Rv2, respectively, in the case of FL-N/35 cDNA. This second set of PCRs was aimed at increasing the specificity and yield of each specific product, which was then electrophoretically purified (Qiagen, Valencia, CA) and sequenced (at the UTMB Molecular Genomics Core Facility). For the single tg Alb-NS cDNA, gene-specific nested primers NS2-Rv3 and NS2-Rv2 were used for the 5′ RACE. The nucleotide sequences of the primers were as follows: QT 5′-(GAGCAGAGTGACGAGGACTCGAGCTCAAGCTTTTTTTTTTTTTTTTTTVN)-3′, QO 5′-(GAGCAG AGTGACGAGGAC)-3′, QI 5′-(GAGGACTCGAGCTCAAGC)-3′, NS2Rv 5′-(CCACAAAAACCGCGCCTCCG)-3′, RevNS8 5′-(AGTATTGCTGCACACACGAC)-3′, NSR-3′ 5′-(ACCAGCCGGATAAGTCCAAC)-3′, Seq1R 5′ -(GTGACAGGAGCCATCCTG)-3′, NS2-Rv3 5′- (ACTGCACACGTGAGGAGGATG)-3′, NSR2 5′-(GGTCCTCGGCAGAGGGCGCCTCGCC)-3′, NS2-Rv2 5′- (ATTGTAACCACCATAGGAGC)-3′, HCV3A 5′(ATAGGCTGTCGCCTTCCACG)-3′.
For the 3′ RACE, the RNA was reverse transcribed to cDNA by using a 3′ RACE cDNA synthesis (3CDS) primer, following which, amplification was carried out by using an NS5B-specific forward primer NS5B-FR1 and a universal primer mix comprising 0.04-μM long primer UPL and 0.2-μM short primer UPS. The product was then further amplified by using a nested forward primer NS5B-FR2 and primer UPS. The nested PCR product was electrophoretically purified prior to sequencing. The nucleotide sequences of the primers were as follows: 3CDS 5′-(AAGCAGTGGTAACAACGCAGAGTACTTTTTTTTTTTTTTTTTTTTTTVN)-3′, UPL 5′-(CTAATACGACTCACTATAGGGCAAGCAGTGGTAACAACGCAGAGT)-3′, UPS 5′-(CTAATACGACTCACTATAGGGC)-3′, NS5B-FR1 5′-(GTTGGACTTATCCGGCTGGT)-3′ and NS5B-FR2 5′-(TCTCGTGCCCGACCCCGCTG)-3′.
Results
Transgene expression in cell culture
The Alb-NS construct was designed to establish an immunocompetent tg mouse model with moderate-to-high levels of HCV NS protein expression in the liver (Fig. 1a). The construct was transfected into the human hepatocyte cell line Huh-7 and its expression was induced with a replication-deficient adenovirus expressing Cre recombinase (Kanegae et al. 1995) at 24 h post-transfection. Efficient recombination of the transgene (Fig. 1b) was detected following induction with AdCre. NS3 and NS5A proteins were detected in the Huh-7 protein extracts by western blot following transgene recombination (Fig. 1c).
Transgenic mice
Since the construct could express NS proteins in Huh-7 cells, it was used to generate tg mice. A total of 17 Alb-NS founders were obtained. Of these 3 animals were infertile, while 8 failed to transmit the transgene to the N1 offspring. Of the remaining 6 founder lines, all except lineages 679 and 740 were found to carry a single copy of the integrated transgene as determined by slot blot analysis. Lineage 679 and 740 had 2 and 3 integrated transgene copies, respectively. We confirmed these results by Southern blot analysis of liver DNA from lineage 679 and 740 in which we used several restriction enzymes that cut different segments of the transgene. Our findings further demonstrated that the entire transgene was present in both lineages (Fig. 2a, b). When founder genomic DNA was cut with XbaI, a 6.8-kb band was detected in both the founder lines consistent with the size of the NS. On the other hand, double digestion with XhoI and EcoRV generated two distinct patterns in the two individual founder lines. In the case of lineage 679, a single 11.8-kb band was detected, consistent with the expected fragment generated through double digestion of a double copy transgene concatemer. Double digestion of lineage 740, on the other hand, yielded two bands of 20.1 and 6.4 kb, a finding indicating that the three transgene copies may not have integrated in tandem with each other, and, instead, generated different restriction fragments according to their individual integration site. For the purpose of further characterization, these lines were expanded by successive backcrossing with Alb-Cre partners to generate double transgenic mice (Alb-NS × Alb-Cre) that constitutively mediated transgene recombination (Fig. 2c).
Fig. 2.
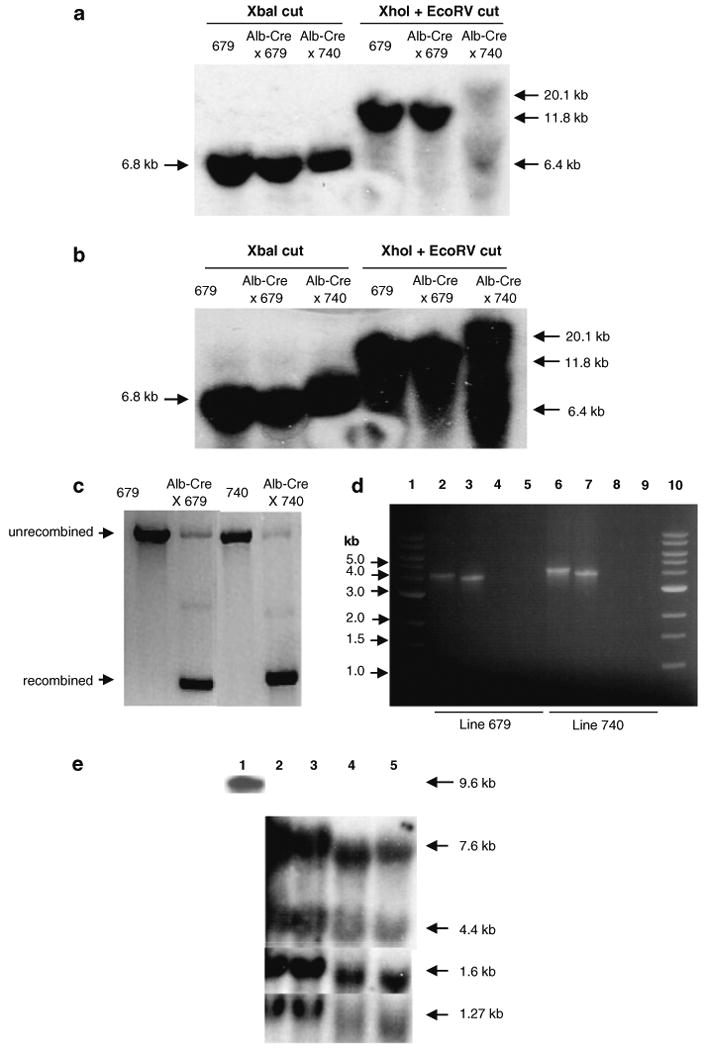
The Alb-NS transgene construct was used to generate multiple mouse lineages. Two of the female founders, lineage 679 and 740, were bred with a male mouse transgenic for liver-specific expression of Cre-recombinase (Alb-Cre) to generate double transgenic (Alb-NS × Alb-Cre) mice. (a and b) Southern blot showing integration of the 13.6 kb Alb-NS transgene into mouse genomic DNA. Single and double transgenic liver DNA from line 679 and double transgenic liver DNA from line 740 was digested with XbaI and XhoI + EcoRV and probed with NS3-4A-(a) and NS5B-(b) specific probes. c Recombination of the transgene in single (Alb-NS) and double (Alb-NS × Alb-Cre) tg mice detected by PCR. The efficiency of constitutive recombination is greater than 90%. d Detection of transgene-specific mRNA transcript in double tg mouse liver from line 679 (lanes 2–5) and line 740 (lanes 6–9) by RT–PCR. Lanes 1 and 10: 1 kb DNA ladder; Lane 2 and 6: 4.2-kb RT–PCR fragment spanning NS2 to NS5A gene region; Lanes 3 and 7: 3.9-kb RT–PCR fragment spanning NS3 to NS5B gene region; Lanes 4 and 8: PCR without RT and with NS2 and NS5A-specific forward and reverse primers, respectively (negative controls); Lanes 5 and 9: PCR without RT and with NS3 and NS5B-specific forward and reverse primers, respectively (negative controls). e Detection of NS-specific mRNA transcript in tg mouse liver by northern blot. Lane 1: In vitro transcribed synthetic HCV RNA (5 ng) (positive control); Lane 2: FL-N/35 liver RNA (20 μg); Lane 3: Line 679 double tg (Alb-NS × Alb-Cre) mouse liver RNA (20 μg); Lane 4: Line 740 double tg (Alb-NS × Alb-Cre) mouse liver RNA (20 μg); Lane 5: Line 740 single tg (Alb-NS) mouse liver RNA (20 μg). The exposure was for 6 h (1.27-kb GAPDH mRNA) or overnight (in vitro transcribed synthetic HCV RNA) or 5–7 days (1.6-kb RNA) or 14-18 days (4.4 and 7.6-kb RNA)
Transgene transcription analysis
To confirm the presence of a transgene-specific RNA transcript in lineage 679 and 740, we extracted RNA from liver tissues of mice from the respective lines and amplified it with two independent RT–PCR reactions by using primer pairs spanning overlapping segments of the transgene sequence, namely, NS2 to NS5A (4.2 kb) and NS3 to NS5B (3.9 kb) (Fig. 1a). The results indicated the presence of an RNA transcript (or transcripts) spanning the complete NS transgene sequence (Fig. 2d). No specific product was obtained by excluding the reverse transcription step, which demonstrated the absence of contaminating DNA in the RNA samples.
Northern blot analysis of both lineages with an NS5B-specific probe unexpectedly revealed the presence of only an HCV transcript of approximately 1.6 kb, from which it was concluded that a major species of the transgene-specific mRNA in the liver tissues of these mice was truncated (Fig. 2e). When the membrane was probed with a mixture of two probes, NS4B-5A-specific and NS5B-specific, two additional bands of approximately 7.6 and 4.4 kb were detectable in both Alb-NS tg lineages. Similar bands were also detected with the FL-N/35 RNA. Interestingly, the longer RNAs (7.6 and 4.4 kb) were detectable only when the two probes were applied together and after prolonged exposure, a result leading us to conclude they were present as relatively minor species. The sizes of the individual RNA bands were estimated based on the RF with reference to the respective GAPDH (1.27 kb) band for each RNA sample. All three northern bands were consistently detected in three independent animals from each founder lineage.
Sequencing of the mRNA 5′ and 3′ ends
As mentioned above, RT–PCR analysis by a long-range PCR pointed to the presence of a complete HCV RNA sequence, perhaps in one contiguous molecule, in the liver tissues of the two Alb-NS lineages 679 and 740 (Fig. 2d). Earlier, the presence of a full-length transcript was inferred for the FL-N/35 line by using a comparable strategy, but with a shorter-range PCR method (Lerat et al. 2002). Considering that all three (7.6, 4.4 and 1.6 kb) RNA species observed in the northern blot analysis were detectable by the NS5B-specific probe, it should be assumed that the 3′-terminal sequence of all three RNA transcripts, from both the Alb-NS and the FL-N/35 lineage, corresponded to the 3′-terminal sequence of the respective transgene. To prove this point, we performed a classic 3′-RACE procedure and confirmed that the 3′ -terminal sequence of each of the transcripts was tagged with the polyA, which was engineered to be part of the transgene construct.
We then employed a 5′-RACE procedure and nested PCR, by using three HCV gene-specific primers in three separate reactions, to amplify the 5′-terminal sequence of each RNA species, and sequenced the products. In all, four independent RNA species were actually identified in the Alb-NS livers. Figure 3a–c and g represent the immediate 5′-terminal sequences of the four HCV NS-specific RNA species detected in a lineage 679 Alb-NS × Alb-Cre mouse liver following transgene recombination. Three of these RNAs (Fig. 3a–c) represent contiguous sequences which have different transcription start sites, but terminate at the common 3′-terminal region of the transgene. They all contain an extra, non-templated 5′ G residue, a finding suggesting that all the three RNAs are capped, as reverse transcriptases are known to transcribe the 5′-terminal cap nucleotide of RNA. Since their upstream DNA sequences do not meet the description of a consensus splice junction, these three RNA species seem to have initiated from alternate transcription start sites in the HCV NS genomic region. Two of these sequences start in the NS5B genomic region, one (Fig. 3b) being shorter than the other by 147 bases (Fig. 3a). These may have been represented as a single 1.6-kb band, not necessarily of equal abundance, in the northern blot. The 4.4-kb northern fragment begins in the NS4B genomic region (Fig. 3c).
Fig. 3.
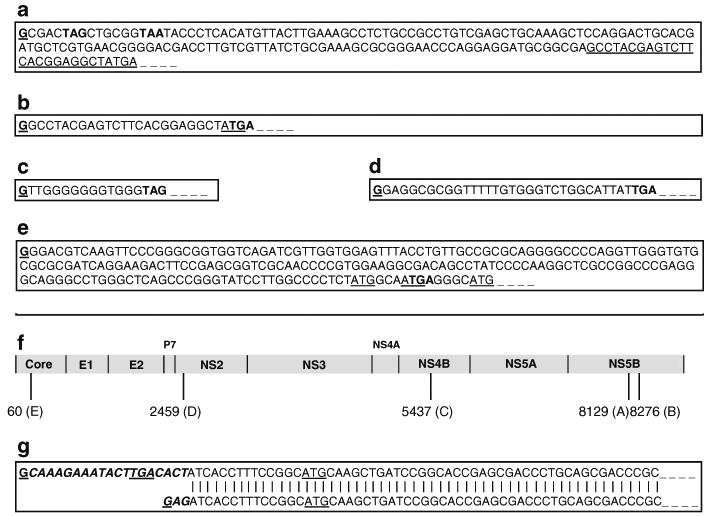
Sequencing of the 5′ ends of the truncated transgene RNAs by 5′ RACE revealed alternate transcription starts within the HCV ORF. a 5′ nucleotide sequence of the longer mRNA transcript (presumably 1.6 kb) with the transcription start site in the middle of the NS5B region. Underlined bases are the beginning of the sequence b. b 5′ nucleotide sequence of the mRNA transcript, shorter by 147 bases as compared to (a) above, with the transcription start site also in the middle of the NS5B region. Note that the first G of this sequence is the 147th base of the above. c 5′ nucleotide sequence of the mRNA transcript (presumably 4.4 kb) with transcription start site in the NS4B region. d 5′ nucleotide sequence of the longest mRNA transcript (presumably 7.6 kb) from an unrecombined Alb-NS transgene and a NS-derived transcript from the FL-N/35 line. e 5′ nucleotide sequence of the longest mRNA transcript (predicted size, 9 kb, and undetectable on northern) from an FL-N/35 tg liver. All five mRNA species begin with a nontemplate-specific 5′ G base and appear to be capped. Stop codons are indicated in boldface triplets, in Fig. 3a–e. f HCV map (genotype 1b) showing nucleotide positions of the alternate transcription start sites along the HCV ORF represented by the above sequences. g Trans-splicing of the Alb-NS transcript. Upper sequence: Actual nucleotide sequence of the Alb-NS transgene mRNA, which includes the upstream spliced leader sequence that ends in a 5′ G base and appears to be capped. Note that the spliced leader sequence contains a potential stop codon (TGA). Lower sequence: Predicted nucleotide sequence of a recombined Alb-NS transgene mRNA containing a splice acceptor (AG) site at its 5′ end and a hypothetical G cap preceding it
To our surprise, the 5′ sequence of the 7.6-kb RNA which represents the complete, recombined Alb-NS transcript, contains an additional 19 non-complementary bases in place of the 5′ AG residues on the transgene that represent the predicted transcription start site of the complete Alb-NS transcript (Fig. 3g upper sequence). Moreover, the 5′ end of the predicted Alb-NS transcript contains a consensus splice acceptor sequence of an intron with no functional 5′ splice donor site upstream, characteristic of a trans-splicing signal or an outron (Fig. 3g lower sequence). Therefore, we speculate that the 19-base, 5′ terminal sequence of the RNA transcript, which is not featured anywhere in the entire transgene construct and contains a 5′ cap, is derived from a spliced leader of an unknown cellular gene that could have been transferred to the HCV trangene pre-mRNA, along with its methylguanosine cap (Stover et al. 2006). In addition, this spliced leader-derived sequence is most likely to hinder the translation of the transgene, as it contains a potential stop codon in frame and upstream of the ATG translation start of the transcript.
In addition, we also sequenced the 5′ ends of the 7.6-kb northern fragments from a single tg Alb-NS mouse (without transgene recombination) and an FL-N/35 mouse. Both lineages had the identical 5′ capped RNA originating from a common transcription start site within the HCV NS2 transgene region (Fig. 3d). Further, we sequenced the 5′ end of the complete FL-N/35 transcript and found that it too was represented by a truncated, albeit 5′ capped RNA derived from an alternate transcription start site in the HCV core transgene region (Fig. 3e). This particular RNA species was not detectable in the northern blot possibly due to rapid degradation. Notably, as in the case of the trans-spliced RNA, three of the five identified truncated transcripts contain a potential stop codon upstream of the first ATG (potential translation start) triplet (Fig. 3a, c, d). In the remaining two transcripts, the earliest potential translation start signal occurs in a reading frame that is variant from that of the HCV coding sequence (Fig. 3b, e).
Discussion
The strict host specificity of HCV has been a major impediment for study of the pathobiology of the infection, as well as for the preclinical evaluation of potential therapeutic candidates. It would be expected that livers of animals that are not inherently susceptible to HCV infection should be permissive to the expression of virus genes in the transgenic milieu. As cited earlier, several tg mice expressing HCV structural and/or NS genes, either individually or in various combinations have been generated (Pasquinelli et al. 1997; Wakita et al. 1998; Sun et al. 2001; Lerat et al. 2002; Majumder et al. 2002; Alonzi et al. 2004; Sun et al. 2005; Frelin et al. 2006; Koike et al. 2010). Among these, those expressing HCV structural genes have been shown to express these proteins in the tg livers at levels similar to those in hepatitis C patients (Pasquinelli et al. 1997; Wakita et al. 1998; Sun et al. 2001; Lerat et al. 2002; Sun et al. 2005). On the other hand, transgenic mice specifically carrying an entire set of the viral NS genes have failed to definitively show a satisfactory expression of the relevant proteins. An obvious explanation is that the NS proteins, with such activities as serine protease and RNA helicase, are deleterious to embryonic development. We thus designed a conditional HCV NS transgene which expresses only when Cre recombinase is present in the cells. The mice carrying the transgene, described here, were normal in growth and development as well as in fertility and fecundity. Yet, they did not express any of the NS proteins when the transgene was induced to undergo recombination mediated by Cre, which was expressed from another transgene in the cell (Fig. 2c) or from injected AdCre (unpublished observation).
Although RT–PCR analysis of the total liver RNA by using two independent primer pairs spanning overlapping segments of the transgene provided evidence for the presence of a full-length RNA transcript, the northern blot data showed the presence of additional, shorter RNAs. In fact, these shorter transcripts were detected as the major species, whereas the longer (7.6-kb) transcript and an intermediate-size RNA (4.4 kb) were detected in much smaller amounts. Sequence analysis of the truncated RNAs revealed contiguous sequences corresponding with the 3′-terminal end of the transgene. Earlier, it has been referred that inappropriately spliced transcripts have been noted in the case of transgenes derived from HCV sequences (Lemon et al. 2000). However, the absence of the universally conserved splice junctional sequence GU-AG at the 5′ end of the shorter RNAs rules out the possibility of these truncated transcripts being cryptic splice products. Instead, they appear to be initiating from alternate transcription start sites within the chromosomally integrated HCV sequence that are recognized in the mouse liver. This interpretation is supported by the presence of a capped G at the 5′ termini of these RNAs (Fig. 3).
In natural HCV infection, viral RNA replication takes place in the cytoplasm through the virally encoded NS5B RNA-dependent RNA polymerase (RDRP), and all viral proteins are translated from the viral RNA using the IRES (Internal Ribosomal Entry Site) mechanism. While the Alb-NS construct expressed protein efficiently in Huh-7 cells (Fig. 1c), which involves transcription by Pol II and cap-dependent translation by ribosomes, it failed to do so in the murine Hepa 1-6 cell line (data not shown). This unusual occurrence is not unique to the vector used for the preparation of the Alb-NS construct or the Cre-loxP methodology because both the vector and the method were used earlier by us and others to successfully generate HCV structural transgenic lines (Wakita et al. 1998; Sun et al. 2001; Lerat et al. 2002; Sun et al. 2005). Moreover, another HCV NS transgene construct prepared by us, using an independent liver-specific vector, pLive (Mirus, Madison, WI) also failed to show expression of the viral proteins both in vitro and in vivo (data not shown). Though in vitro findings seemed to support species-specific differences in transgene expression, it was conceivable that expression of an HCV NS transgene could be differentially regulated in vivo. We, therefore, compared the transcripts produced in our Alb-NS tg lines with those from the independently generated FL-N/35 line (Lerat et al. 2002). The detection of identical truncated RNAs in both these lines (Figs. 2e and 3a–d) leads us to suggest that the alternate transcription starts in the tg livers may be due to the presence of mouse transcription factor binding sites and initiator (Inr) elements within the HCV ORF (Smale 2001). The relative abundance of the different truncated HCV transcripts indicates that the mouse transcriptional apparatus recognizes the individual transcription start signals at different efficiencies. Identification of the specific promoter elements within the HCV sequence via a comprehensive CAGE (cap analysis gene expression) study can perhaps help throw more light on this issue (Frith et al. 2006).
On the other hand, the positioning of a stop codon preceding the earliest translation initiation signal (Fig. 3a, c, d) or the shift in reading frame (Fig. 3b) that ensued through the alternate transcription start is striking and appears to function towards obstructing translation of the viral genes. It is also interesting to note that the longest RNA species identified from the FL-N/35 line (Fig. 3e), that originated from inside the core gene from an alternate transcription start, could possibly encode for the HCV ARFP (alternate reading frame protein) (Vassilaki and Mavromara 2009) and/or the p8 minicore protein (Eng et al. 2009), and this may account for some of the phenotype reported for this lineage.
From the evidence presented in this paper, the complete Alb-NS RNA that was transcribed off the recombined transgene appears to have undergone trans-splicing, with a 19-base leader sequence spliced onto the 5′ end of the transcript (Fig. 3g). Trans-splicing refers to the recombination of sequences from two independently transcribed pre-mRNAs to form a composite RNA (Stover et al. 2006). A nucleotide blast search revealed that though the 19-base sequence is not featured in the Alb-NS transgene construct, it is present at more than one location in the mouse genome and, therefore, should be originating from an independently transcribed mouse pre-mRNA. The sequence it replaces at the 5′ end of the Alb-NS mRNA represents a splice acceptor site (AG nucleotides). The trans-splicing also appears to be occurring in a specific manner that can possibly block the translation of the transcript through the insertion of a stop codon upstream of, and in frame with, its translation start site. Moreover, this could render the transcript susceptible to nonsense-mediated decay, which might explain its presence as a low abundance species in the mouse liver RNA population. Trans-splicing in mammals has been shown to be contributory towards proteome diversity and as a mechanism of inducing transgression of host RNA processing by an infecting virus through heterologous trans-splicing (Mayer and Floeter-Winter 2005). To our knowledge, this is the first reported demonstration of trans-splicing of an HCV transgenic transcript in vivo, even though Pasquinelli et al. (1997) in their core-317 transgenic line reported the presence of a longer, 1.7-kb mRNA in addition to the expected 1.4 kb transcript, hinting at a mechanism of RNA recombination. Since they failed to detect transgene-specific sequences either upstream or downstream of their MUP (mouse urinary protein promoter)-HCV transcription unit, they concluded that the longer transcript may have originated through splicing of the SV40 portion of the transcript and transcription read-through of mouse sequences past the transgene. Possible remedies to overcome the trans-splicing could be to delete or modify the potential trans-splice signals in individual HCV NS or full-length tg constructs or to introduce an intronic sequence 3′ of the potential trans-splice site, so as to guide the mouse cellular splicing machinery to cis splice the mRNA instead of trans-splicing.
Overall, our analysis of these HCV tg mouse lines brings to light two independent veritable phenomena that result in the positioning of a translation stop codon prior to the contextual translation start or lead to a shift in reading frame. Both of these beget restriction on the translation of a combination of HCV NS sequences, transgenically introduced in the murine liver. A more comprehensive investigation of these phenomena could provide important clues and give impetus to the development of tg mouse models of HCV infection.
Acknowledgments
This work was supported by National Institutes of Health Grant R01 AI069142 (to J.S.) and John Sealy Memorial Endowment Fund Development Grant (to LLC). M.D. is a James W. McLaughlin Postdoctoral Fellow and a Hans Popper Postdoctoral Fellow of the American Liver Foundation. We thank Dr. James Ellis of NIH for the SAGE analysis, Dr. Stanley Lemon for the FL-N/35 mice, Dr. Junhui Jia for technical support and Dr. Thomas Wood for helpful discussion.
Contributor Information
Mayura M. Desai, Department of Microbiology and Immunology, University of Texas Medical Branch at Galveston, Galveston, TX 77555-1019, USA
Batbayar Tumurbataar, Department of Microbiology and Immunology, University of Texas Medical Branch at Galveston, Galveston, TX 77555-1019, USA.
Yueqing Zhang, Department of Microbiology and Immunology, University of Texas Medical Branch at Galveston, Galveston, TX 77555-1019, USA.
Lee-Nien Lillian Chan, Department of Biochemistry and Molecular Biology, University of Texas Medical Branch at Galveston, Galveston, TX 77555-0645, USA.
Jiaren Sun, Email: jisun@utmb.edu, Department of Microbiology and Immunology, University of Texas Medical Branch at Galveston, Galveston, TX 77555-1019, USA.
Teh-sheng Chan, Email: tchan@utmb.edu, Department of Microbiology and Immunology, University of Texas Medical Branch at Galveston, Galveston, TX 77555-1019, USA.
References
- Alonzi T, Agrati C, Costabile B, et al. Steatosis and intrahepatic lymphocyte recruitment in hepatitis c virus transgenic mice. J Gen Virol. 2004;85:1509–1520. doi: 10.1099/vir.0.19724-0. [DOI] [PubMed] [Google Scholar]
- Bartenschlager R. Hepatitis C virus molecular clones: from cDNA to infectious virus particles in cell culture. Curr Opin Microbiol. 2006;9:416–422. doi: 10.1016/j.mib.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Beard MR, Abell G, Honda M, et al. An infectious molecular clone of a Japanese genotype 1b hepatitis c virus. Hepatology. 1999;30:316–324. doi: 10.1002/hep.510300137. [DOI] [PubMed] [Google Scholar]
- Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis c virus RNA replication. J Virol. 2002;76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duverlie G, Wychowski C. Cell culture systems for the hepatitis C virus. World J Gastroenterol. 2007;13:2442–2445. doi: 10.3748/wjg.v13.i17.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng FJ, Walewski JL, Klepper AL, et al. Internal initiation stimulates production of p8 minicore, a member of a newly discovered family of hepatitis C virus core protein isoforms. J Virol. 2009;83:3104–3114. doi: 10.1128/JVI.01679-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frelin L, Brenndorfer ED, Ahlen G, et al. The hepatitis C virus and immune evasion: non-structural 3/4A transgenic mice are resistant to lethal tumour necrosis factor alpha mediated liver disease. Gut. 2006;55:1475–1483. doi: 10.1136/gut.2005.085050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Ponjavic J, Fredman D, et al. Evolutionary turnover of mammalian transcription start sites. Genome Res. 2006;16:713–722. doi: 10.1101/gr.5031006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanegae Y, Lee G, Sato Y, et al. Efficient gene activation in mammalian cells by using recombinant adenovirus expressing site-specific Cre recombinase. Nucleic Acids Res. 1995;23:3816–3821. doi: 10.1093/nar/23.19.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike K, Moriya K, Matsuura Y. Animal models for hepatitis C and related liver disease. Hepatol Res. 2010;40:69–82. doi: 10.1111/j.1872-034X.2009.00593.x. [DOI] [PubMed] [Google Scholar]
- Lemon SM, Lerat H, Weinman SA, et al. A transgenic mouse model of steatosis and hepatocellular carcinoma associated with chronic hepatitis C virus infection in humans. Trans Am Clin Climatol Assoc. 2000;111:146–156. discussion 156–147. [PMC free article] [PubMed] [Google Scholar]
- Lerat H, Honda M, Beard MR, et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology. 2002;122(2):352–365. doi: 10.1053/gast.2002.31001. [DOI] [PubMed] [Google Scholar]
- Majumder M, Ghosh AK, Steele R, et al. Hepatitis C virus NS5a protein impairs TNF-mediated hepatic apoptosis, but not by an anti-FAS antibody, in transgenic mice. Virology. 2002;294:94–105. doi: 10.1006/viro.2001.1309. [DOI] [PubMed] [Google Scholar]
- Mayer MG, Floeter-Winter LM. Pre-mRNA transsplicing: from kinetoplastids to mammals, an easy language for life diversity. Mem Inst Oswaldo Cruz. 2005;100:501–513. doi: 10.1590/s0074-02762005000500010. [DOI] [PubMed] [Google Scholar]
- McGivern DR, Lemon SM. Tumor suppressors, chromosomal instability, and hepatitis C virus-associated liver cancer. Annu Rev Pathol. 2009;4:399–415. doi: 10.1146/annurev.pathol.4.110807.092202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson CE, Shim EY, Friedman DS, et al. An active tissue-specific enhancer and bound transcription factors existing in a precisely positioned nucleosomal array. Cell. 1993;75:387–398. doi: 10.1016/0092-8674(93)80079-t. [DOI] [PubMed] [Google Scholar]
- Pasquinelli C, Shoenberger JM, Chung J, et al. Hepatitis C virus core and E2 protein expression in transgenic mice. Hepatology. 1997;25:719–727. doi: 10.1002/hep.510250338. [DOI] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- Scotto-Lavino E, Du G, Frohman MA. 3′ end cDNA amplification using classic RACE. Nat Protoc. 2006a;1:2742–2745. doi: 10.1038/nprot.2006.481. [DOI] [PubMed] [Google Scholar]
- Scotto-Lavino E, Du G, Frohman MA. 5′ end cDNA amplification using classic RACE. Nat Protoc. 2006b;1:2555–2562. doi: 10.1038/nprot.2006.480. [DOI] [PubMed] [Google Scholar]
- Smale ST. Core promoters: active contributors to combinatorial gene regulation. Genes Dev. 2001;15:2503–2508. doi: 10.1101/gad.937701. [DOI] [PubMed] [Google Scholar]
- Stover NA, Kaye MS, Cavalcanti AR. Spliced leader trans-splicing. Curr Biol. 2006;16:R8–R9. doi: 10.1016/j.cub.2005.12.019. [DOI] [PubMed] [Google Scholar]
- Sun JR, Bodola F, Fan XG, et al. Hepatitis C virus core and envelope proteins do not suppress the host's ability to clear a hepatic viral infection. J Virol. 2001;75:11992–11998. doi: 10.1128/JVI.75.24.11992-11998.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Tumurbaatar B, Jia J, et al. Parenchymal expression of CD86/b7.2 contributes to hepatitis C virus-related liver injury. J Virol. 2005;79:10730–10739. doi: 10.1128/JVI.79.16.10730-10739.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Grise H. Cellular and molecular biology of HCV infection and hepatitis. Clin Sci (Lond) 2009;117:49–65. doi: 10.1042/CS20080631. [DOI] [PubMed] [Google Scholar]
- Vassilaki N, Mavromara P. The HCV ARFP/F/core + 1 protein: production and functional analysis of an unconventional viral product. IUBMB Life. 2009;61:739–752. doi: 10.1002/iub.201. [DOI] [PubMed] [Google Scholar]
- Wakita T, Taya C, Katsume A, et al. Efficient conditional transgene expression in hepatitis C virus cDNA transgenic mice mediated by the Cre/loxP system. J Biol Chem. 1998;273:9001–9006. doi: 10.1074/jbc.273.15.9001. [DOI] [PubMed] [Google Scholar]
- Weber F. Interaction of hepatitis C virus with the type I interferon system. World J Gastroenterol. 2007;13:4818–4823. doi: 10.3748/wjg.v13.i36.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]