

Abstract
The aging-related decline in cardiac function is an important public health problem. The molecular basis of age-dependent loss of cardiac function is largely unknown and there are no effective therapies addressing this important form of heart disease. This study evaluates the role of the cytoskeletal protein dystrophin in the process of normal cardiac aging. Here, we show that the cytoskeletal protein dystrophin in the hearts of old mice is significantly decreased to a level of 36% that of young mice, whereas other key members of the dystrophin complex are unchanged. Age-dependent decreased ejection fraction was rescued by systemic delivery of an adeno-associated viral vector harboring a functional micro-dystrophin cassette (48.9 ± 2.5% in untreated aged vs. 61.6 ± 7.4% in treated aged mice, compared to 67.1 ± 2.6% in young mice). These data provide the first direct evidence that decreased dystrophin levels are an important modulator of cardiac function in the aged heart.
Introduction
The molecular processes of aging are complex and incompletely understood. Regardless of the mechanism, aging at the cell, tissue, organ and organism levels is associated with a decline in function. While all body systems are affected, the loss of heart function is of particular importance. Heart disease is the leading cause of death in the United States.1 Furthermore, as the demographics of the nation's populace shift toward the elderly, age-related heart disease is increasingly contributing to death and morbidity of millions of people. Understanding the basic processes and consequences of myocardial aging will facilitate the development of specific therapies designed to counteract the age-related decline in cardiac function. Compared to young hearts, old myocardium displays declines in both systolic and diastolic performance.2 These deficits are also evident in isolated cardiac myocytes.3 These functional losses undoubtedly result from disruption of a large number of processes within the cardiovascular system, including changes in the properties of the vasculature and the myocardium itself.4
In addition to cardiac senescence, or perhaps because if it, age is an independent risk factor for heart failure.1 Reductions in cardiac function observed in the senescent heart mirror those observed in failing myocardium.5 During the development of heart failure many aspects of myocardial biology are disturbed. Among these is the loss of dystrophin from cardiac myocyte membranes.6 Furthermore, the proteolytic degradation of dystrophin in the heart by enteroviruses is implicated in the pathophysiology of viral myocarditis.7 Dystrophin is a large cytoskeletal protein that forms a mechanical connection between the internal cytoskeleton and the extracellular matrix.8,9,10 This connection is mediated through interactions with a complex of glycoproteins that span the sarcolemmal membrane and bind directly to laminin.10 Dystrophin is thought to buffer the sarcolemmal membrane from the forces generated by the underlying sarcomeres and in the absence of dystrophin the sarcolemmal membrane becomes unstable.11
The global loss of dystrophin results in Duchenne muscular dystrophy (DMD), a progressive X-linked disease of skeletal and cardiac muscle that is uniformly fatal.9 Cardiac specific loss of dystrophin results in X-linked dilated cardiomyopathy,12 a disease characterized by a severe early onset heart failure. Female carriers of mutant dystrophin have approximately half the normal levels of dystrophin and are at increased risk of developing significant and progressive cardiac disease during their lifetimes.13,14,15 These genetic diseases demonstrate that the absence of dystrophin in the heart is sufficient to cause significant cardiac disease and failure.
The role of dystrophin loss in the pathophysiology of heart failure in aging is not clear. Loss of dystrophin is correlated with symptoms of heart failure, but its function in the pathophysiology of disease remains undefined. Functional similarities between senescent and failing hearts raise the possibility of shared molecular underpinnings. The present study provides the first direct evidence of the loss of dystrophin protein, but not closely associated dystrophin binding partners, in the senescent rodent heart. Importantly, evidence of causality is provided by reconstitution of dystrophin via gene delivery of a highly functional micro-dystrophin leading to improved cardiac hemodynamic performance in the aged heart.
Results
The loss of dystrophin accompanies many forms of acquired and inherited cardiac disease. To determine whether this pathway is evident in the senescent heart, dystrophin levels were determined in microsomal membranes isolated from both young and aged hearts by quantitative western blotting. Full-length dystrophin protein was significantly reduced in the myocardium of old animals to a value of 36% of that observed in young animals (P < 0.0001) (Figure 1). There was no evidence of degradation products of dystrophin, suggesting that the protein is either completely degraded or being produced at a lower rate than in young myocytes. In old myocytes, the residual dystrophin remained properly localized to the membrane (Figure 1c). This loss of dystrophin appears to be selective as levels of the closely associated α- and β-dystroglycan were maintained in the aged heart (Figure 2).
Figure 1.

Quantification of dystrophin levels in membrane preparations from young and old mouse hearts. (a) Representative blot demonstrating dystrophin (C-terminal) and Na-K ATPase expression. (b) Summary of blot quantification, dystrophin levels from old hearts are 36% that of young hearts. Data is from 9 young hearts and 17 old hearts. (c) Immunofluorescent assessment of dystrophin subcellular localization in young versus old hearts. Bar represents 100µm, *Indicates a significant difference of P < 0.0001 by t-test.
Figure 2.

Western blotting following administration of AAV-µ-dystrophin (AAV-µ-Dys). Representative blot demonstrating the expression of both (a) full-length dystrophin (C-terminal directed antibody) and (b) truncated dystrophin (N-terminal directed antibody). The dystrophin associated (c) α-dystroglycan (α-DG) and (d) β-dystroglycan (β-DG) are over expressed in samples from mice receiving AAV-µ-Dys. (e,f) Quantification of (e) α-DG and (f) β-DG levels, data from 5 to 13 hearts. *Indicates significant difference from young samples (P < 0.05, one-way analysis of variance, Dunnett's post-test).
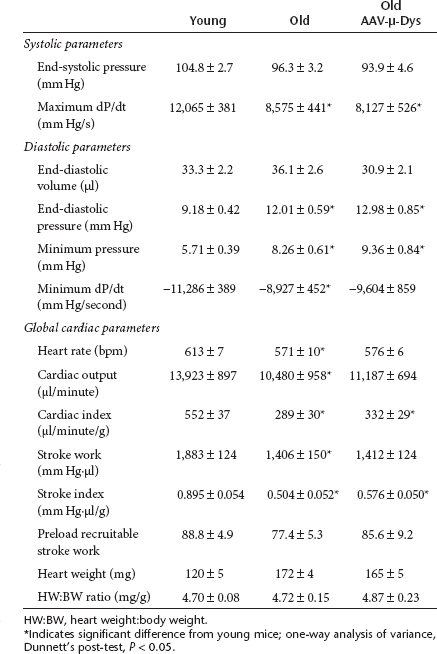
Assessment of cardiac function revealed significant elevations in left ventricular end-diastolic pressures in aged mice. This diastolic dysfunction was accompanied by significant reductions in heart rate and in measures of systolic function (Figure 3; Table 1). The maximum rate of pressure development, stroke work index, cardiac index, and ejection fraction were all significantly depressed in aged mice (Table 1). All of these parameters could potentially be affected by a reduction in dystrophin, which places a large metabolic load on already poorly functioning cardiac myocytes, by destabilizing the cellular membranes.11
Figure 3.

Hemodynamic assessment of old mice ten weeks after administration of AAV-µ-dystrophin (AAV-µ-Dys). (a) Representative pressure-volume loops from injected (Old µ-Dys, gray) and uninjected (Old, black). (b) Summary of the end-systolic volumes and (c) ejection fraction derived from pressure-volume analysis of cardiac function. Data from 6 to 30 mice, *Indicates significant difference from young (P < 0.05, one-way analysis of variance, Dunnett's post-test).
Table 1. Summary of hemodynamics derived from young mice, old mice, and old mice treated with AAV-µ-dystrophin (AAV-µ-Dys).
To test for the first time whether dystrophin deficiency has a direct effect depressing hemodynamic performance in the aging heart, we utilized a systemic in vivo gene transfer approach to replace the missing dystrophin with a highly functional truncated form of dystrophin; µ-dystrophin (µ-Dys).13 Ten weeks following intravenous administration of AAV-µ-Dys to 23-month-old mice, heart muscle membrane composition was assessed. Microsomal membranes from these injected mice revealed the expression of the truncated dystrophin (MW ≈137 kD; Figure 2b). Compared to age matched untreated mice, there was no significant difference in the amount of full-length dystrophin present in the AAV-µ-Dys treated hearts. Results showed significant increases in α-sarcoglycan and α- and β-dystroglycan, the central components of the dystrophin associated glycoprotein complex, suggesting that the total dystrophin level (full length plus µ-Dys) following AAV-µ-Dys administration is increased (Figure 2c–f and Supplementary Figure S1).
Real-time hemodynamic assessment of old mice receiving AAV-µ-Dys was performed utilizing pressure-volume loop analysis (Table 1). The age-dependent decrease in ejection fraction (young mice versus untreated old mice; P < 0.0001) was corrected by µ-Dys gene delivery in old mice Figure 3. Ejection fraction, a measure of global heart function, is influenced by both vascular function and myocardial contractility. It is likely that these effects are mediated primarily by changes within the cardiac myocytes, as AAV shows particular tropism for this tissue.13,14
Discussion
This study demonstrates for the first time that levels of dystrophin protein in the aged heart are significantly lower in comparison to the myocardium of young mice. The correlation of decreased dystrophin expression and reduced cardiac function in the aged heart does not, in itself, demonstrate cause and effect. Accordingly, a main finding of this study is demonstrating a significant improvement in ejection fraction in old mice following the cardiac expression of a micro-dystrophin molecule by systemic gene delivery. Considering that gene delivery was initiated in old animals (23 months) that already had age-related cardiac dysfunction,2 we take the micro-dystrophin functional improvement as evidence of a reversal, at least in part, of age-dependent hemodynamic deficits. The results of this gene replacement experiment further indicate that the loss of dystrophin is at least one of the key components of age-related cardiac dysfunction.
We found that dystrophin content in the hearts of old mice is reduced to nearly one third that observed in young controls. A partial loss in dystrophin is also present in female carriers of mutated dystrophin alleles. Female carriers of Duchenne muscular dystrophy mutations have a greater risk of developing severe cardiac disease during their lifetime.13,14,15 In mice, mdx carriers have a 50% reduction in dystrophin content.15 Despite the similar reductions in total dystrophin observed here, the distribution of dystrophin is quite different in mdx carriers and aged mouse hearts. Carriers of mutated dystrophin alleles demonstrate a mosaic pattern of dystrophin expression, due to X-chromosomal inactivation. In mice, this results in a heart with about half of the myocytes with no dystrophin and the other half with a full complement of dystrophin.15 In contrast, the reduction of dystrophin protein with aging is characterized by an apparent uniform dystrophin staining throughout the myocardium, suggesting that all of the cardiac myocytes have reduced dystrophin content. We hypothesize that this global reduction in myocyte dystrophin content results in all cells having increased susceptibility to membrane damage.
In the absence of dystrophin, the sarcolemma of cardiac myocytes is highly susceptible to mechanical stress leading to membrane instability. It is hypothesized transient micro-tears in the sarcolemma during stress allows, among other things, calcium to traverse the membrane.11 We propose that the partial loss of dystrophin in the aged heart causes a degree of membrane instability paralleling what is observed in the completely dystrophin deficient heart. In the aged heart, we propose that calcium overload/metabolic stress resulting from membrane instability causes dysfunction in both systolic and diastolic myocardial performance. This process, in conjunction with other aging-related deficits in the myocardium,16,17,18,19 results in the poor contractile function observed in the senescent heart. We further propose that the expression of µ-Dys in old myocardium works in tandem with residual full-length endogenous dystrophin to reinforce the connection between the internal cytoskeletal network and the membrane by increasing the expression of dystrophin associated glycoprotein complex. The increased frequency of contacts between the cytoskeleton and the extracellular matrix would be expected to improve membrane stability, thus reducing the metabolic stress placed on the senescent myocyte, allowing it to regain calcium homoeostasis and improve contractile function.
Micro-dystrophin expression in old hearts did not fully normalize hemodynamic performance to that found in young mice, specifically diastolic function remains poor in old mice with or without µ-dystrophin treatment. This is likely due to the combination of the multiple processes involved in age-related decline in cardiac function, the noted decreases in function between micro- and full-length dystrophin, and the relatively short duration of treatment.14,20 This report represents the first evidence that reductions in dystrophin content significantly contribute to myocardial senescence. The mechanism underlying this loss of dystrophin is unknown. Previous studies have shown that dystrophin transcript levels are reduced suggesting a transcriptional mechanism,21 although increased degradation of the dystrophin protein cannot be ruled out. This study also demonstrates the effectiveness of gene transfer utilizing adeno-associated viral (AAV) vectors in old animals. Previous studies have used adenoviral vectors to produce short-term (<1 week) gene transfer in old rats.22,23,24,25 In comparison, the current study utilizing AAV, the exogenous µ-Dys gene product was expressed for 10 weeks. The prolonged expression provided by AAV was likely an important factor contributing to the recovery of cardiac function observed in this study.
The restoration of dystrophin expression in the aged myocardium is a new molecular intervention that restores, at least in part, systolic function of the senescent heart. These results suggest that stabilizing dystrophin levels is important for maintenance of cardiac function during aging. This can be done either by reintroducing dystrophin, as done in this study, or by preventing the initial loss of dystrophin. While the process of cardiac aging is complex, and it is unlikely that any single process will completely restore heart function, these results underscore the importance of dystrophin to cardiac function and its therapeutic potential.
Materials and Methods
Animals. Mice of both sexes were either obtained from Jackson Labs (Bar Harbor, ME) and housed locally or obtained through National Institute of Aging aged mouse colony. The genetic background of the mice was either C57BL/6 or C57BL/10. Previous studies have found no significant hemodynamic differences between these two closely related mouse strains.26 Mice were grouped into young (10 C57BL/6 and 20 C57BL/10) and old (17 C57BL/6 and 17 C57BL/10), with average ages of 4.3 ± 0.1 months and 22.9 ± 0.5 months, respectively.
Virus production and delivery. AAV vectors pseudotyped with the AAV6 capsid and carrying a truncated dystrophin construct (µ-Dys) were generated and purified using methods previously described.13,14,27 Briefly, HEK293 cells were transfected with genomic (pAAV-CMV-µDys) and packaging plasmids.13 Pseudotyped AAV6 viruses were purified via heparin affinity column. Twenty three-month-old mice were anesthetized and injected intravenously with 1E12 vector genomes in a final volume of 300 µl. A cohort of control 23-month-old animals was injected with the same volume of saline. The hemodynamic properties of these saline injected mice were not significantly different from noninjected aged mice, so these data were merged into a single control group. Ten weeks following the injection catheter based hemodynamic measurements were performed.
Western blotting. Crude microsomal preparations were isolated from hearts were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and western blot analysis as outlined previously.14 The antibodies utilized in this study were anti-dystrophin (C-terminal, 1:1000; Abcam, Cambridge, MA), anti-human dystrophin (N-terminal, 1:100; DY10 Vector Labs, Burlingame, CA), anti-α-dystroglycan (1:1,000, IIH6C4; Millipore, Billerica, MA), and anti-Na-K ATPase (1:10,000, α-F6 from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA). Secondary antibodies labeled with infra-red fluorophores were used to quantify protein levels using an Odyssey scanner (Li-Cor, Lincoln, NE). The intensity of dystrophin was normalized to the Na-K ATPase to control for differences in loading. To control for inter blot variability, samples were further normalized to dystrophin levels from young on the same blot.
Immunofluorescence. Hearts were flash frozen in OTC and 7 µm sections were cut on a cryostat. Sections were washed with phosphate-buffered saline to remove residual OTC (Tissue-Tek, Torrence, CA) and subjected to standard immunofluorescent methodologies. Dystrophin was detected utilizing a polyclonal antibody from Abcam (1:100).
Hemodynamics. Mice were induced and maintained at a surgical plane of anesthesia with isoflurane delivered in 100% oxygen. Mice were ventilated and a thoracotomy performed to expose the heart. A 1.4 Fr Millar PV catheter (Millar Instruments, Houston, TX) was inserted through an apical stab into the left ventricular cavity. Absolute volumes were calibrated utilizing a hypertonic saline injection and a Lucite cuvette. Data were collected and analyzed by a Ponemah acquisition device and the LV Pressure and Volume analysis modules (DSI, St Paul, MN). Procedures performed in this study were reviewed and approved by the institutional animal care and use committee.
SUPPLEMENTARY MATERIAL Figure S1. Western blotting following administration of AAV-µ-dystrophin shows markedly increased expression in α-sarcoglycan levels compared to untreated aged mouse hearts. Data from 6-8 hearts. *Indicates a significant difference of P<0.001 by T-test.
Acknowledgments
This work was supported by grants from National Institutes of Health (J.M.M, J.S.C, D.T.), the Lillehei Heart Institute (M.D.) and the Muscular Dystrophy Association (D.T.).
Supplementary Material
Western blotting following administration of AAV-µ-dystrophin shows markedly increased expression in α-sarcoglycan levels compared to untreated aged mouse hearts. Data from 6-8 hearts. *Indicates a significant difference of P<0.001 by T-test.
REFERENCES
- Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G.et al. (2009Heart disease and stroke statistics--2010 update: a report from the American Heart Association Circulation 121e46–e215. [DOI] [PubMed] [Google Scholar]
- Yang B, Larson DF., and, Watson R. Age-related left ventricular function in the mouse: analysis based on in vivo pressure-volume relationships. Am J Physiol. 1999;277 5 Pt 2:H1906–H1913. doi: 10.1152/ajpheart.1999.277.5.H1906. [DOI] [PubMed] [Google Scholar]
- Howlett SE, Grandy SA., and, Ferrier GR. Calcium spark properties in ventricular myocytes are altered in aged mice. Am J Physiol Heart Circ Physiol. 2006;290:H1566–H1574. doi: 10.1152/ajpheart.00686.2005. [DOI] [PubMed] [Google Scholar]
- Lakatta EG., and, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation. 2003;107:346–354. doi: 10.1161/01.cir.0000048893.62841.f7. [DOI] [PubMed] [Google Scholar]
- Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part III: cellular and molecular clues to heart and arterial aging. Circulation. 2003;107:490–497. doi: 10.1161/01.cir.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]
- Vatta M, Stetson SJ, Perez-Verdia A, Entman ML, Noon GP, Torre-Amione G.et al. (2002Molecular remodelling of dystrophin in patients with end-stage cardiomyopathies and reversal in patients on assistance-device therapy Lancet 359936–941. [DOI] [PubMed] [Google Scholar]
- Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE.et al. (1999Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy Nat Med 5320–326. [DOI] [PubMed] [Google Scholar]
- Abmayr S., and, Chamberlain J.2006The Structure and Function of Dystrophin Winder SJ.ed). Molecular Mechanisms of Muscular Dystrophies Landes Bioscience: Georgetown, TX.14–34. [Google Scholar]
- Koenig M, Monaco AP., and, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–228. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- Ervasti JM., and, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda S, Townsend D, Michele DE, Favre EG, Day SM., and, Metzger JM. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature. 2005;436:1025–1029. doi: 10.1038/nature03844. [DOI] [PubMed] [Google Scholar]
- Towbin JA, Hejtmancik JF, Brink P, Gelb B, Zhu XM, Chamberlain JS.et al. (1993X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus Circulation 871854–1865. [DOI] [PubMed] [Google Scholar]
- Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG.et al. (2004Systemic delivery of genes to striated muscles using adeno-associated viral vectors Nat Med 10828–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend D, Blankinship MJ, Allen JM, Gregorevic P, Chamberlain JS., and, Metzger JM. Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine-induced cardiac pump failure. Mol Ther. 2007;15:1086–1092. doi: 10.1038/sj.mt.6300144. [DOI] [PubMed] [Google Scholar]
- Bostick B, Yue Y, Long C., and, Duan D. Prevention of dystrophin-deficient cardiomyopathy in twenty-one-month-old carrier mice by mosaic dystrophin expression or complementary dystrophin/utrophin expression. Circ Res. 2008;102:121–130. doi: 10.1161/CIRCRESAHA.107.162982. [DOI] [PubMed] [Google Scholar]
- Kakarla SK, Rice KM, Katta A, Paturi S, Wu M, Kolli M.et al. (2010Possible molecular mechanisms underlying age-related cardiomyocyte apoptosis in the F344XBN rat heart J Gerontol A Biol Sci Med Sci 65147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitahara JA, Cheng W, Liu Y, Li B, Leri A, Li P.et al. (1998Intracellular calcium, DNase activity and myocyte apoptosis in aging Fischer 344 rats J Mol Cell Cardiol 30519–535. [DOI] [PubMed] [Google Scholar]
- Matsuzaki S., and, Szweda LI. Inhibition of complex I by Ca2+ reduces electron transport activity and the rate of superoxide anion production in cardiac submitochondrial particles. Biochemistry. 2007;46:1350–1357. doi: 10.1021/bi0617916. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Ocorr K, Bodmer R., and, Cartry J. Drosophila as a model to study cardiac aging. Exp Gerontol. 2011;46:326–330. doi: 10.1016/j.exger.2010.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF.et al. (2002Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy Nat Med 8253–261. [DOI] [PubMed] [Google Scholar]
- Bodyak N, Kang PM, Hiromura M, Sulijoadikusumo I, Horikoshi N, Khrapko K.et al. (2002Gene expression profiling of the aging mouse cardiac myocytes Nucleic Acids Res 303788–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huq F, Lebeche D, Iyer V, Liao R., and, Hajjar RJ. Gene transfer of parvalbumin improves diastolic dysfunction in senescent myocytes. Circulation. 2004;109:2780–2785. doi: 10.1161/01.CIR.0000131764.62242.96. [DOI] [PubMed] [Google Scholar]
- Michele DE, Szatkowski ML, Albayya FP., and, Metzger JM. Parvalbumin gene delivery improves diastolic function in the aged myocardium in vivo. Mol Ther. 2004;10:399–403. doi: 10.1016/j.ymthe.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Schmidt U, del Monte F, Miyamoto MI, Matsui T, Gwathmey JK, Rosenzweig A.et al. (2000Restoration of diastolic function in senescent rat hearts through adenoviral gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase Circulation 101790–796. [DOI] [PubMed] [Google Scholar]
- Schmidt U, Zhu X, Lebeche D, Huq F, Guerrero JL., and, Hajjar RJ. In vivo gene transfer of parvalbumin improves diastolic function in aged rat hearts. Cardiovasc Res. 2005;66:318–323. doi: 10.1016/j.cardiores.2004.06.028. [DOI] [PubMed] [Google Scholar]
- Barnabei MS, Palpant NJ., and, Metzger JM. Influence of genetic background on ex vivo and in vivo cardiac function in several commonly used inbred mouse strains. Physiol Genomics. 2010;42A:103–113. doi: 10.1152/physiolgenomics.00071.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankinship MJ, Gregorevic P, Allen JM, Harper SQ, Harper H, Halbert CL.et al. (2004Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6 Mol Ther 10671–678. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Western blotting following administration of AAV-µ-dystrophin shows markedly increased expression in α-sarcoglycan levels compared to untreated aged mouse hearts. Data from 6-8 hearts. *Indicates a significant difference of P<0.001 by T-test.